
Abstract
To sustainably increase wheat yield to meet the growing world population’s food demand in the face of climate change, Conservation Agriculture (CA) is a promising approach. Still, there is a lack of genomic studies investigating the genetic basis of crop adaptation to CA. To dissect the genetic architecture of 19 morpho-physiological traits that could be involved in the enhanced adaptation and performance of genotypes under CA, we performed GWAS to identify MTAs under four contrasting production regimes viz., conventional tillage timely sown (CTTS), conservation agriculture timely sown (CATS), conventional tillage late sown (CTLS) and conservation agriculture late sown (CALS) using an association panel of 183 advanced wheat breeding lines along with 5 checks. Traits like Phi2 (Quantum yield of photosystem II; CATS:0.37, CALS: 0.31), RC (Relative chlorophyll content; CATS:55.51, CALS: 54.47) and PS1 (Active photosystem I centers; CATS:2.45, CALS: 2.23) have higher mean values in CA compared to CT under both sowing times. GWAS identified 80 MTAs for the studied traits across four production environments. The phenotypic variation explained (PVE) by these QTNs ranged from 2.15 to 40.22%. Gene annotation provided highly informative SNPs associated with Phi2, NPQ (Quantum yield of non-photochemical quenching), PS1, and RC which were linked with genes that play crucial roles in the physiological adaptation under both CA and CT. A highly significant SNP AX94651261 (9.43% PVE) was identified to be associated with Phi2, while two SNP markers AX94730536 (30.90% PVE) and AX94683305 (16.99% PVE) were associated with NPQ. Identified QTNs upon validation can be used in marker-assisted breeding programs to develop CA adaptive genotypes.
Similar content being viewed by others

Introduction
Bread wheat (Triticum aestivum L.) stands as the most extensively grown and second most consumed crop globally which provides a fifth of global food calories and proteins1. With the world population expected to reach 9 billion by 2050, there is a pressing need to enhance the annual grain yield growth from the current 1% per year to 1.6% per year to meet the demands of such a rapidly increasing population2,3. However, achieving this advancement is hindered by significant challenges posed by both biotic and abiotic stress factors. Consequently, directing efforts towards enhancing wheat in specific mega-environments becomes crucial to address the unique requirements of different populations. Challenges in wheat production arise from abiotic stress factors like heat, drought, and unexpected rainfall during flowering and grain maturity. Of these, heat stress proves to be the most formidable, significantly diminishing grain yield4. According to Tesfaye et al.5, the annual average maximum temperature may rise by 1.4–1.8 degrees Celsius in 2030 and 2.1–2.6 ℃ in 2050, increasing the number of heat-stressed places by 12% in 2030 and 21% in 2050 in South Asia. According to projections made by Ortiz et al.6, by 2050, approximately half of India’s Indo-Gangetic Plains (IGP) may no longer be suitable for growing wheat. Therefore, in the IGP region, the most important element in determining wheat productivity is the unpredictable variation of temperature in March. Warming causes anthesis and maturity to occur earlier, which decreases crop output by shortening the growing season, decreasing the number of kernels per spike, and reducing the weight of the kernels7,8. The most delicate plant growth stages occur around anthesis in several crops, including wheat9,10. Recurrence of extreme weather patterns, such as sudden downpours of rain and hailstorms that cause crop lodging, has made the issues even more difficult. According to a thorough analysis of wheat yield from 208 districts conducted by Gupta et al.11, between 1981 and 2009, wheat yield decreased by 5.2% due to global warming, and wheat yields decreased by 2–4% for each 1℃ increase in the average daily maximum and lowest temperatures. Most wheat fields in India currently experience somewhat above-average temperatures, particularly during the important grain-filling stage of growth; even a tiny temperature increase will substantially impact yield.
Crop development that is moderated by climatic fluctuations can be substantially addressed by either modifying the production environment via the use of stress-reduction techniques or by choosing varieties with appropriate flowering times and duration12. There is a need to integrate agronomic management and responsive genotypes to rectify the problems. Although there has been progress in understanding the phenological traits under unfavourable temperature conditions, knowledge of crop plants’ overall behaviour under ideal conditions is still lacking13. Extreme events are predicted to occur more frequently as a result of climate change, making this understanding crucial. Reminders of these events can be found in the wheat-growing seasons of 2014–15, 2015–16, and 2021–2214. Predictive breeding strategies and production technologies become increasingly complex and challenging to design under such circumstances. Identification of physiological and/or phenological variables that contribute to yield improvement over time can shed some information on the pattern of adaptation to changing climatic conditions.
One effective strategy is conservation agriculture (CA), which involves retaining crop residue and practising zero tillage15,16,17. This approach ensures enhanced soil structure, optimal soil temperature, preserved soil moisture, and improved soil biodiversity18,19, increased soil organic carbon, root penetration, and reduced subsoil compaction20,21. Studies indicate that CA consistently yields better grain productivity compared to conventional tillage (CT) practices, both in stress and non-stress environments. Devkota et al.22 reported a substantial increase of 62% in wheat grain production under CA compared to CT in their study. CA-based cropping systems also ensure increased economic profitability, better adaptation to heat and water stress, and reduced emissions of greenhouse gases23. However, there is a notable gap in research regarding how plants adapt to this optimal environment at the genetic level and the lack of genotypes with specific adaptability for CA, since genotypes bred for CT practices do not always perform equally under CA19. Consequently, there is a pressing need for comprehensive investigations to unravel the phenological and morpho-physiological changes occurring in plants that facilitate their adaptation to conservation agriculture, leading to improved grain yields. For yield under zero-tillage conditions, three specific regions were identified on chromosomes 2D (wPt3728-cfd44 and gmw484-wmc27) and one on 5B (wmc99-wPt2373) in a study by Trethowan et al.24.
To deepen our understanding of such adaptation mechanisms, a thorough examination of the genetic basis, involving the dissection of traits and identification of chromosomal regions controlling their expression, would be highly valuable in the current era of molecular breeding14. A key QTL on chromosome 7D (7D-acc/cat-10) boosts yield and weight in wheat, showing resilience to late-season heat and drought stress. It accounts for 19.6% yield variation in stress trials, making the linked marker a potential candidate for selection in other populations25. Significant marker-trait associations (MTAs) were found on chromosomes 2A and 2B, explaining 7–25% of trait variation26. GWAS conducted for irrigated, stressed, and combined environments, along with trait per se and stress indices, revealed QTL hotspots on chromosomes 2A (54–70 cM) and 2B (75–82 cM). QTLs previously identified for grain filling rate (GFR) and grain filling duration (GFD) were validated in Indian breeding material under conservation agriculture. Notably, two SSR markers, XCfd42 and Xwmc500, accounted for approximately 6% and 1% variation in GFR, respectively27. Genome-wide association studies (GWAS) have enabled the identification of MTAs for various phenological and morpho-physiological traits such as heading date, grain filling duration, plant height, spike traits, kernel weight, grain yield, canopy temperature, chlorophyll content and normalised difference vegetative index (NDVI) in wheat28,29,30,31,32,33. Keeping previous knowledge and current circumstances in consideration, the current study was aimed at finding important morpho-physiological traits that increase genotype adaptation for CA environments, as well as MTAs for the traits under differing production regimes, using GWAS . It will provide new insights into the mechanisms by which wheat adapts to conservation agriculture (CA), ultimately leading to an improved grain yield compared to conventional tillage (CT). This will be the first report where we have identified key physiological traits and putative candidate genes responsible for the adaptation of wheat to CA environments.
Materials and methods
Experimental materials
The plant material used in the current study comprised 183 advanced breeding lines with diverse parentages (Supplementary Table 1) along with five commercially cultivated varieties viz., HD3226, DBW187, HD3086, HD3298 and HI1621. The varieties HD3226, DBW187 and HD3086 were used as checks for timely sown conditions and varieties HD3298 and HI1621 for late sown conditions. The use of advanced breeding lines for association studies also enables their direct use in the transfer of identified QTNs and their relevance in the cultivar development process34.
Location, experimental site and production environments
The field experiments were conducted at the Research Fields of ICAR-Indian Agricultural Research Institute, New Delhi. The location is situated at N28°38′24.0252″, E77°10′26.328″, with an altitude of 228.61 m (750 ft) above sea level in the subtropical region of the Northwestern Plain Zone of India. The experiment was designed with four contrasting production environments: conventional tillage timely sown (CTTS), conventional tillage late sown (CTLS), conservation agriculture timely sown (CATS), and conservation agriculture late sown (CALS). The experimental field is maintained under CA (zero tillage with residue retention) since 2008. For timely sown conditions, the trials were planted in the second week of November in the Rabi season 2022–23. In contrast, for late sown conditions, the planting was done in the third week of December. This scheduling aimed to subject the genotypes to terminal heat stress during their growth.
Field experiment and phenotyping
The experiment was laid in an augmented block design comprising all 183 advanced breeding lines and 5 checks in 8 blocks in all four production environments. Augmented block design is very helpful in experiments where a huge number of genotypes need to be screened but land resources are scarce due to which replication of all genotypes is not possible. In an augmented block design, only the checks are replicated across the blocks while test genotypes are planted without replication as new entries. The experimental materials were planted when adequate moisture was available in the soil. In each production environment, genotypes were grown with a plot size of 4.8m2 (6 rows of 4 m length). The fields were irrigated in regular intervals depending upon rainfall and moisture availability in the fields. The cultivation of the crop adhered to recommended standard cultural and agronomic practices to ensure proper growth and development of the crop.
Physiological traits included chlorophyll fluorescence parameters viz., ϕII quantum yield (Phi2) and non-photochemical quenching (NPQ), active photosystem I center (PS1), relative chlorophyll (RC, as SPAD measurement), canopy temperature differential (CTD, in ℃), leaf thickness (LT, in mm) and leaf angle (LA, in °). All the physiological traits were observed using PhotosynQ MultispeQ v2.035 in a non-destructive way in the field. The agro-morphological traits observed in the present study included days to heading (DTH, when 50% of plants in the plot achieve heading, Zadoks stage 5936), days to maturity (DTM, when 50% of plants in the plot achieve physiological maturity, Zadoks stage 8936), plant height (PH, in cm), tiller count (TC, number of productive tillers per mt row), spike length (SL, in cm), spikelets per spike (SPS), grains per spike (GPS), thousand-grain weight (TGW, in g) and grain yield (GY, in q/ha). Phenological observations were recorded on five randomly selected plants from each plot in all four production environments and grain yield was taken by harvesting of whole 4.8m2 plot. In addition to the morpho-physiological traits, grain geometric parameters viz., grain length (GL, in mm), grain breadth (GB, in mm) and grain surface area (GSA, in mm2) were also measured using scanned images of threshed grains obtained from two randomly picked spikes by analysing the images in SmartGrain software v1.337.
