
Abstract
In this study, we reported the ammonium metavanadate (NH4VO3) as an efficient, cost-effective, and mild catalyst for the synthesis of substituted pyridines via a one-pot pseudo four-component reaction. Furthermore, we investigated Hantzsch 1,4-dihydropyridines (1,4-DHPs) synthesis and oxidation of 1,4-DHPs to their corresponding pyridines. The present approach offers a rapid methodology for accessing various pyridines with broad functional group tolerance and good yields using NH4VO3 catalyst as a green catalyst.
Similar content being viewed by others

Introduction
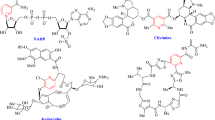
For several decades nitrogen-containing six-membered heterocyclic compounds have attracted the interest of synthetic organic chemists due to their pharmaceutical and biological properties. Among the nitrogen heterocycles, pyridine derivatives are well known as calcium channel blockers and exhibit therapeutic effects, such as vasodilator, bronchodilator, geroprotective, hepatoprotective, neuroprotective, and anti-tumor activity1,2,3,4. For example, there are many pharmaceutical pyridine compounds (Fig. 1) such as (A) and (B), as selective modulators of human adenosine receptors implicated in asthma, Parkinson’s disease, epilepsy, kidney disease, and cancer, as well as cerivastatin (C) for the treatment of atherosclerosis and other coronary diseases5,6,7,8. Pyridine derivatives are not only privileged scaffolds for drug discovery but also used as building blocks reagents in organic synthesis and ligands in coordination chemistry9. Due to their importance, the development of novel synthetic methods for the preparation of pyridine derivatives is of interest10,11.

Substituted pyridines as privileged structures.
The traditional so-called Hantzsch synthesis of 1,4-DHPs includes one-pot cyclocondensation of a β-ketoester with an aldehyde and a nitrogen source, which occurs either in acetic acid at room temperature or by refluxing in alcohols; this protocol has some drawbacks such as prolonged reaction times and low yields12. Therefore, numerous modifications have been made to the original Hantzsch reaction, such as using microwave radiation13,14, ionic liquid15, SiO2/NaHSO416, metal triflates17, I218, ceric ammonium nitrate (CAN)19 and ZnO20.
Recently, the oxidation of 1,4-DHPs was successfully carried out by using various oxidants, such as peroxydisulfate-Co(II)21, silica-modified sulfuric acid/NaNO222, Co-naphthenate23, KBrO3/SnCl4.5H2O24, MnO225, silica chromate26, urea- hydrogen peroxide catalyzed by molecular iodine27, b-cyclodextrin28, silica-sulfuric acid and Al(NO3)3·9H2O or Fe(NO3)3·9H2O29.
In recent years, the application of the bifunctional solid acid/ noble metal Pd/C/K-10 catalyst was reported for the one-pot synthesis of pyridine derivatives30,31. In addition, Khaskel and Barman reported the one-pot synthesis of pyridines in ethanol by benzyltrimethylammoniumfluoride hydrate (BTMAFH) and K2S2O832. Ghosh et al. reported the direct synthesis of pyridine derivatives using visible light in aqueous media catalyzed by non-ionic surfactant Triton-X-10033. Although, many of the reported methods for synthesis of pyridine derivatives offer distinct benefits, some of them still have some drawbacks, such as long reaction times, expensive reagents, harsh conditions, low product yields, tedious work-up, and by-products formation.
Hence, the development of a new procedure for the one-pot synthesis of pyridine derivatives would be highly desirable. Recently, NH4VO3 has been utilized as an inorganic acid and economical catalyst in organic synthesis34,35,36. Furthermore, to the best of our knowledge the use of NH4VO3 in the synthesis of pyridine derivatives has been never reported before. In continuation of our previous works on the introduction of new catalysts in organic synthesis37,38,39,40,41,42,43, herein, we report the use of NH4VO3 without any post-modification as an efficient, inexpensive, and eco-friendly catalyst for the synthesis of substituted pyridines via one-pot pseudo four-component reaction, including a combination of the Hantzsch synthesis and the subsequent oxidation step for the first time (Fig. 2).

One-pot synthesis of pyridines, 1,4-DHPs, and the oxidation aromatization of 1,4-DHPs to the corresponding pyridines.
Experimental
General
All solvents, chemicals, and reagents were purchased from Merck, Fluka, and Sigma-Aldrich chemical companies. Melting points were measured with an Electrothermal 9100 apparatus and are uncorrected. FT-IR spectra were obtained over 400–4000 cm−1 with a Shimadzu IR-470 spectrometer using KBr pellets. 1H-NMR and 13C-NMR spectra were recorded by a Bruker Avance DRX500 spectrometer. All the synthesized products were known, and the structure of the isolated products was confirmed by previously reported data.
