
Abstract
The individual variation of carcinogenesis and drug response is influenced by the absorption, distribution, metabolism, and excretion (ADME) of drugs. The utilization of signatures derived from ADME-related genes holds potential for predicting prognosis and treatment response across diverse cancer types. Further investigation is required to completely understand the role of ADME-associated genes in breast cancer. A signature was constructed through the application of a least absolute shrinkage and selection operator regression model, employing prognostic differentially expressed genes found in both cancer tissue and normal tissue. To assess the robustness of the signature, verification analyses were carried out. RT-qPCR was utilized for the validation of gene expression related to risk. Subsequently, a nomogram was developed to enhance the clinical utility of our prognostic tool. The ADME signature, comprising four genes, was established and exhibited a robust association with the prognoses of individuals diagnosed with breast cancer. The nomogram was created by fusing the clinicopathological characteristics with the ADME signature. The ADME signature demonstrated remarkable superiority when compared to the performance of the other individual predictors. Additionally, the analysis of the immune microenvironment revealed that the ImmuneScores of the low-risk group were elevated. The variation in both the infiltration of immune cells and the expression of immune-related genes in the tissues differed among the two groups. For patients with breast cancer, the utilization of ADME signatures as biomarkers presents a significant reference point for prognosis and individualized treatment strategies.
Similar content being viewed by others

Introduction
Breast cancer is acknowledged as a prevalent malignancy that impacts women on a global scale, with an increasing incidence rate documented each year1,2,3. The disease is characterized by a high degree of heterogeneity, resulting in variations in clinical presentation, morphology, molecular profile, treatment response, and prognosis among individual cases4,5. Clinically, tumor stage, histological grade and molecular subtype are commonly used as prognostic indicators for breast cancer patients6. However, accurate prediction of patient prognosis based on clinicopathological characteristics remains challenging. This limitation may result in misjudgments of prognosis, potentially leading to inappropriate treatment decisions that could increase the risk of metastasis or recurrence in high-risk patients and unnecessary or overly aggressive treatment in low-risk patients7. In order to optimize patient treatment for individuals diagnosed with breast cancer, it is imperative to identify innovative molecular markers that can reliably predict the prognosis.
ADME-related genes refer to the genes involved in the absorption, distribution, metabolism, and excretion of drugs, which are composed of 266 extended genes and 32 core genes8,9,10. Based on their roles in the pharmacokinetic process, these genes can be classified into various categories such as drug transporters, modified proteins, and drug metabolic enzymes11,12,13. Increasing evidence suggests that polymorphisms in ADME-related genes play a significant role in individual differences in carcinogenesis and drug response14,15. Likewise, population variations in the regulation of ADME-related genes are observed at the transcriptional, translational, and epigenetic levels16,17. ADME-related genes have been identified as significant contributors to various cancer types. In the context of gastric cancer, the decreased expression of ALDH6A1 may serve as a prognostic indicator and a determinant of response to immunotherapy18. Furthermore, the pivotal roles of ADH1B and its associated network in immune modulation and therapeutic efficacy suggest their potential as therapeutic targets in ovarian cancer, as demonstrated in a study by Xu et al.19. Nan et al. found that the PLD/PA lipid signal transduction pathway, facilitated by D-AS2/FAM3D, plays a crucial role in conferring chemical resistance in squamous cell carcinoma. This suggests that targeting D-AS2 may enhance the sensitivity of squamous cell carcinoma to cytotoxic chemotherapy agents20. However, the impact of ADME-related genes on breast cancer remains unclear.
In this research, gene expression data obtained from The Cancer Genome Atlas (TCGA) was utilized to identify ADME-associated genes with unique expression patterns in breast cancer tissue compared to normal tissue. The least absolute shrinkage and selection operator (LASSO) method was then employed to develop a prognostic signature comprising four genes for predicting the survival outcomes of breast cancer patients. To ensure the accuracy of our model, we performed validation utilizing the TCGA-test dataset and five external datasets. Subsequently, we assessed functional enrichment, immune cell infiltration, response to immunotherapy, and drug sensitivity to compare high-risk and low-risk groups. Our results indicated that a gene-based signature related to ADME was associated with immune cell infiltration, and could predict patient prognosis and response to treatment.