Plant adaptation to diverse environments involves a multifaceted interplay of physiological and metabolic adjustments orchestrated by alterations in gene expression. Among these adaptations, photosynthetic efficiency emerges as a critical trait profoundly influenced by environmental factors, ultimately dictating the performance of plant genotypes. The quantum yield of photosystem II (Phi2) serves as a pivotal indicator of photosynthetic efficiency, reflecting the extent to which incident light is harnessed in photochemical reactions—a parameter with profound implications for biomass yield optimization. In contrast, the quantum yield of non-photochemical quenching (NPQ) denotes the proportion of incident light dissipated as heat, which is an important photoprotective process that protects the photosystem from damage caused by excess light energy. In addition, a higher value of relative chlorophyll content and active photosystem I reaction centers indicate an optimized light capture and electron transport. Furthermore, a cooler canopy temperature (indicated by higher value of CTD—canopy temperature depression) indicates efficient transpiration and potentially higher stress tolerance, while a warmer canopy temperature (lower CTD value) suggests limitations in water uptake or transpiration. To deepen our comprehension of how these physiological traits influence grain yield, we have selected agro-morphological traits to establish significant correlations between yield component traits and the aforementioned physiological traits.
Statistical analysis
Statistical analyses were performed using the mean values of the observations recorded on ten randomly selected plants from each plot in all four production environments. Analysis of variance (ANOVA) was performed for Augmented block design in R studio software v4.3.1 using the “AugmentedRCBD” package38. The statistical significance for the morpho-physiological traits was analyzed as test and check treatments for block-adjusted ANOVA. To account for the presence of GxE interactions, a combined analysis of variance was performed for the traits in the present study using the linear mixed model analysis from the lme4 package in R embedded in the META-R software39. The analysis was carried out considering the RCBD design and using the restricted maximum likelihood (REML) method assuming fixed effects of the genotypes, while all other terms were treated as random effects. The best linear unbiased prediction (BLUP) was calculated for tested genotypes combined across all environments to determine the variance components and the significance level of genotype, environment and GxE interactions. In addition, the coefficient of variation (CV%), least significance difference (LSD, p ≤ 0.05), broad sense heritability (H2) and the grand mean for each trait were calculated. Mean squares of each source of variation were used to estimate the variance components for genotype (\({\sigma }_{G}^{2}\)), genotype Χ environment interaction (\({\sigma }_{GE}^{2}\)) and residual error (\({\sigma }_{\varepsilon }^{2}\)), respectively, and the heritabilities were estimated using the formula \({H}^{2}={\sigma }_{G}^{2}/({\sigma }_{G}^{2}+\frac{{\sigma }_{GE}^{2}}{e}+\frac{{\sigma }_{\varepsilon }^{2}}{r})\), where e and r were the number of environments and replications in each environment, respectively40. The correlation analysis for the traits was performed using the “corrplot” package41 in R Studio employing Pearson’s correlation test.
Genotyping and quality control
The advanced breeding lines and check varieties in the association panel were genotyped using Breeders’ 35 K Axiom Array which contains 35,143 SNPs. To avoid confounding effects of spurious alleles, quality control of SNPs was performed by removing SNPs with missing data of > 20%, MAF < 5% and heterozygosity > 15% to generate the filtered SNP data using TASSEL v5.0 (Trait Analysis by Association, Evolution and Linkage) software42.
Population structure and linkage disequilibrium
To determine the presence of any population stratification in the association panel, a SNP-based principal component analysis (PCA) was performed using the filtered SNP data in TASSEL v5.0. The population structure within the group of 188 genotypes was also determined using the Bayesian inference program STRUCTURE v2.3.443. This software utilises a model-based clustering approach, leveraging genotype information from independent markers to infer population structure. The optimal number of populations (denoted as K) was identified using the ad-hoc delta K method44. Linkage disequilibrium (LD) was evaluated by calculating the correlation (r2) in allele frequency between marker pairs, using TASSEL v5.0 software. The LD decay was assessed by plotting pairwise LD values (r2) against genetic distance for the A, B, and D subgenomes, as well as the whole genome of bread wheat genotypes in our association panel using the “LDheatmap package”45 in R Studio.
Genome-wide association studies and in-silico analysis
To identify significant MTAs in each production environment using the phenotypic and genotypic data, GAPIT3 package46 was used in R Studio applying the BLINK model47 for its high computing efficiency and statistical power. Besides its ability to include principal components as covariates to mitigate the risk of false positives resulting from population stratification, BLINK methodically integrates linked markers as covariates during the testing of other markers. This process is designed to unravel their link to any cryptic relationships among individuals. The markers that are linked to each other are chosen based on linkage disequilibrium, optimized concerning Bayesian information content (BIC), and subject to repeated scrutiny across multiple tests to minimize the occurrence of false negatives. Only such QTNs that had a -log10P value above the Bonferroni threshold of 5 were identified as significant MTAs. To identify candidate genes associated with the identified QTNs through GWAS analysis, an in-silico analysis was conducted. Genes located within a 1 Mb interval centred on the identified QTN were identified by searching the EnsemblPlants wheat database with the IWGSC Reference Sequence v1.0 assembly (https://plants.ensembl.org/Triticum_aestivum/Info/Index). While researchers typically search the entire linkage disequilibrium (LD) region to find candidate genes linked to important QTNs, we narrowed our search area to 1 Mb due to the unusually large LD blocks in our association panel. Information regarding the proteins encoded by the genes identified through gene annotation and their respective functions was sourced from the Triticeae-Gene Tribe (http://wheat.cau.edu.cn/TGT/). Only high-confidence protein-coding genes, accompanied by their Gene ID, were considered. These genes were then analyzed for their protein functions to establish a connection with the trait of interest. Subsequently, only protein-coding genes highly relevant to the expression of the specific traits under investigation were selected. The workflow for quality control of SNP data and subsequent utilisation of filtered genotypic data and phenotypic to identify MTAs followed by the in-silico analysis to identify putative candidate genes linked with the identified MTAs is represented in Fig. 1.

Workflow to identify MTAs for morpho-physiological traits through GWAS.
Plant guideline statement
The use of plant and plant material in the study is in compliance with relevant guidelines and regulations.
Results
Phenotypic variation for morpho-physiological traits
The genotypes in the association panel were analysed in all four production environments. The phenotypic distribution of the studied morpho-physiological traits across the four production environments is depicted as ridgeline plots in Fig. 2. The summary of descriptive statistics and block-adjusted ANOVA for the studied traits under all four production environments is given in Supplementary Tables S2, S3a–d, respectively. Through ANOVA, significant variations (**p ≤ 0.01) were observed among the genotypes for all the morpho-physiological traits in each production environment. CT was found to be the environment with the highest variability for the studied traits as compared to CA emphasizing the ability of genotypes to adapt to CA and retain optimum and stable yields. The combined ANOVA revealed significant differences (*p ≤ 0.05) for all test traits across environments for genotype (except PS1) as shown in Table 1. Grand means, coefficient of variation (CV), least significance difference (LSD) and broad-sense heritability (H2) are given in Table 1. The CV values ranged from 1.37% for GY to 5.30% for NPQ. For H2, the highest estimates were produced for GL (93.81%) followed by DTM and DTH at 85.94% and 85.87%, respectively. Furthermore, highly significant differences (***p ≤ 0.001) for all tested traits (except GL) were observed due to the GxE interactions (Table 1).

Frequency distribution of the studied morpho-physiological traits in the four contrasting production environments (CTTS, CTLS, CATS and CALS). The vertical black lines indicate the mean trait value in the respective production environment. Phi2 PS II quantum yield, NPQ non-photochemical quenching, RC relative chlorophyll, CTD canopy temperature depression, PS1 active PS I center, LT leaf thickness, LA leaf angle, DTH days to heading, DTM days to maturity, PH plant height, TC tiller count, SL spike length, SPS spikelets per spike, GPS grains per spike, TGW thousand-grain weight, GY grain yield, GL grain length, GB grain breadth, GSA grain surface area.
Correlations among traits
Pairwise correlation based on Pearson’s coefficients was employed to examine the correlations between the morpho-physiological traits under each production environment separately to understand how the correlation patterns change under differing production conditions (Fig. 3). Significant positive correlations (p ≤ 0.05) were identified between DTH and DTM (0.83 to 0.97) across the production environments. The negative correlation (−0.07) between NPQ and GY observed under timely-sown conditions turned positive (0.35) under late-sown environments, while a consistent negative relation between NPQ and Phi2 (− 0.88 to -0.90) across the environments was observed. PH showed a positive correlation with GY across the environments (0.22 to 0.49) except CTTS (− 0.25). Both DTH and DTM showed negative correlations with TGW, GY, GB, GL, and GSA across the environments. Significant positive correlations were found between Phi2 with DTH (0.24 to 0.30) and DTM (0.24 to 0.27) under CA environments but not in CT conditions. Interestingly, a positive correlation (0.10 to 0.24) was identified between NPQ and CTD with the highest value observed under CTLS (0.24). Furthermore, LT which showed a positive correlation with NPQ, had a negative correlation with Phi2 under timely-sown environments whereas the reverse relationship was observed under late-sown conditions. TC specifically showed a consistent positive correlation (0.28) with GY under both CA environments while no such relation was observed under CT environments. The correlation matrices for the 19 studied traits have been provided in the supplementary information (Supplementary Tables S4–S7) showing the significance of the correlations based on p-values from Pearson’s correlation test.

Correlation among the morpho-physiological traits under the four production environments (CTTS, conventional tillage timely sown; CTLS, conventional tillage late sown; CATS, conservation agriculture timely sown; CALS, conservation agriculture late sown). The pair plot shows Pearson’s correlation estimates among each trait pair below the diagonal, traits on the diagonal, and proportional correlation coefficients depicted as the colour and size of circles above the diagonal. Phi2 PS II quantum yield, NPQ non-photochemical quenching, RC relative chlorophyll CTD canopy temperature depression PS1 active PS I center LT leaf thickness LA, leaf angle, DTH days to heading, DTM days to maturity, PH plant height, TC tiller count, SL spike length, SPS spikelets per spike GPS grains per spike TGW thousand-grain weight, GY grain yield, GL grain length, GB grain breadth, GSA grain surface area.
Quality control, population structure and linkage disequilibrium
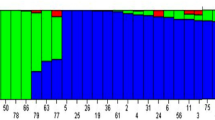
The initial genotypic data contained 31,433 SNPs which were filtered to obtain a subset of 9,771 highly informative SNPs that had MAF > 0.05, heterozygosity < 0.15 and missing data < 0.20. The highest number of SNPs were found in the B genome (3633; 37.18%) followed by the D (3174; 32.48%) and A (2964; 30.33%) genomes. Chromosome 2D had the highest number of SNPs (699), while chromosome 4D had the lowest (192) as depicted in Fig. 4. Our analysis using Principal Component Analysis (PCA) revealed the presence of two distinct sub-populations within our data set. This separation is visualized by the first two principal components(Fig. 5a). The entire population was segregated into two sub-populations, as the ΔK value was maximized when K was set to two. (Fig. 5b). Also, using Evanno’s method in STRUCTURE Harvester, the optimal number of clusters (K) was determined to be two (Fig. 5c), which was consistent across both the STRUCTURE analysis and the graphing of ΔK values. R2 values for SNP pairs were utilised to assess LD decay by plotting against genetic distances. LOESS regression characterized LD decay, with the curve depicted in red on LD decay plots. A threshold was identified at r2 = 0.10, based on the fitted model. LD decay was fastest in sub-genome A, followed by D and B. Decay occurred at 4.63 Mb (A), 5.45 Mb (D), 7.41 Mb (B), and 5.77 Mb (genome-wide) as depicted in Fig. 6.