General procedure for one-pot synthesis of pyridines
A mixture of an aldehyde 1 (1.0 mmol), ammonium acetate 2 (2.0 mmol), ethyl acetoacetate 3 (2.0 mmol), and ammonium metavanadate (NH4VO3) (117.0 mg) in 3.0 mL acetic acid was stirred under reflux condition for the appropriate time (Table 7). After completion of the reaction, as indicated by thin-layer chromatography (TLC), the catalyst (NH4VO3) was separated by filtration. Then, products afforded by evaporation of the solvent, and recrystallized from diethyl ether to give the pure desired pyridines (5).
General procedure for preparation of 1,4-DHPs
A mixture of an aldehyde 1 (1.0 mmol), ammonium acetate 2 (2.0 mmol), ethyl acetoacetate 3 (2.0 mmol), and ammonium metavanadate (NH4VO3) (15.0 mg) in 3.0 mL ethanol was stirred under reflux condition for the appropriate time (Table 2). After completion of the reaction, as indicated by thin-layer chromatography (TLC), the catalyst (NH4VO3) was separated by filtration, washed with ethanol, and reused five times in other fresh reactions without a considerable loss of activity. Then, products (4) are afforded by evaporation of the solvent, followed by recrystallization from ethanol.
General procedure for oxidative aromatization of 1,4-DHPs
To a solution of 1,4-DHPs 4 (1.0 mmol) in 3.0 mL of acetic acid, ammonium metavanadate (NH4VO3) (117.0 mg) was added. The resulting mixture was refluxed for an appropriate time (Table 5). After completion of the reaction (monitored by TLC), the mixture was cooled to room temperature and the catalyst was filtered off. Then the filtrate was evaporated and recrystallized from diethyl ether to give the pure desired pyridines (5).
Spectral data
Diethyl 4-(4-methoxyphenyl)-2,6-dimethyl-3,5-pyridinedicarboxylate (5d): FT-IR (KBr: υ/cm−1): 2985, 2929, 1724, 1558, 1510, 1488, 1294, 1232, 1107, 1045, 860, 792; 1H NMR (500 MHz, CDCl3): δH (ppm) = 1.08 (t, 6H, J = 7.1 Hz, CH3), 2.68 (s, 6H, CH3), 3.92(s, 3H, OCH3) 4.14 (q, 4H, J = 7.1 Hz, CH2), 6.99 (d, 2H, J = 8.7 Hz, H-Ar), 7.29 (d, 2H, J = 8.7 Hz, H-Ar).
Diethyl 4-(4-bromophenyl)-2,6-dimethyl-3,5-pyridinedicarboxylate (5e): FT-IR (KBr: υ/cm−1): 2981, 2931, 1726, 1556, 1488, 1446, 1292, 1232, 1211, 1103, 1043, 860, 829 ; 1H NMR (500 MHz, CDCl3): δH (ppm) = 0.97 (t, 6H, J = 7.1 Hz, CH3), 2.58 (s, 6H, CH3), 4.03 (q, 4H, J = 7.1 Hz, CH2), 7.12 (d, 2H, J = 8.4 Hz, H-Ar), 7.50 (d, 2H, J = 8. 4 Hz, H-Ar).
Diethyl 4-(4-chlorophenyl)-2,6-dimethyl-3,5-pyridinedicarboxylate (5f.): FT-IR (KBr: υ/cm−1): 2983, 1724, 1554, 1292, 1232, 1097, 1043, 860, 665; 1H NMR (500 MHz, DMSO): δH (ppm) = 0.97 (t, 6H, J = 7.1 Hz, CH3), 2.59 (s, 6H, CH3), 4.10 (q, 4H, J = 7.1 Hz, CH2), 7.27 (d, 2H, J = 8. 4 Hz, H-Ar), 7.61 (d, 2H, J = 8. 7 Hz, H-Ar).
Diethyl 2,6-dimethyl-4-(thiophen-2-yl)pyridine-3,5-dicarboxylate (5 m): FT-IR (KBr: υ/cm−1): 2981, 2933, 1728, 1558, 1444, 1288, 1234, 1099, 1041, 860, 705; 1H NMR (500 MHz, CDCl3): δH (ppm) = 1.17 (t, 6H, J = 7.1 Hz, CH3), 2.68 (s, 6H, CH3), 4.23 (q, 4H, J = 7.1 Hz, CH2), 7.15 (bs, 2H, H-Ar), 7.50 (bs, 1H, H-Ar).
4-(4-methoxy-phenyl)-2,6-dimethyl-1,4-dihydro-pyridine-3,5-dicarboxylic acid diethyl ester (4d): FT-IR (KBr: υ/cm−1): 682, 838, 1026, 1209, 1496, 1650, 1689, 2974, 3340; 1H NMR (500 MHz, CDCl3): δH (ppm) = 7.31 (d, 2H, J = 8.5 Hz, H-Ar), 1.33 (t, 6H, J = 7.1 Hz, CH3), 6.86 (d, 2H, J = 8.5 Hz, H-Ar), 6.01 (s, 1H, NH), 5.04 (s, 1H, CH), 4.20 (m, 4H, CH2), 3.86 (s, 3H, OCH3), 2.41 (s, 6H, CH3).
4-(4-bromo-phenyl)-2,6-dimethyl-1,4-dihydro-pyridine-3,5-dicarboxylic acid diethyl ester (4e): FT-IR (KBr: υ/cm−1): 780, 1012, 1217, 1377, 1488, 1652, 1693, 2989, 3357; 1H NMR (500 MHz, DMSO): δH (ppm) = 8.92 (s, 1H, NH), 7.22–7.32 (m, 4H, H-Ar), 4.90 (s, 1H, CH), 4.90 (s, 1H, CH), 4.16–4.24 (m, 4H, CH2, broad), 2.32 (m, 6H, CH3, broad), 1.18 (s, 6H, CH3); 13C NMR (125 MHz, DMSO): δC (ppm) = 166.8, 147.1, 145.6, 130.4, 129.2, 127.8, 101.5, 59.0, 38,5, 18.2, 14.1.
4-(4-chloro-phenyl)-2,6-dimethyl-1,4-dihydro-pyridine-3,5-dicarboxylic acid diethyl ester (4f.): FT-IR (KBr: υ/cm−1): 1213, 1371, 1487, 1652, 1695, 3357; 1H NMR (500 MHz, DMSO): δH (ppm) = 8.92 (s, 1H, NH), 7.22–7.32 (m, 4H, H-Ar), 4.90 (s, 1H, CH), 4.04 (m, 4H, CH2, broad), 2.32 (s, 6H, CH3), 1.18 (s, 6H, CH3); 13C NMR (125 MHz, DMSO): δC (ppm) = 166.8, 14.1, 147.1, 145.6, 130.4, 129.2, 127.8, 101.5, 59.0, 38,5, 18.2.
2,6-dimethyl-4-(3-nitro-phenyl)-1,4-dihydro-pyridine-3,5-dicarboxylic acid diethyl ester (4j): FT-IR (KBr: υ/cm−1): 1118, 1213, 1348, 1487, 1647, 1704, 2987, 3346; 1H NMR (500 MHz, DMSO): δH (ppm) = 8.94 (s, 1H, NH), 7.47 (d, 2H, J = 8. 4 Hz, H-Ar), 7.17 (d, 2H, J = 8. 4 Hz, H-Ar), 4.90 (s, 1H, CH), 4.01–4.11 (m, 4H, CH2), 2.33 (s, 6H, CH3), 1.20 (t, 6H, J = 7.1 Hz, CH3).
2,6-dimethyl-4-(thiophen-2-yl)-1,4-dihydro-pyridine-3,5-dicarboxylic acid diethyl ester (4 m): FT-IR (KBr: υ/cm−1): 719, 1124, 1209, 1299, 1371, 1487, 1652, 1695, 2985, 3344; 1H NMR (500 MHz, CDCl3): δH (ppm) = 7.47 (dd, 1H, J = 1.2 Hz, J = 3.9 Hz, H-Ar), 6.90–6.97 (m, 2H), 5.97 (s, 1H, NH), 5.46 (s, 1H, CH), 4.25–4.32 (m, 4H, CH2), 2.45 (s, 6H, CH3), 1.38 (t, 6H, J = 7.1 Hz, CH3).
Results and discussion
Regarding the fact that the one-pot approach to the synthesis of substituted pyridines through Hantzsch synthesis is hardly carried out and there are only a few literatures reported in this field. Hence, the efficiency of ammonium metavanadate (NH4VO3) was investigated in the one-pot synthesis of pyridine derivatives. In an initial attempt, the condensation of 4-chlorobenaldehyde (1.0 mmol) with ethyl acetoacetate (2.0 mmol) and ammonium acetate (2.0 mmol) as a model reaction (Fig. 3) was examined in the presence of different catalytic amounts of NH4VO3 in acetic acid for the one-pot synthesis of pyridine derivatives. Surprisingly, when NH4VO3 was used as the catalyst in acetic acid under reflux conditions, the reaction went to completion in 10 min and 96% of the pyridine (product 8f.) was isolated as the desired product.

One-pot synthesis of pyridine derivatives.
To optimize the amount of catalyst and reaction conditions for the one-pot synthesis of pyridines, the model reaction was examined in acetic acid (Table 1). As shown in Table 1, the best results were achieved when the reaction was carried out in the presence of 117.0 mg of NH4VO3 as the catalyst in acetic acid under reflux conditions (entry 1, Table 1). Increasing the amount of catalyst (117.0–120.0 mg) did not improve the yield of the desired product (entries 1–5, Table 1). In the absence of NH4VO3 catalyst, the reaction was not successful (entry 11, Table 1).