Bar graph depicting the chromosome-wise distribution of filtered SNPs across the genome.

Population structure analysis. (a) PCA performed using the filtered genotypic data indicates the presence of two groups in the association panel; (b) Evanno plot depicting estimated ΔK values for a given K identified max ΔK value at K = 2; (c) Barplot of the population structure showing the presence of two sub-populations.

LD decay plots of (a) A genome; (b) B genome; (c) D subgenome; and (d) whole genome. The LD decay was the fastest in subgenome A.
Genome-wide association studies and gene annotation
To detect significant MTAs within each production environment using both phenotypic and genotypic data, the GAPIT3 package was employed in R Studio using the BLINK model. GWAS was performed for individual traits under each environment to uncover the presence of MTA*Environment interaction as many MTAs found in one environment could not be observed under other environments. To identify significant MTAs, a Bonferroni threshold was set at a -log10(p) value of 5.0. A total of 80 MTAs were identified through GWAS across all environments for all the traits, and their chromosome-wise and trait-wise distributions are depicted in Supplementary Figs. S1, S2, respectively. The highest number of significant MTAs were identified in the D subgenome (30) followed by the B subgenome (28) and A subgenome (22). The maximum number of significant MTAs were detected for DTH (11) followed by TGW (8), while LA (1) had the lowest. Upon a detailed investigation, it was observed that the A subgenome had the highest number of identified MTAs for LT (2), PH (3) and TC (2). In contrast, the B subgenome had the highest number of MTAs for DTH (8) and RC (3) while the D subgenome harboured the most MTAs for Phi2 (3), DTM (3), GB (2), GSA (3), GY (3) and SPS (3). In addition, Manhattan plots and QQ plots were created using the R package “GAPIT3” to detect significant MTAs by plotting SNPs that had a -log10(p) value of more than 5.0. The Manhattan plots demonstrating the identification of significant QTNs discovered by the BLINK model approach are depicted in Fig. 7.

Manhattan plots showing identified MTAs through BLINK model for the morpho-physiological traits under (a) CTTS; (b) CTLS; (c) CATS; and (d) CALS. Phi2 PS II quantum yield, NPQ non-photochemical quenching, RC relative chlorophyll, CTD canopy temperature depression, PS1 active PS I center, LT leaf thickness, LA leaf angle, DTH days to heading, DTM days to maturity, PH plant height, TC tiller count, SL spike length, SPS spikelets per spike, GPS grains per spike, TGW thousand-grain weight, GY grain yield, GL grain length, GB grain breadth, GSA grain surface area.
The highest number of MTAs were identified in the CTTS (35) production environment followed by CALS (25) and CATS (11) while the lowest numbers were identified in CTLS (9). The Manhattan plots and QQ plots for the morpho-physiological traits in the present study under each production environment are shown in Supplementary Figs. S3–S21. Out of the total 80 significant MTAs initially identified, further analysis focused on the 48 most highly significant SNPs as shown in Table 2. Notably, SNP AX95138506, situated on Chromosome 2B, played a pivotal role by explaining 40.2% of the phenotypic variance for CTD under the CTTS condition. Similarly, SNP AX94525104, located on Chromosome 3A, accounted for a noteworthy 31.3% of the variance for GL under CALS. In addition to these, 9 other SNPs were identified, each contributing more than 20% to the phenotypic variance in various morpho-physiological traits. Interestingly, SNP AX94803899, located on chromosome 3B had the highest -log10(p) at 12.68 explaining a phenotypic variation of 12.98% for GY. Another SNP AX95219528 on chromosome 1B was observed to be associated with DTH (PVE 11.88–21.42%) and was found to be consistent under both CATS and CALS environments. The remaining 36 SNPs, subjected to further gene annotation through in-silico analysis, elucidated phenotypic variances ranging from 2.15 to 19.45% for the different traits under study. Furthermore, it was observed that MTAs for Phi2 and NPQ were identified under CA and CT environments, respectively.
Out of the total 80 SNP markers associated with 19 morpho-physiological traits, 48 SNPs were found to be located within and contiguous to genes, and they are listed in Table 2. The detailed investigation focused solely on high-confidence protein-coding genes that play a role in the expression and development of the associated traits. Genes identified through an in-silico analysis were mostly transcription factors that have been reported to overexpress in stress conditions, peroxidases, kinases, pentatricopeptide repeat proteins, splicing factors, proteins involved in photophosphorylation and electron transport chain, various kinds of domain-containing proteins and sugar transporters. Details of the candidate genes identified through gene annotation are provided in Table 3.
Discussions
In the Indo-Gangetic plain zone, the prevalent cropping system is the rice–wheat rotation. A significant factor contributing to the low yield of wheat in this system is the delayed sowing of wheat, often caused by the late transplanting and harvesting of the preceding rice crop. To address this issue and establish rice–wheat as a sustainable system, the adoption of zero tillage techniques for wheat cultivation can be highly beneficial48. With delayed wheat sowing in India, the occurrence of high-temperature events during the grain-filling stage becomes more frequent. Conservation agriculture (CA) represents a valuable approach to combat the impacts of climate change on agriculture. By minimizing soil disturbance, promoting soil health, and reducing greenhouse gas emissions, CA practices contribute to climate change mitigation49. Simultaneously, CA’s ability to enhance water conservation, improve crop resilience, and diversify agricultural systems aids in adapting to the changing climate. Breeding wheat varieties optimized for Conservation Agriculture (CA) involves a multifaceted approach, targeting traits such as drought tolerance, disease resistance, improved root architecture, shorter growth cycles, stress tolerance, nutrient efficiency, and lodging resistance19,50. These traits align with CA principles by enhancing crop resilience and adaptability to changing environmental conditions, improving soil health, and promoting sustainable agricultural practices.
The advanced breeding lines were observed to have ample variation for the studied morpho-physiological traits in the present study within each production environment as well as across them which creates a possibility to target these materials in various breeding programmes (Table 1). The variability for the studied traits among the genotypes was higher in CT environments than in CA environments signifying the role of management practices on crop performance. Among the physiological traits, the heritability estimates were higher for RC (0.71) and LT (0.62) while moderate heritability was observed for NPQ (0.46) and LA (0.32), and low heritability in the case of Phi2 (0.26) and CTD (0.24). Although GY (0.31) had a lower heritability estimate, moderate (0.45 for GPS and 0.60 for SPS) to high heritability (0.63 for TGW, 0.66 for SL, 0.67 for TC, 0.71 for PH, and 0.85 for both DTH and DTM) were observed among the agro-morphological traits. The seed geometric parameters had high heritability estimates (0.71 for GB, 0.72 for GSA and 0.94 for GL). Similar heritability estimates for physiological and agro-morphological traits were reported in previous studies51,52,53. The moderate to high heritability estimates for most of the studied traits across the production environments suggest that these traits can be utilised to understand the genetic control of these traits upon grain yield and their utilisation in improving grain yield in wheat.
Complex correlation patterns were identified for the studied traits due to the influence of change in production regimes. Among the two chlorophyll fluorescence parameters, Phi2 was positively correlated and NPQ was negatively correlated with GY under timely-sown (TS) conditions whereas the relationships changed under late-sown (LS) conditions as the correlation between NPQ and Phi2 with GY turned positive and negative, respectively. This signifies that plants grown under LS conditions are exposed to more light stress due to the coincidence of high temperature and light intensity with the reproductive growth stages of the crop. Thus, such genotypes which can maintain optimum Phi2 against NPQ, as plants use photoprotective mechanisms to reduce photoinhibition of both PS II and PS I complexes, are supposedly more tolerant to such high light and heat stress54,55,56. The consistent negative correlations (− 0.11 to − 0.37) of GY with DTH and DTM across the four production environments except for CATS wherein no significant negative correlation of GY with these phenological traits was observed indicates that plants grown under CATS get an opportunity to accumulate more photosynthetic reserves during the vegetative stage as the stress factors are less prominent in this ideal environment50,57. Additionally, in CA, there is a shift from a negative correlation (− 0.10 to − 0.17) between RC and NPQ in conventional tillage (CT) to a positive one (0.07 to 0.10), indicating that CA’s reduced soil disturbance and improved structure offer resilience against stress, unlike the variable responses in CTLS conditions. Under TS conditions, PH was negatively correlated (−0.25)with GY in CT while in CA, the relationship was observed to be positive (0.20). PH and GY are negatively correlated traits58,59 as taller plants are more prone to lodge due to heavy wind during the maturity stages but the positive relationship in CA represents an opportunity to target increased PH for improved GY as plants grown under CA have better anchorage due to less soil disturbance and better soil structure18. Kernel traits such as TGW, GL, GB and GSA showed moderate to high positive correlations (ranging from 0.14 to 0.92) which corroborate with previous studies60,61. Furthermore, CTD showed a significant positive correlation with GY (0.17) only under CTLS out of the four production environments as also reported in previous studies62,63,64. This suggests that plants sown under TS conditions produce optimum GY by completing most of the grain filling before heat stress coincides when compared to crops grown under LS conditions wherein reproductive growth stages coincide with high temperature which hampers GY. Interestingly, under LS conditions, only CT showed a positive correlation between CTD and GY whereas no such relationship was observed in CA which indicates plants grown in CA are less exposed to high-temperature stress as lower soil temperatures are offered by CA compared to CT19.
The adoption of marker-assisted breeding approaches holds the potential to empower wheat breeders in selecting promising genotypes that demonstrate improved performance under CA practices. DNA-based markers, including abundant SNPs present within the wheat genome, can be harnessed for this purpose, and be identified using high-throughput techniques such as GWAS. GWAS involves the integration of phenotypic data related to the traits of interest with genotypic data generated through sequencing for the identification of significant MTAs based on a Bonferroni threshold65, aiding breeders in their selection efforts. Contemplating the considerations mentioned earlier, the primary emphasis of this study was placed on identifying MTAs for the 19 morpho-physiological traits under contrasting production regimes through GWAS. The effectiveness of GWAS in uncovering significant MTAs hinges significantly on the marker distribution within the genotypic dataset. GWAS is more likely to identify MTAs with greater precision when markers are evenly distributed across the genome. Conversely, the presence of rare alleles diminishes GWAS’s resolution power. Additionally, the existence of population structure, if not properly addressed, can lead to unrelated associations and false positive results. To account for relatedness among the genotypes used in association mapping, kinship analysis is employed. Among all these factors, LD plays a crucial role in MTA identification66. High LD requires fewer markers, while low LD necessitates a larger number of markers for GWAS. Ensuring the quality of both phenotypic and marker genotypic data is paramount in the association mapping process. In the present study, we observed rapid Linkage Disequilibrium (LD) decay in the A subgenome (4.63 Mb), followed by the D subgenome (5.45 Mb) and the B subgenome (7.41 Mb), with a whole-genome LD decay rate of 5.77 Mb. A similar trend for subgenome-wise distribution of markers and rate of LD decay has been reported by Krishnappa et al.67. 48 MTAs out of the total 80 MTAs were selected for detailed study after gene annotation that identified putative candidate genes that are related to the development of the target traits.