After optimizing the reaction conditions, to explore the scope of the reaction, a series of pyridine derivatives were synthesized by various aldehydes including both electron-donating and electron-withdrawing substituents (Table 7). All the aldehydes with both electron-withdrawing groups and electron-donating groups reacted very well, giving high yields of the desired products in short reaction times. Based on the results, we propose a plausible mechanism for the one-pot synthesis of pyridines (Fig. 4). This mechanistic pathway includes a combination of the Hantzsch synthesis and the subsequent oxidation step. First, the ammonium (NH4+) group in the structure of NH4VO3 activates the carbonyl functional groups of aldehyde and ethyl acetoacetate by hydrogen bonding. Therefore, it increases the carbonyl activity to Knoevenagel condensation with enol form of ethyl acetoacetate to give the corresponding Knoevenagel intermediate (I). In the next step, the reaction of the second molecule of ethyl acetoacetate with ammonium acetate gives the imine intermediate (II). The Michael addition of I with enamine form of II occurs to form intermediate III, which is activated through hydrogen bonding from NH4VO3 to facilitate cyclization and elimination of water, affording the desired 1,4-DHP derivatives. In continue, acetic acid using NH4VO3 as a catalyst is converted into acetate ion which is leading to an acid–base reaction with 1,4-DHPs. In the following, the negative charge of nitrogen of intermediate (IV) binds with the vacant “d” orbital of transition metal vanadium to achieve the stable oxidation state of vanadium. The last step might be progressed through unusual hydride transfer and H2 releasing from (V). For proving this opinion, the reaction was evaluated under a nitrogen atmosphere (entry 2, Table 1). The results show that the oxidation reaction progressed in an atmosphere of nitrogen similar to the air or oxygen atmosphere condition (entries 1–3, Table 1). Due to electron-donating from the nitrogen lone pairs into the anti-bonding orbital of C–H (s*C–H orbital), the C–H bond is easily broken by reaction with a proton to afford molecular hydrogen. This phenomenon has been known as the anomeric effect.

Proposed mechanism for the one-pot synthesis of pyridines by NH4VO3.
Although there are a few literatures that reported on the direct approach for the one-pot synthesis of pyridines, this method is superior to the earlier methods in terms of yields, reaction time, and mild reaction conditions (Table 2).
To further confirm the possible mechanism, we also examined the efficiency of NH4VO3 as a catalyst for the one-pot synthesis of 1,4-DHPs. To optimize the reaction conditions. The condensation of 4-chlorobenaldehyde (1.0 mmol) with ethyl acetoacetate (2.0 mmol) and ammonium acetate (2.0 mmol) as a model reaction (Fig. 5) was chosen and the effect of different catalytic amounts of NH4VO3 in a wide variety of solvents and under reflux condition were investigated (Table 3).

Hantzsch synthesis of 1,4-DHPs catalyzed by NH4VO3.
In the absence of NH4VO3 as the catalyst, the reaction proceeded slowly with a low yield (entry 16, Table 3). As seen in Table 3 (entries 7–12) using 15.0–23.0 mg of the catalyst (NH4VO3) showed higher activity for the synthesis of 1,4-DHPs. However, when the amount of catalyst increased to 18.0–23.0 mg (entries 10–12, Table 3) the yield of the desired product (93%) did not improve. Among the investigated solvents, ethanol is the best choice with its short reaction time, high yield, cheapness, and being environmentally friendly for this reaction. According to the results in Tables (1,3), it is obvious that in the absence of acetic acid and using other solvents the 1,4-DHPs form as the desired products. After optimizing the reaction conditions, the effect of substitution on the aldehydes has also been studied. As shown in Table 7 all the aromatic aldehydes with both electron-withdrawing groups and electron-donating groups reacted very well, giving high yields of the desired products. As expected substituted aldehydes with electron-withdrawing groups require a shorter reaction time in comparison to those with electron-donating groups.
Moreover, the catalytic activity of the NH4VO3 for the synthesis of 1,4-DHPs was compared to the other reported catalysts in Table 4.
We also extended our study to the oxidation of the synthesized 1,4-DHPs. Compound 4f. (diethyl 4-(4-chloro phenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate) was used as a model substrate to optimize the oxidation reaction conditions (Fig. 6).

Oxidation of 1,4-DHPs by using NH4VO3.