The ϕII quantum yield (Phi2) is essentially the percentage of incoming light (excited electrons) that goes into the Photosystem II (Photophosphorylation), where the light energy is converted into chemical energy. Thus, increasing the proportion of Phi2 can improve the photosynthetic efficiency thereby increasing the grain yield. Two known genes “Photosystem II reaction center W protein, chloroplastic”68,69 on Chr1B and “Cytochrome P450 89A9”70,71,72 on Chr2D were found to be linked to markers AX94651261 and AX95228767, respectively for Phi2 that are known to play pivotal roles in photophosphorylation and can be targeted to improve the photosynthetic efficiency. The identification of these significant MTAs under CALS emphasizes their role in the enhanced physiological response of genotypes towards adaptation to CA with improved stress tolerance. Due to its ideal environment, the CA offers a chance for crops to reach their maximum potential. Better and longer-lasting soil moisture availability and temperature modulation have produced this environment73. In a late-sown scenario with CT conditions, the crop experiences stress due to the rapid depletion of soil moisture. On the other hand, crops may withstand stress for a longer period under CA. While Phi2 relates to the proportion of absorbed light that goes into photochemical reactions, ϕNPQ (NPQ) is the proportion of the excitement energy of the PSII complex that is dissipated as heat to protect the plant against photooxidative damage and is referred to as non-photochemical quenching. Other than these two known regulated processes, the excess absorbed energy is expressed as ϕNO (non-regulated NPQ processes) and since these three processes of energy dissipation are proportions of the absorbed light energy, their sum is equal to 1. As NPQ is considered a photoprotective mechanism of plants, several stress-responsive genes are assumed to be involved in the process. We identified three MTAs for NPQ and through gene annotation, it was found that SNP AX94730536 (PVE 30.90%) on Chr4B was linked to “UDP-glucose 6-dehydrogenase 4” gene while “25.3 kDa vesicle transport protein” gene was linked to SNP AX94477203 (6.75% PVE) on Chr3D. In addition, SNP AX94683305 (16.99% PVE) was found to be associated with three genes viz., TraesCS5A02G314800, TraesCS5A02G314700 and TraesCS5A02G314600 for “UDP-glucose 4-epimerase”, “Heat stress transcription factor B-2c” and “Ethylene-responsive transcription factor ERF071”, respectively. All these proteins have been reported to be differentially expressed under stress conditions74,75,76 and induce tolerance77,78,79,80,81. According to our data, NPQ under CT (0.54) was higher for both TS and LS than it was for CA (0.49). Due to the fast loss of soil water and the rising temperatures of the soil and air, the impact of stress is greater in CTLS conditions than in CALS. The fact that no QTNs for NPQ were found in the study under CA conditions indicates that CA is a superior environment when stress levels are high. Under stress conditions, plants initiate the accumulation of reactive oxygen species (ROS) within chloroplasts as a result of excess excitation energy, and the primary sites for the generation of these ROS are the reaction centers of Photosystem I (PS I) and Photosystem II (PS II) in the thylakoids82. Due to such ROS production, photo-inhibitive damage occurs in the PS I centers thereby reducing the number of PS I active reaction centers. The PS1 Active Centers (PS1) was found to be associated with SNP AX95178106 (23.05% PVE) on Chr2D which is linked with three genes viz., TraesCS2D02G404800, TraesCS2D02G404600 and TraesCS2D02G404500 that codes for “Aquaporin PIP1-2”, “Cytochrome P450 86A22” and “Thioredoxin-like 3-3”, respectively. All three proteins are related to Photosystem I activity, as supported by evidence presented by Kromdijk et al.83; Zhang et al.84 and Zhu et al.85. Among the four production environments, CATS had an enhanced PS I activity (mean value of 2.45) as evidenced by the identification of all QTNs for PS1 in this environment, resulting in improved photophosphorylation.
Since, enhanced photosynthetic efficiency is a function of increased amount of chlorophyll content both under stress and non-stress conditions, targeting relative chlorophyll content has been a regular practice of breeders to select genotypes as a proxy trait for improved grain yield86,87. For relative chlorophyll content (RC), two genes TraesCS1D02G355200 and TraesCS1D02G354100 associated with SNP AX95247332 (6.89% PVE) on Chr1D that translates into “Hexokinases-2” and “Peroxidase 1”, respectively have been reported to catalyze chlorophyll destruction leading to leaf senescence88,89,90. Also, SNP AX94449793 (3.38% PVE) on Chr5B associated with TraesCS5B02G368900 that translates into “Chloroplastic Chaperon 60 subunit α-2” has been reported to play an important role in protein transport under stress conditions91,92. Another SNP AX95249924 on Chr6D was found to be present in close proximation with genes TraesCS6D02G042400 and TraesCS6D02G042300 that codes for “Putative wall-associated receptor kinase-like 16” and “Wall-associated receptor kinase 2” have been reported to negatively regulate leaf senescence and help plants maintain higher relative chlorophyll content even under stress conditions. Similar, findings were presented by Li et al.93; Riou et al.94; Zhao et al.95. The expression of these genes in the CT environment makes it more evident that these QTNs can be targeted to maintain relative chlorophyll contents under stress conditions. In contrast, no QTNs could be identified for RC under CA environments implying that plants thrive without the necessity for differential gene expression to uphold optimal chlorophyll content. This suggests that under CA conditions, where plant stress is minimized, maintaining ideal chlorophyll levels does not heavily depend on significant changes in gene expression. A “Cyclin-dependent kinase B1-1” protein coding gene TraesCS4A02G192300 was found to be associated with SNP AX94985892 (19.45% PVE) for leaf thickness (LT). The “Cyclin-dependent kinase B1-1” has been reported to promote Anaphase and is involved in increased growth rate and organ size96.
Crop phenology is highly influenced by the agrometeorological parameters and the production environment in which they are grown. Early heading, late maturity and increased plant height can be considered important non-grain parameters to select genotypes for improved grain yield under both timely and late sown conditions97. The highest number (8) of MTAs were found for days to heading (DTH). Notably, SNP AX94476007 (19.45% PVE) on Chr3A was found to be linked to three genes viz., TraesCS3A02G421900, TraesCS3A02G421400 and TraesCS3A02G422600 for “26S proteasome regulatory subunit 6A homolog A”, “Transcription factor bHLH49” and “Mitochondrial Glycine-rich RNA-binding protein 4”, respectively. The role played by these genes in regulating the flowering time has been reported by Liu et al.98; Ahmad et al.99; Li et al.100; Alptekin et al.101; Ma et al.102. As described earlier in this section, CATS (97.71 and 150.47) was found to be the ideal production environment that had early heading and late maturity as compared to CTTS (97.96 and 145.64). Another SNP AX94950339 (7.24% PVE) on Chr 6B was linked to TraesCS6B02G402700 translates for “BTB/POZ and MATH domain-containing protein 1” which plays a role in vernalisation and regulates heading time under stress conditions as reported by Byrne et al.103 and Strejčková et al.104. Notably, SNP AX95219528 (11.88–21.42% PVE) linked with TraesCS1B02G246600 on Chr1B translates “Protein SRC2” was found in both CATS and CALS production environments, which has been reported to be upregulated during vernalisation105. 6 MTAs were found to be associated with days to maturity (DTM). Remarkably, SNP AX94521760 (7.89% PVE) linked with TraesCS1A02G433400 on Chr1A that translates into “Mitochondrial Pentatricopeptide repeat-containing protein At2g15690” which is reported to play a role in controlling the maturity time106,107. A “MADS-box transcription factor PHERES 2” coding gene TraesCS6B02G028300 was found to be linked to SNP AX94658713 (22.87%PVE) on Chr6B. The MADS-box genes belong to a well-known protein family that regulates floral transition, floral patterning, normal growth and development of carpels and fruits, and embryo and seed growth108,109,110. The identification of “MADS-box transcription factor PHERES 2” (upregulated under stress condition) associated SNP under CALS indicates its role in providing extended duration for grain filling111. SNP AX94544614 (7.85% PVE) on Chr1D was found to be linked with genes TraesCS1D02G066400 coding for “Polygalacturonase QRT2” regulating grain maturity112,113,114. For plant height (PH), SNP AX95153371 (3.95% PVE) on Chr2A was linked with two genes TraesCS2A02G041300 and TraesCS2A02G041500 which translates for “Fatty acid amide hydrolase” and “Ethylene-responsive transcription factor 1B”, respectively both of which have been reported to play key roles in regulating plant growth and height115,116,117. Two SNPs viz., AX95181791 (29.47% PVE) on Chr5A and AX95110699 (10.72% PVE) on Chr1D linked with genes TraesCS5A02G386600 and TraesCS5B02G252500 which code for proteins “YTH domain-containing protein ECT2” and “Serine / threonine-protein kinase D6PKL2” were found to be associated with number of productive tillers (TC). Arabidopsis ECT2/3/4 proteins are reported to be important for organogenesis and cell proliferation through binding with the m6A site via the YTH domain and affecting mRNA stability, and have been reported to play a role in modifying tiller numbers118.
Grain yield depends on various spike-related traits such as the spike length, number of spikelets per spike, grains per spike and thousand-grain weight119. Targeting such traits has shown fruitful results in improving grain yield over the last decades. Two QTNs (AX94650173 and AX94707304) were identified for spike length (SL) that were linked to three putative high-confidence protein-coding genes. SNP AX94650173 (21.89% PVE) on Chr2A was found to be linked to gene TraesCS2A02G212200 translating for “Transcription factor UDT1” and SNP AX94707304 (26.90% PVE) on Chr6B was linked with two genes TraesCS6B02G360300 and TraesCS6B02G360500 which code for proteins “Chloroplastic Soluble inorganic pyrophosphatase 6” and “WAT1-related protein At2g39510”. The WALLS ARE THIN 1 (WAT1) gene has been reported to play a key role in cell elongation through auxin transport, which signifies its effect on spike length modification120. Interestingly, many QTLs for spike length have been reported to be present in the 2AS region where SNP AX94650173 was found to be associated with SL in our present study121. The role played by these proteins in regulating spike length has been reported by Ko et al.122; Müller et al.123 and Nazari et al.124. The identification of SNP AX94650173 under CTLS may indicate its role in regulating SL under stress conditions. Genes TraesCS6B02G360300 and TraesCS6B02G360500 were also found to be linked to SNP AX94669346 (11.10% PVE) on Chr6B regulating the number of spikelets per spike (SPS). SNP AX94882016 (17.17% PVE) on Chr3B was found to be associated with grains per spike (GPS) linked with genes TraesCS3B02G303600, TraesCS3B02G304300 and TraesCS3B02G304200 which translates for proteins “Aldehyde oxidase GLOX1”, “Protein MALE DISCOVERER 1” and “Rop guanine nucleotide exchange factor 11”. It has been demonstrated that mutation and overexpression of an aldehyde oxidase gene OsAO3 increased and decreased grain yield, respectively, by affecting panicle number per plant, spikelet number per panicle and spikelet fertility in rice125 and plants tend to downregulate the expression of GLOX1 gene under stress condition as it has detrimental effects on pollen and pistil126. The MALE DISCOVERER 1 (MDIS1) gene is a receptor-like kinase that has been reported to play a key role in guiding the pollen tube growth by sensing the LURE signals during fertilisation in Arabidopsis127. A Rop guanine nucleotide exchange factor (OsRopGEF7B) was demonstrated to affect floral organ development and seed setting rate in Rice128. This SNP being identified under CTLS and the linked genes being expressed under stress conditions further signifies and makes their utilisation more evident in targeting stress tolerance and grain yield improvement in wheat.