As revealed in Table 5 (entries 1–8), the nature of the solvent is an important factor in the oxidation of 1,4-DHPs to the corresponding pyridines. The effect of the solvent in the oxidation reaction, in dichloromethane, ethanol, chloroform, H2O, acetonitrile, formic acid, and tetrahydrofuran was investigated; no oxidation occurred in these solvents. While by addition of acetic acid as the solvent to the reaction mixture, the yield of the desired product reached 96% under reflux conditions (entry 8, Table 5), this observation suggests that acetic acid is essential for the oxidation reaction. Additionally, the model substrate converts into the corresponding pyridine in acetic acid at room temperature (entry 9, Table 5). The model substrate was treated with 58.0–180.0 mg of NH4VO3 in the presence of acetic acid under reflux conditions (entries 10–16, Table 5). The satisfactory yield of the desired product can be obtained with 117.0 mg of NH4VO3 (entry 8, Table 5). The experiment was conducted in the oxygen, nitrogen, and air atmosphere (entries 8–11, Table 5), the oxidation reaction progressed in the nitrogen atmosphere the same as in normal reaction conditions using air or oxygen atmosphere.
Under the optimized reaction conditions, the catalytic performance of NH4VO3 was further evaluated for the oxidation reaction of various 1,4-DHPs containing electron-withdrawing and donating substituents (Table 7). The Hantzsch 1,4-DHPs including a variety of substituents were converted to the corresponding pyridines in excellent yield (Table 7). Based on the results for the oxidation of 1,4-DHPs by other catalysts reported previously (Table 6), the NH4VO3 can act as a highly efficient heterogeneous catalyst in oxidation reaction through a facile method (Table 7).

Conclusion
In conclusion, a novel and convenient approach for the one-pot synthesis of pyridine derivatives through the one-pot pseudo four-component reaction, and oxidation of 1,4-DHPs by using NH4VO3 as the catalyst has been developed. NH4VO3 is an efficient, commercially available, inexpensive, and eco-friendly catalyst for these reactions. These methods involve several remarkable advantages, such as simple procedure, mild reaction conditions, short reaction times, high yields, and ease of product isolation.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information file. The data is also available through request from corresponding author.
References
Triggle, D. J. 1,4-dihydropyridine calcium channel ligands: Selectivity of action. The roles of pharmacokinetics, state-dependent interactions, channel isoforms, and other factors. Drug Dev. Res. 58, 5–17 (2003).
Liang, J.-C. et al. The new generation dihydropyridine type calcium blockers, bearing 4-phenyl oxypropanolamine, display α-/β-Adrenoceptor antagonist and long-Acting antihypertensive activities. Bioorg. Med. Chem. 10, 719–730 (2002).
Edraki, N., Mehdipour, A. R., Khoshneviszadeh, M. & Miri, R. Dihydropyridines: Evaluation of their current and future pharmacological applications. Drug Discov. Today 14, 1058–1066 (2009).
Nasr-Esfahani, M., Hoseini, S. J., Montazerozohori, M., Mehrabi, R. & Nasrabadi, H. Magnetic Fe3O4 nanoparticles: Efficient and recoverable nanocatalyst for the synthesis of polyhydroquinolines and Hantzsch 1,4-dihydropyridines under solvent-free conditions. J. Mol. Catal. A Chem. 382, 99–105 (2014).
Beukers, M. W. et al. New, non-adenosine, high-potency agonists for the human adenosine A2B receptor with an improved selectivity profile compared to the feference agonist N-ethylcarboxamidoadenosine. J. Med. Chem. 47, 3707–3709 (2004).
Chang, L. C. et al. A series of ligands displaying a remarkable agonistic−antagonistic profile at the adenosine A1 receptor. J. Med. Chem. 48, 2045–2053 (2005).
Fredholm, B. B., Ijzerman, A. P., Jacobson, K. A., Klotz, K.-N. & Linden, J. International union of pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 53, 527–552 (2001).
Bischoff, H. et al. Cerivastatin: Pharmacology of a novel synthetic and highly active HMG-CoA reductase inhibitor. Atherosclerosis 135, 119–130 (1997).
Kwong, H.-L. et al. Chiral pyridine-containing ligands in asymmetric catalysis. Coord. Chem. Rev. 251, 2188–2222 (2007).
Sepehrmansourie, H., Zarei, M., Zolfgol, M., Babaee, S. & Rostamnia, S. Application of novel nanomagnetic metal–organic frameworks as a catalyst for the synthesis of new pyridines and 1,4-dihydropyridines via a cooperative vinylogous anomeric based oxidation. Sci. Rep. 11, 5279 (2021).
Elnaggar, D. H. et al. Antiproliferative activity of some newly synthesized substituted nicotinamides candidates using pyridine-2(1H) thione derivatives as synthon. ACS Omega 7, 10304–10316 (2022).
Hantzsch, A. Ueber die synthese pyridinartiger verbindungen aus acetessigäther und aldehydammoniak. Eur. J. Org. Chem. 215, 1–82 (1882).