Another key component trait that decides the grain yield is the thousand-grain weight (TGW) which highly depends upon the grain filling rate129. Two QTNs for TGW were identified where SNP AX94879209 on Chr3D was found to be linked with “Sodium/calcium exchanger NCL1” and “Basic leucine zipper 2” genes which play important functions in calcium homeostasis and abiotic stress tolerance130,131,132,133 while SNP AX94543129 on Chr6D was linked to a “SAP-like protein BP-73” which is reported to regulate cell proliferation in developing seeds134,135. The identification of NCL1 and bZIP gene associated QTN in CTTS makes it more evident that plants grown in CA environment are less exposed to stress compared to CT environment. For grain yield (GY), three SNPs viz., AX94514123, AX94803899 and AX94819762 were found to be linked to genes that code for proteins and protein families such as “NAC domain-containing protein 22”, “Beta-1,2-xylosyltransferease XAX1”, “Protein PSK SIMULATOR 1” and “Transcription factor MYB44” under CTTS environment. The roles of these proteins in regulating the grain yield under abiotic stress through modifying root architecture136,137,138, by regulating the xylan biosynthesis in the cell wall139, cell proliferation and light stress139,140, and stress tolerance by regulating stomatal development142,143,144. SNP AX94525104 (31.33% PVE) on Chr3A was found to be linked to genes TraesCS3A02G408200 and TraesCS3A02G408000 which translates for proteins “Bidirectional sugar transporter SWEET1a” and “Scarecrow-like protein 1”, respectively which play a key role in regulating the grain length (GL) through enhanced grain filling rates145,146,147 and promoting gibberellin signalling through repression of DELLA repressor protein148,149,150. The identification of this QTN under the CALS environment and the genes annotated being reported to play a pivotal role in regulating grain yield under stress conditions signifies the importance of this QTN in improving grain yield under stress conditions. Two SNPs viz., AX95194941 (6.56% PVE) on Chr5B and AX94857819 (27.66% PVE) on Chr7D which translates for “Oil body-associated protein 1A” and “Bifunctional monodehydroascorbate reductase and carbonic anhydrase nectarin-3”, respectively were found to be associated with grain breadth (GB) and play roles in regulating grain yield151,152,153,154. The SNP AX94857819 being identified under CALS emphasizes its importance in grain filling and stress tolerance.
In conclusion, this is the first report where we have identified key physiological traits viz., Phi2, NPQ, RC and PS1 which are responsible for improved performance of the genotypes under CA as compared to CT. These traits are the traits of adaptability under CA. Out of the 80 MTAs identified through GWAS, 48 were found to be highly associated with our traits of the present study as identified through in-silico gene annotation studies. The candidate genes with pivotal roles in trait development such as Chloroplastic Photosystem II reaction center W protein for Phi2 and Bidirectional sugar transporter SWEET1a for GL were identified through gene annotation studies. Upon validation, the identified QTNs hold potential for integration into marker-assisted breeding programs, contributing towards the development of wheat genotypes adapted to Conservation Agriculture.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Code availability
Publicly available statistical tools and databases were used for various analyses in the present study.
References
Erenstein, O. et al. Global trends in wheat production, consumption and trade. In Wheat Improvement (eds Reynolds, M. P. & Braun, H.-J.) (Springer International Publishing, 2022).
Mittal, S. Wheat and barley production trends and research priorities: A global perspective. In New Horizons in Wheat and Barley Research (eds Gupta, V. et al.) (Springer Singapore, 2022).
Sendhil, R. et al. Wheat in Asia: Trends, challenges and research priorities. In New Horizons in Wheat and Barley Research (eds Kashyap, P. L. et al.) (Springer Singapore, 2022).
Asseng, S. et al. Rising temperatures reduce global wheat production. Nat. Clim. Chang. 5(2), 143–147. https://doi.org/10.1038/nclimate2470 (2015).
Tesfaye, K. et al. Climate change impacts and potential benefits of heat-tolerant maize in South Asia. Theor. Appl. Climatol. 130(3–4), 959–970. https://doi.org/10.1007/s00704-016-1931-6 (2017).
Ortiz, R. et al. Climate change: Can wheat beat the heat?. Agric. Ecosyst. Environ. 126(1–2), 46–58. https://doi.org/10.1016/j.agee.2008.01.019 (2008).
Rahman, M. A., Chikushi, J., Yoshida, S. & Karim, A. Growth and yield components of wheat genotypes exposed to high temperature stress under control environment. Bangladesh J. Agric. Res. 34(3), 360–372. https://doi.org/10.3329/bjar.v34i3.3961 (2009).
Prasad, P. V. V., Pisipati, S. R., Ristic, Z., Bukovnik, U. & Fritz, A. K. Impact of nighttime temperature on physiology and growth of spring wheat. Crop Sci. 48(6), 2372–2380. https://doi.org/10.2135/cropsci2007.12.0717 (2008).
Jagadish, S. V. K., Craufurd, P. Q. & Wheeler, T. R. Phenotyping parents of mapping populations of rice for heat tolerance during anthesis. Crop Sci. 48(3), 1140–1146. https://doi.org/10.2135/cropsci2007100559 (2008).
Wheeler, T. R., Craufurd, P. Q., Ellis, R. H., Porter, J. R. & Vara Prasad, P. V. Temperature variability and the yield of annual crops. Agric. Ecosyst. Environ. 82(1–3), 159–167. https://doi.org/10.1016/S0167-8809(00)00224-3 (2000).
Gupta, R., Somanathan, E. & Dey, S. Global warming and local air pollution have reduced wheat yields in India. Clim. Chang. 140(3–4), 593–604. https://doi.org/10.1007/s10584-016-1878-8 (2017).
Richards, R. A. Physiological traits used in the breeding of new cultivars for water-scarce environments. Agric. Water Manag. 80(1–3), 197–211. https://doi.org/10.1016/j.agwat.2005.07.013 (2006).
Craufurd, P. Q. & Wheeler, T. R. Climate change and the flowering time of annual crops. J. Exp. Bot. 60(9), 2529–2539. https://doi.org/10.1093/jxb/erp196 (2009).
Gaikwad, K. B., Babu, P., Kumar, M., Kumar, N. & Yadav, R. Breeding wheat for conservation agriculture (ca) in the era of climate change. In New Horizons in Wheat and Barley Research (eds Gupta, V. et al.) (Springer Singapore, 2022).
Jat, R. K. et al. Seven years of conservation agriculture in a rice–wheat rotation of Eastern Gangetic Plains of South Asia: Yield trends and economic profitability. Field Crop. Res. 164(1), 199–210. https://doi.org/10.1016/J.FCR.2014.04.015 (2014).
Aryal, J. P. et al. Conservation agriculture-based wheat production better copes with extreme climate events than conventional tillage-based systems: A case of untimely excess rainfall in Haryana, India. Agric. Ecosyst. Environ. 233, 325–335. https://doi.org/10.1016/j.agee.2016.09.013 (2016).
Pradhan, A., Chan, C., Roul, P. K., Halbrendt, J. & Sipes, B. Potential of conservation agriculture (CA) for climate change adaptation and food security under rainfed uplands of India: A transdisciplinary approach. Agric. Syst. 163, 27–35. https://doi.org/10.1016/j.agsy.2017.01.002 (2018).
Herrera, J. M., Verhulst, N., Trethowan, R. M., Stamp, P. & Govaerts, B. Insights into genotype × tillage interaction effects on the grain yield of wheat and maize. Crop Sci. 53(5), 1845–1859. https://doi.org/10.2135/cropsci2013.01.0071 (2013).
Yadav, R., Gaikwad, K. B. & Bhattacharyya, R. Breeding wheat for yield maximization under conservation agriculture. Indian J. Genet. Plant Breed. 77(2), 185. https://doi.org/10.5958/0975-6906.2017.00026.8 (2017).
Singh, A., Phogat, V. K., Dahiya, R. & Batra, S. D. Impact of long-term zero till wheat on soil physical properties and wheat productivity under rice–wheat cropping system. Soil Tillage Res. 140, 98–105. https://doi.org/10.1016/j.still.2014.03.002 (2014).
Bhattacharyya, R. et al. Conservation agriculture effects on soil organic carbon accumulation and crop productivity under a rice–wheat cropping system in the western Indo-Gangetic Plains. Eur. J. Agron. 70, 11–21. https://doi.org/10.1016/j.eja.2015.06.006 (2015).
Devkota, M., Patil, S. B., Kumar, S., Kehel, Z. & Wery, J. Performance of elite genotypes of barley, chickpea, lentil, and wheat under conservation agriculture in Mediterranean rainfed conditions. Exp. Agric. 57(2), 126–143. https://doi.org/10.1017/S0014479721000107 (2021).
Sapkota, T. B., Jat, M. L., Aryal, J. P., Jat, R. K. & Khatri-Chhetri, A. Climate change adaptation, greenhouse gas mitigation and economic profitability of conservation agriculture: Some examples from cereal systems of Indo-Gangetic Plains. J. Integr. Agric. 14(8), 1524–1533. https://doi.org/10.1016/S2095-3119(15)61093-0 (2015).
Trethowan, R. M., Mahmood, T., Ali, Z., Oldach, K. & Garcia, A. G. Breeding wheat cultivars better adapted to conservation agriculture. Field Crop. Res. 132, 76–83. https://doi.org/10.1016/j.fcr.2011.10.015 (2012).
Tahmasebi, S., Heidari, B., Pakniyat, H. & McIntyre, C. L. Mapping QTLs associated with agronomic and physiological traits under terminal drought and heat stress conditions in wheat (Triticum aestivum L.). Genome 60(1), 26–45. https://doi.org/10.1139/gen-2016-0017 (2017).
Sukumaran, S., Reynolds, M. P. & Sansaloni, C. Genome-wide association analyses identify QTL hotspots for yield and component traits in durum wheat grown under yield potential, drought, and heat stress environments. Front. Plant Sci. https://doi.org/10.3389/fpls.2018.00081 (2018).
Yadav, R. et al. Identification of genotypes and marker validation for grain filling rate and grain filling duration in wheat under conservation agriculture. Indian J. Genet. Plant Breed. https://doi.org/10.31742/IJGPB.78.3.1 (2018).
Liu, J. et al. A genome-wide association study of wheat spike related traits in China. Front. Plant Sci. https://doi.org/10.3389/fpls.2018.01584 (2018).
Pradhan, S. et al. Genetic dissection of heat-responsive physiological traits to improve adaptation and increase yield potential in soft winter wheat. BMC Genom. 21(1), 315. https://doi.org/10.1186/s12864-020-6717-7 (2020).
Kumar, S. et al. Genome-wide association study reveals genomic regions associated with ten agronomical traits in wheat under late-sown conditions. Front. Plant Sci. https://doi.org/10.3389/fpls.2020.549743 (2020).
Pankaj, Y. K., Kumar, R., Gill, K. S. & Nagarajan, R. Unravelling QTLs for non-destructive and yield-related traits under timely, late and very late sown conditions in wheat (Triticum aestivum L.). Plant Mol. Biol. Report. https://doi.org/10.1007/s11105-022-01357-3 (2022).
Pankaj, Y. K. et al. Mapping QTLs for morpho-physiological traits related to grain yield under late sown conditions in wheat (Triticum aestivum L.). Cereal Res. Commun. 50(4), 779–788. https://doi.org/10.1007/s42976-021-00234-1 (2022).
Khan, H. et al. Genome-wide association study for grain yield and component traits in bread wheat (Triticum aestivum L.). Front. Genet. https://doi.org/10.3389/fgene.2022.982589 (2022).
Gill, H. S. et al. Whole-genome analysis of hard winter wheat germplasm identifies genomic regions associated with spike and kernel traits. Theor. Appl. Genet. 135(9), 2953–2967. https://doi.org/10.1007/s00122-022-04160-6 (2022).
Kuhlgert, S. et al. MultispeQ beta: A tool for large-scale plant phenotyping connected to the open PhotosynQ network. Royal Soc. Open Sci. 3(10), 160592. https://doi.org/10.1098/rsos.160592 (2016).
Zadoks, J. C., Chang, T. T. & Konzak, C. F. A decimal code for the growth stages of cereals. Weed Res. 14(6), 415–421. https://doi.org/10.1111/j.1365-3180.1974.tb01084.x (1974).
Tanabata, T., Shibaya, T., Hori, K., Ebana, K. & Yano, M. SmartGrain: High-throughput phenotyping software for measuring seed shape through image analysis. Plant Physiol. 160(4), 1871–1880. https://doi.org/10.1104/pp.112.205120 (2012).
Aravind J, Mukesh Sankar S, Wankhede DP, Kaur V (2023) augmentedRCBD: Analysis of augmented randomised complete block designs. https://doi.org/10.5281/ZENODO.8015094
Alvarado, G. et al. META-R: A software to analyze data from multi-environment plant breeding trials. Crop J. 8(5), 745–756. https://doi.org/10.1016/j.cj.2020.03.010 (2020).
Holland, J. B., Nyquist, W. E. & Cervantes-Martínez, C. T. Estimating and interpreting heritability for plant breeding: An Update. In Plant Breeding Reviews (ed. Janick, J.) 9–112 (Wiley, 2002).
Wei T, Simko V (2021) R package ‘corrplot’: Visualization of a correlation matrix. (Version 0.92). https://github.com/taiyun/corrplot
Bradbury, P. J. et al. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 23(19), 2633–2635 (2007).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155(2), 945–959. https://doi.org/10.1093/genetics/155.2.945 (2000).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 14(8), 2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x (2005).
Shin, J. H., Blay, S., Graham, J. & McNeney, B. LDheatmap: An R function for graphical display of pairwise linkage disequilibria between single nucleotide polymorphisms. J. Stat. Softw. https://doi.org/10.1863/jss.v016.c03 (2006).
Wang, J. & Zhang, Z. GAPIT Version 3: Boosting power and accuracy for genomic association and prediction. Genom. Proteom. Bioinform. 19(4), 629–640. https://doi.org/10.1016/j.gpb.2021.08.005 (2021).
Huang, M., Liu, X., Zhou, Y., Summers, R. M. & Zhang, Z. BLINK: A package for the next level of genome-wide association studies with both individuals and markers in the millions. GigaScience https://doi.org/10.1093/gigascience/giy154 (2019).
Singh, S. K., Singh, G., Singh, V. P. & Singh, A. Effect of establishment methods and weed management practices on weeds and rice in rice-wheat cropping system. Indian J. Weed Sci. 37, 51–57 (2005).
Bhatia, A., Ghosh, A., Kumar, A. & Bhattacharyya, R. Greenhouse gas emission and carbon sequestration in conservation agriculture. In Conservation Agriculture in India (ed. Sharma, A. R.) 223–242 (Routledge, 2022).
Ranjan, R. et al. Genetic variability for root traits and its role in adaptation under conservation agriculture in spring wheat. Indian J. Genet. Plant Breed. 81(01), 24–33. https://doi.org/10.31742/IJGPB.81.1.2 (2021).
Silva-Pérez, V. et al. Genetic variation for photosynthetic capacity and efficiency in spring wheat. J. Exp. Bot. 71(7), 2299–2311. https://doi.org/10.1093/jxb/erz439 (2020).
Narendra, M. C., Roy, C., Kumar, S., Virk, P. & De, N. Effect of terminal heat stress on physiological traits, grain zinc and iron content in wheat (Triticum aestivum L.). Czech J. Genet. Plant Breed. 57(2), 43–50. https://doi.org/10.17221/63/2020-CJGPB (2021).
Shirvani, F., Mohammadi, R., Daneshvar, M. & Ismaili, A. Genetic variability, response to selection for agro-physiological traits, and traits-enhanced drought tolerance in durum wheat. Acta Ecol. Sin. 43(5), 810–819. https://doi.org/10.1016/j.chnaes.2022.10.009 (2023).
Chen, J., Burke, J. J. & Xin, Z. Chlorophyll fluorescence analysis revealed essential roles of FtsH11 protease in regulation of the adaptive responses of photosynthetic systems to high temperature. BMC Plant Biol. 18(1), 11. https://doi.org/10.1186/s12870-018-1228-2 (2018).
Guidi, L., Lo Piccolo, E. & Landi, M. Chlorophyll fluorescence, photoinhibition and abiotic stress: Does it make any difference the fact to be a C3 or C4 species?. Front. Plant Sci. https://doi.org/10.3389/fpls.2019.00174 (2019).
Robson, J. K. et al. Chlorophyll fluorescence-based high-throughput phenotyping facilitates the genetic dissection of photosynthetic heat tolerance in African (Oryza glaberrima) and Asian (Oryza sativa) rice. J. Exp. Bot. 74(17), 5181–5197. https://doi.org/10.1093/jxb/erad239 (2023).
Sattar, A., Nanda, G., Singh, G., Jha, R. K. & Bal, S. K. Responses of phenology, yield attributes, and yield of wheat varieties under different sowing times in Indo-Gangetic Plains. Front. Plant Sci. https://doi.org/10.3389/fpls.2023.1224334 (2023).
Gao, Z. et al. Plant height and its relationship with yield in wheat under different irrigation regimes. Irrig. Sci. 38(4), 365–371. https://doi.org/10.1007/s00271-020-00678-z (2020).
Baye, A., Berihun, B., Bantayehu, M. & Derebe, B. Genotypic and phenotypic correlation and path coefficient analysis for yield and yield-related traits in advanced bread wheat (Triticum aestivum L.) lines. Cogent Food Agric. 6(1), 1752603. https://doi.org/10.1080/23311932.2020.1752603 (2020).
Rasheed, A. et al. Genome-wide association for grain morphology in synthetic hexaploid wheats using digital imaging analysis. BMC Plant Biol. 14(1), 128. https://doi.org/10.1186/1471-2229-14-128 (2014).
Gao, Y. et al. Dissecting the genetic basis of grain morphology traits in Chinese wheat by genome-wide association study. Euphytica 217(4), 56. https://doi.org/10.1007/s10681-021-02795-y (2021).
Saxena, D. C. et al. Evaluation of root characteristics, canopy temperature depression and stay green trait in relation to grain yield in wheat under early and late sown conditions. Indian J. Plant Physiol. 19(1), 43–47. https://doi.org/10.1007/s40502-014-0071-1 (2014).
Thapa, S. et al. Canopy temperature depression at grain filling correlates to winter wheat yield in the U.S Southern High Plains. Field Crop. Res. 217, 11–19. https://doi.org/10.1016/j.fcr.2017.12.005 (2018).
Khan, A., Ahmad, M., Shah, M. K. N. & Ahmed, M. Performance of wheat genotypes for morpho-physiological traits using multivariate analysis under terminal heat stress. Pakistan J. Bot. https://doi.org/10.30848/PJB2020-6(30) (2020).
Gupta, P. K., Kulwal, P. L. & Jaiswal, V. Association Mapping in Plants in the Post-GWAS Genomics Era (Elsevier, 2019).
Flint-Garcia, S. A., Thornsberry, J. M. & Buckler, E. S. Structure of linkage disequilibrium in plants. Annu. Rev. Plant Biol. 54(1), 357–374. https://doi.org/10.1146/annurev.arplant.54.031902.134907 (2003).
Krishnappa, G. et al. Genetic dissection of grain iron and zinc, and thousand kernel weight in wheat (Triticum aestivum L.) using genome-wide association study. Sci. Rep. 12(1), 12444. https://doi.org/10.1038/s41598-022-15992-z (2022).
Gilmore, A. M., Hazlett, T. L., Debrunner, P. G. & Govindjee,. Comparative time-resolved photosystem II chlorophyll a fluorescence analyses reveal distinctive differences between photoinhibitory reaction center damage and xanthophyll cycle-dependent energy dissipation. Photochem. Photobiol. 64(3), 552–563. https://doi.org/10.1111/j.1751-1097.1996.tb03105.x (1996).
Murphy, C. D. et al. Quantitating active photosystem II reaction center content from fluorescence induction transients. Limnol. Oceanogr. Methods 15(1), 54–69. https://doi.org/10.1002/lom3.10142 (2017).
Süssenbacher, I. et al. Cryptic chlorophyll breakdown in non-senescent green Arabidopsis thaliana leaves. Photosynth. Res. 142(1), 69–85. https://doi.org/10.1007/s11120-019-00649-2 (2019).
Gorfer, L. M. et al. Chlorophyll breakdown in fruits: The relevance of the senescence-related pheophorbide a oxygenase/phyllobilin (PaO/PB) pathway in ripe ‘Gala’ apples (Malus domestica). Acta Hortic. 1353, 9–18. https://doi.org/10.17660/ActaHortic.2022.1353.2 (2022).
Liu, Y. et al. A novel single-base mutation in CaSGR1 confers the stay-green phenotype in pepper. Hortic. Plant J. 9(2), 293–305. https://doi.org/10.1016/j.hpj.2022.04.002 (2023).
Gaikwad, K. B. et al. Synthetic hexaploid wheat as a source of variation for the traits specific to conservation agriculture. Indian J. Genet. Plant Breed. 83(01), 32–40. https://doi.org/10.31742/ISGPB.83.1.5 (2023).