Khadilkar, B. M., Gaikar, V. G. & Chitnavis, A. A. Aqueous hydrotrope solution as a safer medium for microwave enhanced hantzsch dihydropyridine ester synthesis. Tetrahedron Lett. 36, 8083–8086 (1995).
Agarwal, A. & Chauhan, P. M. Solid supported synthesis of structurally diverse dihydropyrido[2,3-d]pyrimidines using microwave irradiation. Tetrahedron Lett. 46, 1345–1348 (2005).
Ji, S.-J., Jiang, Z.-Q., Lu, J. & Loh, T.-P. Facile ionic liquids-promoted one-pot synthesis of polyhydroquinoline derivatives under solvent free conditions. Synlett 2004, 0831–0835 (2004).
Chari, M. A. & Syamasundar, K. Silica gel/NaHSO4 catalyzed one-pot synthesis of Hantzsch 1,4-dihydropyridines at ambient temperature. Catal. Commun. 6, 624–626 (2005).
Wang, L.-M. et al. Facile Yb(OTf)3 promoted one-pot synthesis of polyhydroquinoline derivatives through Hantzsch reaction. Tetrahedron 61, 1539–1543 (2005).
Ko, S., Sastry, M., Lin, C. & Yao, C.-F. Molecular iodine-catalyzed one-pot synthesis of 4-substituted-1,4-dihydropyridine derivatives via Hantzsch reaction. Tetrahedron Lett. 46, 5771–5774 (2005).
Ko, S. & Yao, C.-F. Ceric Ammonium Nitrate (CAN) catalyzes the one-pot synthesis of polyhydroquinoline via the Hantzsch reaction. Tetrahedron 62, 7293–7299 (2006).
Moghaddam, F. M., Saeidian, H., Mirjafary, Z. & Sadeghi, A. Rapid and efficient one-pot synthesis of 1,4-dihydropyridine and polyhydroquinoline derivatives through the Hantzsch four component condensation by zinc oxide. J. Iran. Chem. Soc. 6, 317–324 (2009).
Anniyappan, M., Muralidharan, D. & Perumal, P. T. A novel application of the oxidizing properties of urea nitrate and peroxydisulfate-cobalt(II): Aromatization of NAD(P)H model Hantzsch 1,4-dihydropyridines. Tetrahedron 58, 5069–5073 (2002).
AliáZolfigol, M. & GhorbaniáChoghamarani, A. Silica modified sulfuric acid/NaNO2 as a novel heterogeneous system for the oxidation of 1,4-dihydropyridines under mild conditions. Green Chem. 4, 562–564 (2002).
Chavan, S. P., Kharul, R. K., Kalkote, U. R. & Shivakumar, I. An efficient Co(II) catalyzed auto oxidation of 1,4- dihydropyridines. Synth. Commun. 33, 1333–1340 (2003).
Zeynizadeh, B., Dilmaghani, K. A. & Roozijoy, A. Oxidative-aromatization of Hantzsch ester 1,4-dihydropyridines by KBr O3/SnCl4 · 5H2O under mild condition. Synth. Commun. 35, 557–562 (2005).
Bagley, M. C. & Lubinu, M. C. Microwave-assisted oxidative aromatization of Hantzsch 1,4-dihydropyridines using manganese dioxide. Synthesis 2006, 1283–1288 (2006).
Zolfigol, M. A., Salehi, P., Ghorbani-Choghamarani, A., Safaiee, M. & Shahamirian, M. Silica chromate as a novel oxidizing agent for the oxidation of 1,4-dihydropyridines. Synth. Commun. 37, 1817–1823 (2007).
Filipan-Litvić, M., Litvić, M. & Vinković, V. An efficient, metal-free, room temperature aromatization of Hantzsch-1,4-dihydropyridines with urea–hydrogen peroxide adduct, catalyzed by molecular iodine. Tetrahedron 64, 5649–5656 (2008).
Chen, J.-M. & Zeng, X.-M. β-Cyclodextrin-catalyzed mild aromatization of Hantzsch 1,4-dihydropyridines with o-Iodoxybenzoic acid in water/acetone. Synth. Commun. 39, 3521–3526 (2009).
Ghorbani-Choghamarani, A. & Zeinivand, J. Aromatization of Hantzsch 1,4-dihydropyridines with Al(NO3)3·9H2O and/or Fe(NO3)3·9H2O in the presence of silica sulfuric acid under mild and heterogeneous conditions. Synth. Commun. 40, 2457–2463 (2010).
De Paolis, O., Baffoe, J., Landge, S. M. & Toeroek, B. Multicomponent domino cyclization-oxidative aromatization on a bifunctional Pd/C/K-10 catalyst: An environmentally benign approach toward the synthesis of pyridines. Synthesis 2008, 3423–3428 (2008).