Chu, S. et al. Physiological and proteomics analyses reveal low-phosphorus stress affected the regulation of photosynthesis in soybean. Int. J. Mol. Sci. 19(6), 1688. https://doi.org/10.3390/ijms19061688 (2018).
Peng, X., Teng, L., Yan, X., Zhao, M. & Shen, S. The cold responsive mechanism of the paper mulberry: Decreased photosynthesis capacity and increased starch accumulation. BMC Genom. 16(1), 898. https://doi.org/10.1186/s12864-015-2047-6 (2015).
Yang, Z. et al. Systems-level analysis of the metabolic responses of the diatom Phaeodactylum tricornutum to phosphorus stress. Environ. Microbiol. 16(6), 1793–1807. https://doi.org/10.1111/1462-2920.12411 (2014).
Paul, P. et al. Structural and functional heat stress responses of chloroplasts of Arabidopsis thaliana. Genes 11(6), 650. https://doi.org/10.3390/genes11060650 (2020).
Misra, A. N. et al. Action and target sites of nitric oxide in chloroplasts. Nitric Oxide 39, 35–45. https://doi.org/10.1016/j.niox.2014.04.003 (2014).
Ding, W., Ye, Y., Yu, L., Liu, M. & Liu, J. Physiochemical and molecular responses of the diatom Phaeodactylum tricornutum to illumination transitions. Biotechnol. Biofuels Bioprod. 16(1), 103. https://doi.org/10.1186/s13068-023-02352-w (2023).
Qu, J. et al. New insights into the response of maize to fluctuations in the light environment. Mol. Genet. Genom. 296(3), 615–629. https://doi.org/10.1007/s00438-021-01761-6 (2021).
Tamang, B. & Fukao, T. Plant adaptation to multiple stresses during submergence and following desubmergence. Int. J. Mol. Sci. 16(12), 30164–30180. https://doi.org/10.3390/ijms161226226 (2015).
Huseynova, I. M., Suleymanov, S. Y., Rustamova, S. M. & Aliyev, J. A. Drought-induced changes in photosynthetic membranes of two wheat (Triticum aestivum L.) cultivars. Biochemistry 74(8), 903–909. https://doi.org/10.1134/S0006297909080124 (2009).
Kromdijk, J., Głowacka, K. & Long, S. P. Photosynthetic efficiency and mesophyll conductance are unaffected in Arabidopsis thaliana aquaporin knock-out lines. J. Exp. Bot. https://doi.org/10.1093/jxb/erz442 (2019).
Zhang, X. et al. Exogenous strigolactones alleviate the photosynthetic inhibition and oxidative damage of cucumber seedlings under salt stress. Sci. Hortic. 297, 110962. https://doi.org/10.1016/j.scienta.2022.110962 (2022).
Zhu, Y., Liberton, M. & Pakrasi, H. B. A novel redoxin in the thylakoid membrane regulates the titer of photosystem I. J. Biol. Chem. 291(36), 18689–18699. https://doi.org/10.1074/jbc.M116.721175 (2016).
Roy, C. et al. Association of leaf chlorophyll content with the stay-green trait and grain yield in wheat grown under heat stress conditions. Czech J. Genet. Plant Breed. 57(4), 140–148. https://doi.org/10.17221/45/2021-CJGPB (2021).
Zhang, Y. et al. Transfer-learning-based approach for leaf chlorophyll content estimation of winter wheat from hyperspectral data. Remote Sens. Environ. 267, 112724. https://doi.org/10.1016/j.rse.2021.112724 (2021).
Zheng, S. et al. Hexokinase gene OsHXK1 positively regulates leaf senescence in rice. BMC Plant Biol. 21(1), 580. https://doi.org/10.1186/s12870-021-03343-5 (2021).
Dai, N. et al. Overexpression of Arabidopsis hexokinase in tomato plants inhibits growth, reduces photosynthesis, and induces rapid senescence. Plant Cell 11(7), 1253–1266. https://doi.org/10.1105/tpc.11.7.1253 (1999).
Baader, W. J. & Gillo, V. E. Peroxidase-catalysed chlorophyll destruction accompanied by the formation of electronically excited states: Chlorophyll emission not due to energy transfer from excited substrate metabolites. J. Photochem. Photobiol. A Chem. 101(1), 49–55. https://doi.org/10.1016/S1010-6030(96)04424-3 (1996).
Kim, S. R., Yang, J. I. & An, G. OsCpn60α1, encoding the plastid chaperonin 60α subunit, is essential for folding of rbcL. Mol. Cells 35(5), 402–409. https://doi.org/10.1007/s10059-013-2337-2 (2013).
Jiang, Q. et al. Importance of the rice TCD9 encoding α subunit of chaperonin protein 60 (Cpn60α) for the chloroplast development during the early leaf stage. Plant Sci. 215–216, 172–179. https://doi.org/10.1016/j.plantsci.2013.11.003 (2014).
Li, L., Li, K., Ali, A. & Guo, Y. AtWAKL10, a cell wall associated receptor-like kinase, negatively regulates leaf senescence in Arabidopsis thaliana. Int. J. Mol. Sci. 22(9), 4885. https://doi.org/10.3390/ijms22094885 (2021).
Riou, C., Hervé, C., Pacquit, V., Dabos, P. & Lescure, B. Expression of an Arabidopsis lectin kinase receptor gene, lecRK-a1, is induced during senescence, wounding and in response to oligogalacturonic acids. Plant Physiol. Biochem. 40(5), 431–438. https://doi.org/10.1016/S0981-9428(02)01390-6 (2002).
Zhao, M. et al. GmWAK1, novel wall-associated protein kinase, positively regulates response of soybean to Phytophthora sojae infection. Int. J. Mol. Sci. 24(1), 798. https://doi.org/10.3390/ijms24010798 (2023).
Rojas, C. A. et al. Overexpression of the Arabidopsis anaphase promoting complex subunit CDC27a increases growth rate and organ size. Plant Mol. Biol. 71(3), 307–318. https://doi.org/10.1007/s11103-009-9525-7 (2009).
Mohan, D., Mamrutha, H. M., Khobra, R., Singh, G. & Singh, G. P. Relevance of height, heading and maturity in productivity enhancement of wheat. Indian J. Genet. Plant Breed. 82(01), 31–37. https://doi.org/10.31742/IJGPB.82.1.5 (2022).
Liu, L., Xuan, L., Jiang, Y. & Yu, H. Regulation by FLOWERING LOCUS T and TERMINAL FLOWER 1 in flowering time and plant architecture. Small Struct. https://doi.org/10.1002/sstr.202000125 (2021).
Ahmad, S. et al. Transcriptional cascade in the regulation of flowering in the bamboo orchid Arundina graminifolia. Biomolecules 11(6), 771. https://doi.org/10.3390/biom11060771 (2021).
Li, L. et al. Genome wide analysis of flowering time trait in multiple environments via high-throughput genotyping technique in Brassica napus L. PLoS ONE 10(3), e0119425. https://doi.org/10.1371/journal.pone.0119425 (2015).
Alptekin, B. et al. Combined effects of a glycine-rich RNA-binding protein and a NAC transcription factor extend grain fill duration and improve malt barley agronomic performance. Theor. Appl. Genet. 134(1), 351–366. https://doi.org/10.1007/s00122-020-03701-1 (2021).
Ma, L. et al. Roles of plant glycine-rich RNA-binding proteins in development and stress responses. Int. J. Mol. Sci. 22(11), 5849. https://doi.org/10.3390/ijms22115849 (2021).
Byrne, S. L. et al. Identification of genes involved in the floral transition at the shoot apical meristem of Lolium perenne L. by use of suppression subtractive hybridisation. Plant Growth Regul. 59(3), 215–225. https://doi.org/10.1007/s10725-009-9407-7 (2009).
Strejčková, B. et al. Wild emmer wheat, the progenitor of modern bread wheat, exhibits great diversity in the VERNALIZATION1 gene. Front. Plant Sci. https://doi.org/10.3389/fpls.2022.1106164 (2023).
Han, X. et al. Genome-wide detection of genotype environment interactions for flowering time in Brassica napus. Front. Plant Sci. https://doi.org/10.3389/fpls.2022.1065766 (2022).
Zhang, J. et al. PDM3, a pentatricopeptide repeat-containing protein, affects chloroplast development. J. Exp. Bot. 68(20), 5615–5627. https://doi.org/10.1093/jxb/erx360 (2017).
Sahu, T. K. et al. An associative transcriptomics study on rice bean (Vigna umbellata) provides new insights into genetic basis and candidate genes governing flowering, maturity and seed weight. bioRxiv https://doi.org/10.1101/2023.02.25.530014 (2023).
Teo, Z. W. N., Zhou, W. & Shen, L. Dissecting the function of MADS-box transcription factors in orchid reproductive development. Front. Plant Sci. https://doi.org/10.3389/fpls.2019.01474 (2019).
Arora, R. et al. MADS-box gene family in rice: Genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genom. 8(1), 242. https://doi.org/10.1186/1471-2164-8-242 (2007).
Kumar, K. et al. Identification and characterization of MADS box gene family in pigeonpea for their role during floral transition. 3 Biotech 11(2), 108. https://doi.org/10.1007/s13205-020-02605-7 (2021).
Castelán-Muñoz, N. et al. MADS-box genes are key components of genetic regulatory networks involved in abiotic stress and plastic developmental responses in plants. Front. Plant Sci. https://doi.org/10.3389/fpls.2019.00853 (2019).
Ogawa, M., Kay, P., Wilson, S. & Swain, S. M. ARABIDOPSIS DEHISCENCE ZONE POLYGALACTURONASE1 (ADPG1), ADPG2, and QUARTET2 are polygalacturonases required for cell separation during reproductive development in Arabidopsis. Plant Cell 21(1), 216–233. https://doi.org/10.1105/tpc.108.063768 (2009).
Qian, M. et al. Identification and expression analysis of polygalacturonase family members during peach fruit softening. Int. J. Mol. Sci. 17(11), 1933. https://doi.org/10.3390/ijms17111933 (2016).
Ye, J. et al. Comprehensive analysis of polygalacturonase gene family highlights candidate genes related to pollen development and male fertility in wheat (Triticum aestivum L.). Planta 252(2), 31. https://doi.org/10.1007/s00425-020-03435-w (2020).
Teaster, N. D. et al. Overexpression of fatty acid amide hydrolase induces early flowering in Arabidopsis thaliana. Front. Plant Sci. https://doi.org/10.3389/fpls.2012.00032 (2012).
Arias-Gaguancela, O., Adhikari, B., Aziz, M. & Chapman, K. D. Enhanced seedling growth by 3-n-pentadecylphenolethanolamide is mediated by fatty acid amide hydrolases in upland cotton (Gossypium hirsutum L.). Plant Direct https://doi.org/10.1002/pld3.421 (2022).
Xu, Z. et al. An APETALA2/ethylene responsive factor transcription factor GmCRF4a regulates plant height and auxin biosynthesis in soybean. Front. Plant Sci. https://doi.org/10.3389/fpls.2022.983650 (2022).
Ma, W. et al. YTH domain proteins play an essential role in rice growth and stress response. Plants 11(17), 2206. https://doi.org/10.3390/plants11172206 (2022).
Guo, Z. et al. Manipulation and prediction of spike morphology traits for the improvement of grain yield in wheat. Sci. Rep. 8(1), 14435. https://doi.org/10.1038/s41598-018-31977-3 (2018).
Huang, Z. et al. Euryale Small Auxin Up RNA62 promotes cell elongation and seed size by altering the distribution of indole-3-acetic acid under the light. Front. Plant Sci. https://doi.org/10.3389/fpls.2022.931063 (2022).
Yu, Q. et al. Genetic dissection of three major quantitative trait loci for spike compactness and length in bread wheat (Triticum aestivum L.). Front. Plant Sci. https://doi.org/10.3389/fpls.2022.882655 (2022).
Ko, S. S. et al. Rice transcription factor GAMYB modulates bHLH142 and is homeostatically regulated by TDR during anther tapetal and pollen development. J. Exp. Bot. 72(13), 4888–4903. https://doi.org/10.1093/jxb/erab190 (2021).
Müller, B. et al. Amino acid export in developing Arabidopsis seeds depends on Umamit facilitators. Curr. Biol. 25(23), 3126–3131. https://doi.org/10.1016/j.cub.2015.10.038 (2015).
Nazari, M., Moosavi, S. S., Maleki, M. & Jamshidi Goharrizi, K. Chloroplastic acyl carrier protein synthase I and chloroplastic 20 kDa chaperonin proteins are involved in wheat (Triticum aestivum) in response to moisture stress. J. Plant Interact. 15(1), 180–187. https://doi.org/10.1080/17429145.2020.1758812 (2020).
Shi, X., Tian, Q., Deng, P., Zhang, W. & Jing, W. The rice aldehyde oxidase OsAO3 gene regulates plant growth, grain yield, and drought tolerance by participating in ABA biosynthesis. Biochem. Biophys. Res. Commun. 548, 189–195. https://doi.org/10.1016/j.bbrc.2021.02.047 (2021).
Lohani, N., Singh, M. B. & Bhalla, P. L. RNA-Seq highlights molecular events associated with impaired pollen-pistil interactions following short-term heat stress in Brassica napus. Front. Plant Sci. https://doi.org/10.3389/fpls.2020.622748 (2021).
Cui, Y., Lu, X. & Gou, X. Receptor-like protein kinases in plant reproduction: Current understanding and future perspectives. Plant Commun. 3(1), 100273. https://doi.org/10.1016/j.xplc.2021.100273 (2022).
Huang, J. et al. Guanine Nucleotide Exchange Factor 7B (RopGEF7B) is involved in floral organ development in Oryza sativa. Rice 11(1), 42. https://doi.org/10.1186/s12284-018-0235-0 (2018).
Li, L. et al. Physiological and cytological analyses of the thousand-grain weight in ‘Zhongyan96-3’ wheat. J. Plant Growth Regul. 42(4), 2212–2220. https://doi.org/10.1007/s00344-022-10694-2 (2023).
Zeng, D. et al. The memory of rice response to spaceflight stress: From the perspective of metabolomics and proteomics. Int. J. Mol. Sci. 23(6), 3390. https://doi.org/10.3390/ijms23063390 (2022).
Fan, M. et al. Integration of the transcriptome and proteome provides insights into the calcium-regulated mechanism of Ulva prolifera in response to high-temperature stress. Aquaculture 557, 738344. https://doi.org/10.1016/j.aquaculture.2022.738344 (2022).
Kang, J., Choi, H., Im, M. & Kim, S. Y. Arabidopsis basic leucine zipper proteins that mediate stress-responsive abscisic acid signaling. Plant Cell 14(2), 343–357. https://doi.org/10.1105/tpc.010362 (2002).
Wang, J. C., Xu, H., Zhu, Y., Liu, Q. Q. & Cai, X. L. OsbZIP58, a basic leucine zipper transcription factor, regulates starch biosynthesis in rice endosperm. J. Exp. Bot. 64(11), 3453–3466. https://doi.org/10.1093/jxb/ert187 (2013).
Chen, J., Tang, W. H., Hong, M. M. & Wang, Z. Y. OsBP-73, a rice gene, encodes a novel DNA-binding protein with a SAP-like domain and its genetic interference by double-stranded RNA inhibits rice growth. Plant Mol. Biol. 52(3), 579–590. https://doi.org/10.1023/a:1024854101965 (2003).
Lloret, A. et al. Dual regulation of water retention and cell growth by a stress-associated protein (SAP) gene in Prunus. Sci. Rep. 7(1), 332. https://doi.org/10.1038/s41598-017-00471-7 (2017).
Redillas, M. C. F. R. et al. The overexpression of OsNAC9 alters the root architecture of rice plants enhancing drought resistance and grain yield under field conditions. Plant Biotechnol. J. 10(7), 792–805. https://doi.org/10.1111/j.1467-7652.2012.00697.x (2012).
Hu, H. et al. Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc. Natl. Acad. Sci. 103(35), 12987–12992. https://doi.org/10.1073/pnas.0604882103 (2006).
Jeong, J. S. et al. OsNAC5 overexpression enlarges root diameter in rice plants leading to enhanced drought tolerance and increased grain yield in the field. Plant Biotechnol. J. 11(1), 101–114. https://doi.org/10.1111/pbi.12011 (2013).
Xu, Z. et al. Genetic connection between cell-wall composition and grain yield via parallel QTL analysis in indica and japonica subspecies. Sci. Rep. 7(1), 12561. https://doi.org/10.1038/s41598-017-12903-5 (2017).
Jaškūnė, K., Kemešytė, V., Aleliūnas, A. & Statkevičiūtė, G. Genome-wide markers for seed yield and disease resistance in perennial ryegrass. Crop J. 10(2), 508–514. https://doi.org/10.1016/j.cj.2021.07.005 (2022).
Parrotta, L., Mareri, L. & Cai, G. Environmental stress and plants 2.0. Int. J. Mol. Sci. 24(15), 12413. https://doi.org/10.3390/ijms241512413 (2023).
Datta, K. et al. Overexpression of Arabidopsis and Rice stress genes’ inducible transcription factor confers drought and salinity tolerance to rice. Plant Biotechnol. J. 10(5), 579–586. https://doi.org/10.1111/j.1467-7652.2012.00688.x (2012).
Hussain, Q. et al. Transcription factors interact with ABA through gene expression and signaling pathways to mitigate drought and salinity stress. Biomolecules 11(8), 1159. https://doi.org/10.3390/biom11081159 (2021).
Shamloo-Dashtpagerdi, R., Razi, H., Ebrahimie, E. & Niazi, A. Molecular characterization of Brassica napus stress-related transcription factors, BnMYB44 and BnVIP1, selected based on comparative analysis of Arabidopsis thaliana and Eutrema salsugineum transcriptomes. Mol. Biol. Rep. 45(5), 1111–1124. https://doi.org/10.1007/s11033-018-4262-0 (2018).
Jeena, G. S., Kumar, S. & Shukla, R. K. Structure, evolution and diverse physiological roles of SWEET sugar transporters in plants. Plant Mol. Biol. 100(4–5), 351–365. https://doi.org/10.1007/s11103-019-00872-4 (2019).
Kim, J. Y. et al. Cellular export of sugars and amino acids: Role in feeding other cells and organisms. Plant Physiol. 187(4), 1893–1914. https://doi.org/10.1093/plphys/kiab228 (2021).
Liu, M. et al. Characterization and functional analysis of ZmSWEET15a in maize. DNA Cell Biol. 41(6), 564–574. https://doi.org/10.1089/dna.2021.1144 (2022).
Zhang, Z.-L. et al. SCARECROW-LIKE 3 promotes gibberellin signaling by antagonizing master growth repressor DELLA in Arabidopsis. Proc. Natl. Acad. Sci. 108(5), 2160–2165. https://doi.org/10.1073/pnas.1012232108 (2011).
Gao, M. J. et al. SCARECROW-LIKE15 interacts with HISTONE DEACETYLASE19 and is essential for repressing the seed maturation programme. Nat. Commun. 6(1), 7243. https://doi.org/10.1038/ncomms8243 (2015).
Finegan, C. et al. Genetic perturbation of the starch biosynthesis in maize endosperm reveals sugar-responsive gene networks. Front. Plant Sci. https://doi.org/10.3389/fpls.2021.800326 (2022).
Thakur, A. & Bhatla, S. C. Proteomic analysis of oil body membrane proteins accompanying the onset of desiccation phase during sunflower seed development. Plant Signal. Behavior 10(12), e1030100. https://doi.org/10.1080/15592324.2015.1030100 (2015).
Baud, S., Dubreucq, B., Miquel, M., Rochat, C. & Lepiniec, L. Storage reserve accumulation in Arabidopsis: Metabolic and developmental control of seed filling. Arabidopsis Book 6, e0113. https://doi.org/10.1199/tab.0113 (2008).
Singh, B. K. et al. Meta-analysis of microarray data and their utility in dissecting the mapped QTLs for heat acclimation in rice. Plants 12(8), 1697. https://doi.org/10.3390/plants12081697 (2023).
Cheng, Y., Mou, Y., Zhang, X., Liu, C. & Liu, J. iTRAQ protein profiling reveals candidate proteins regulating ovary and ovule differentiation in pistillate inflorescences after pollination in hazel. Tree Genet. Genomes 15(2), 21. https://doi.org/10.1007/s11295-019-1328-7 (2019).
Acknowledgements
The authors would like to thank ICAR-Indian Agricultural Research Institute, New Delhi for providing the resources to conduct the experiments.
Funding
This work was partially supported from the collaborative Project on “Application of Next-Generation Breeding, Genotyping and Digitalization Approaches for Improving the Genetic Gain in Indian Staple Crops” by the Indian Council of Agricultural Research (ICAR) and Bill and Melinda Gates Foundation (BMGF) [ICAR-IARI project code: 12-229].
Author information
Authors and Affiliations
Contributions
AKM and KBG conceptualised the experiment and designed the methodology; RY, KBG and MK developed the experimental material; AKM recorded the trait observations; AKM, MK and KBG curated the data and performed the data analysis and visualisation; AKM prepared the initial draft of the manuscript; KBG, AUS, SKS, PB, NK, SHW, AIA and AA reviewed and edited the previous versions of the final manuscript; All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mazumder, A.K., Yadav, R., Kumar, M. et al. Discovering novel genomic regions explaining adaptation of bread wheat to conservation agriculture through GWAS. Sci Rep 14, 16351 (2024). https://doi.org/10.1038/s41598-024-66903-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-66903-3
Keywords
This article is cited by
-
Genome wide dissection and haplotype analysis to identify candidate loci for harvest index under spot blotch in bread wheat
Molecular Genetics and Genomics (2026)
-
Dissecting the genetic architecture of pre-harvest sprouting tolerance in Indian dwarf wheat (Triticum sphaerococcum) by multi-locus association analysis
Scientific Reports (2025)
-
Evaluating heat and drought resilience in ancient Indian Dwarf wheat Triticum sphaerococcum Percival using stress tolerance indices
Scientific Reports (2025)