Yamada, T., Hashimoto, Y., Tanaka, K., Morita, N. & Tamura, O. Palladium(II)-catalyzed substituted pyridine synthesis from α, β-unsaturated oxime ethers via a C-H alkenylation/aza-6π-electrocyclization approach. Org. Lett. 23, 1659–1663 (2021).
Paplal, B., Nagaraju, S., Sathish, K. & Kashinath, D. One-pot synthesis of 3-hydroxy-2-oxindole-pyridine hybrids via Hantzsch ester formation, oxidative aromatization and sp3 CH functionalization using FeWO4 nanoparticles as recyclable heterogeneous catalyst. Catal. Commun. 103, 110–115 (2018).
Pratim Ghosh, P., Mukherjee, P. & Das, A. R. Triton-X-100 catalyzed synthesis of 1,4-dihydropyridines and their aromatization to pyridines and a new one pot synthesis of pyridines using visible light in aqueous media. RSC Adv. 3, 8220–8226 (2013).
Sonar, S. S. et al. Ammonium metavanadate: An effective catalyst for synthesis of α-hydroxyphosphonates. ARKIVOC 2, 138–148 (2009).
Jadhav, G. R., Shaikh, M. U., Kale, R. P. & Gill, C. H. Ammonium metavanadate: A novel catalyst for synthesis of 2-substituted benzimidazole derivatives. Chin. Chem. Lett. 20, 292–295 (2009).
Niralwad, K. S., Shingate, B. B. & Shingare, M. S. Microwave-assisted one-pot synthesis of octahydroquinazolinone derivatives using ammonium metavanadate under solvent-free condition. Tetrahedron Lett. 51, 3616–3618 (2010).
Rahimi, J., Mirmohammadi, S. S. & Maleki, A. Trihydrazinotriazine-grafting Fe3O4/SiO2 core-shell nanoparticles with expanded porous structure for organic reactions. Front. Chem. Sci. Eng. 15, 1008–1020 (2021).
Rahimi, J., Niksefat, M. & Maleki, A. Fabrication of Fe3O4@PVA-Cu nanocomposite and its application for facile and selective oxidation of alcohols. Front. Chem. 8, 615 (2020).
Rahimi, J. & Maleki, A. Preparation of a trihydrazinotriazine-functionalized core-shell nanocatalyst as an extremely efficient catalyst for the synthesis of benzoxanthenes. Mater. Today Chem. 18, 100362 (2020).
Rahimi, J., Bahrami, N., Niksefat, M., Kamalzare, M. & Maleki, A. A novel biodegradable magnetic bionanocomposite based on tannic acid as a biological molecule for selective oxidation of alcohols. Solid State Sci. 105, 106284 (2020).
Maleki, A., Niksefat, M., Rahimi, J. & Azadegan, S. Facile synthesis of tetrazolo [1, 5-a] pyrimidine with the aid of an effective gallic acid nanomagnetic catalyst. Polyhedron 167, 103–110 (2019).
Maleki, A. & Rahimi, J. Synthesis of dihydroquinazolinone and octahydroquinazolinone and benzimidazoloquinazolinone derivatives catalyzed by an efficient magnetically recoverable GO-based nanocomposite. J. Porous Mater. 25, 1789–1796 (2018).
Maleki, A., Rahimi, J., Demchuk, O. M., Wilczewska, A. Z. & Jasiński, R. Green in water sonochemical synthesis of tetrazolopyrimidine derivatives by a novel core-shell magnetic nanostructure catalyst. Ultrason. Sonochem. 43, 262–271 (2018).
Xia, J.-J. & Wang, G.-W. One-pot synthesis and aromatization of 1,4-dihydropyridines in refluxing water. Synthesis 2005, 2379–2383 (2005).
Shen, L. et al. A revisit to the Hantzsch reaction: Unexpected products beyond 1,4-dihydropyridines. Green Chem 11, 1414–1420 (2009).
Teimouri, A., Ghorbanian, L. & Moatari, A. A simple and efficient approach for synthesis of 1,4-dihydro-pyridines using nano-crystalline solid acid catalyst. Bull. Chem. Soc. Ethiop. 27, 427–437 (2013).
Patil, S., Pawar, P., Jadhav, S. & Deshmukh, M. An efficient one-pot multicomponent synthesis of dihydropyridines by using succinic acid as mild organocatalyst. Asian J. Chem. 25, 9442–9446 (2013).
Debache, A., Boulcina, R., Belfaitah, A., Rhouati, S. & Carboni, B. One-pot synthesis of 1,4-dihydropyridines via a phenylboronic acid catalyzed hantzsch three-component reaction. Synlett 2008, 509–512 (2008).
Debache, A. et al. An efficient one-step synthesis of 1,4-dihydropyridines via a triphenylphosphine-catalyzed three-component Hantzsch reaction under mild conditions. Tetrahedron Lett. 50, 5248–5250 (2009).
Saikh, F., De, R. & Ghosh, S. Oxidative aromatization of Hantzsch 1,4-dihydropyridines by cupric bromide under mild heterogeneous condition. Tetrahedron Lett. 55, 6171–6174 (2014).
Wei, X. et al. Metal-free-mediated oxidation aromatization of 1,4-dihydropyridines to pyridines using visible light and air. Chin. J. Chem. 32, 1245–1250 (2014).
Su, J. et al. Oxidative aromatization of Hantzsch 1,4-dihydropyridines by H2O2/V2O5 at room temperature. Synth. Commun. 40, 595–600 (2010).
Adibi, H. & Hajipour, A. R. A convenient and efficient protocol for oxidative aromatization of Hantzsch 1,4-dihydropyridines using benzyltriphenylphosphonium peroxymonosulfate under almost neutral reaction conditions. Bioorg. Med. Chem. Lett. 17, 1008–1012 (2007).
Han, B. et al. An efficient aerobic oxidative aromatization of Hantzsch 1,4-dihydropyridines and 1,3,5-trisubstituted pyrazolines. Tetrahedron 62, 2492–2496 (2006).
Zolfigol, M. A., Ghaemi, E., Madrakian, E. & Niknam, K. PEG-N2O4 system as an efficient reagent both for the rapid oxidation of urazoles and 1,4-dihydropyridines under nonaqueous conditions. J. Chin. Chem. Soc. 55, 704–711 (2008).
Rekunge, D. S., Khatri, C. K. & Chaturbhuj, G. U. Sulfated polyborate: An efficient and reusable catalyst for one pot synthesis of Hantzsch 1,4-dihydropyridines derivatives using ammonium carbonate under solvent free conditions. Tetrahedron Lett. 58, 1240–1244 (2017).
Ananda Kumar, T. D., Mohan, P., Subrahmanyam, C. & Satyanarayana, K. Comparative study of catalytic potential of TBAB, BTEAC, and CTAB in one-pot synthesis of 1,4-dihydropyridines under aqueous medium. Synth. Commun. 44, 574–582 (2014).
Jahanbin, B., Davoodnia, A., Behmadi, H. & Tavakoli-Hoseini, N. Polymer support immobilized acidic ionic liquid: Preparation and its application as catalyst in the synthesis of hantzsch 1,4-dihydropyridines. Bull. Korean Chem. Soc. 33, 2140–2144 (2012).
Shahabi, D., Amrollahi, M. & Jafari, A. NaI readily mediated oxidative aromatization of Hantzsch 1,4-dihydropyridines with hydrogen peroxide at room temperature: A green procedure. J. Iran. Chem. Soc. 8, 1052–1057 (2011).
Affeldt, R. F., Iglesias, R. S., Rodembusch, F. S. & Russowsky, D. Photophysical properties of a series of 4-aryl substituted 1,4-dihydropyridines. J. Phys. Org. Chem. 25, 769–777 (2012).
Taheri, N., Heidarizadeh, F. & Kiasat, A. A new magnetically recoverable catalyst promoting the synthesis of 1,4-dihydropyridine and polyhydroquinoline derivatives via the Hantzsch condensation under solvent-free conditions. J. Magn. Magn. Mater. 428, 481–487 (2017).
Gan, Z., Okui, A., Kawashita, Y. & Hayashi, M. Convenient synthesis of linear-extended bipyridines involving a central phenyl linking group. Chem. Lett. 37, 1302–1303 (2008).
Salehi, H. & Guo, Q. X. Synthesis of substituted 1,4-dihydropyridines in water using phase-transfer catalyst under microwave irradiation. Synth. Commun. 34, 4349–4357 (2004).
Acknowledgements
The authors gratefully acknowledge the partial support from the Research Council of the Iran University of Science and Technology.
Author information
Authors and Affiliations
Contributions
J.R.: main researcher, first author, main laboratory's performer, wrote the main manuscript and prepared all figures. M.N.: Formal analysis, Visualization, Writing - Review and Editing, Laboratory colleague and prepared all figures. M.H.: co-author in writing the main manuscript. M.N.: Laboratory colleague. H.A.: Laboratory colleague. M.T.I.: Laboratory colleague. A.M.: supervisor and main reviewer.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rahimi, J., Niksefat, M., Heidari, M. et al. Ammonium metavanadate (NH4VO3): a highly efficient and eco-friendly catalyst for one-pot synthesis of pyridines and 1,4-dihydropyridines. Sci Rep 12, 13687 (2022). https://doi.org/10.1038/s41598-022-17378-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-022-17378-7
This article is cited by
-
Synthesis and characterization of palladium nanoparticles modified UiO66 metalorganic framework for the synthesis of pyridine derivatives
Scientific Reports (2025)
-
Construction of 1,4-Dihydropyridines: The Evolution of C4 Source
Topics in Current Chemistry (2023)