Materials and methods
Data obtain
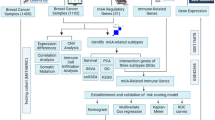
Figure 1 illustrates the research process. The information of gene expression and the associated clinical data was discovered by searching the TCGA database. Using R software, TCGA-Breast invasive carcinoma (BRCA) dataset was divided into train and test subgroups in a 1:1 ratio, as shown in Supplementary Table S1. The GSE20685, GSE7390, GSE20711, GSE25066, GSE58812 dataset was obtained from the Gene Expression Omnibus (GEO) repository. The drug sensitivity validation cohort was obtained from the GSE130787 dataset, which assessed the effectiveness of neoadjuvant docetaxel, carboplatin, trastuzumab, and/or lapatinib treatment in breast cancer patients. The expression data was used for subsequent analysis after standardization. Total 298 ADME-related genes shown in Supplementary Table S2 are obtained from previous literature9.
The creation and validation of the predictive mark
To identify differentially expressed genes (DEGs) associated with prognosis, a Cox regression analysis was conducted using univariate analysis. Subsequently, a signature was developed by incorporating four genes selected through multivariate Cox and LASSO regression analyses21. The individuals with breast cancer were categorized into low-risk and high-risk groups based on the utilization of the median risk score. Afterwards, the overall survival (OS) was evaluated through Kaplan-Meier (KM) analysis. The examination of subgroups’ OS and Receiver Operating Characteristics (ROC) was conducted utilizing the R packages for durations of 1, 3, and 5 years. The R software package ‘ggplot2’ was employed to carry out a principal component analysis (PCA). A nomogram was constructed by integrating risk assessment and clinical data.
Comparative analysis of tumor microenvironment (TME) in comparison to groups at high and low risk
The ESTIMATE22 was used to assess the composition of immune cells (ImmuneScores). To examine the disparities in immune cell infiltration between high-risk and low-risk classifications, we utilized a range of algorithms such as TIMER, CIBERSORT-ABS, QUANTISEQ, EPIC, MCPCOUNTER, CIBERSORT, and XCELL. The presence of different immune cells and their immune activity were examined using single-sample gene set enrichment analysis (ssGSEA). Furthermore, immune-related gene expression patterns were identified. Additionally, we also examined the differences in response to immunotherapy between these two groups.
Analyzing the pathway of the identified signature
An analysis was performed on DEGs within high- and low-risk categories. The enrichment of pathways related to these DEGs was evaluated through the utilization of Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Disease Ontology (DO) analyses23,24,25. Additionally, a gene set variation analysis (GSVA) was conducted to assess potential disparities in biological functioning.
Drug sensitivity analysis
The Genomics of Drugs Sensitivity in Cancer (GDSC) is the largest pharmacogenomics online database. To predict the chemotherapeutic sensitivity of the prognostic model, we assessed the IC50 values of AZ628, CGP-60,474, FTI-277, GSK429286A, IPA-3, KIN001-102, TAE684, XL-184, ZSTK474 drugs from the GDSC database by utilizing the pRRophetic algorithm with ridge regression26.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
BT-549, a tumor cell, and MCF-10 A, a normal cell, were obtained from the Shanghai Institute of Biochemistry and Cell Biology. The cell lines were grown in DMEM, with 10% fetal bovine serum (FBS), 100 µg/ml streptomycin, and 100 U/ml penicillin added to them (Gibco). The extraction of total RNA was conducted using Trizol, which was subsequently followed by reverse transcription to produce cDNA. Subsequently, the RT-qPCR analysis was conducted employing SYBR Green (Toyobo) as a facilitator. The internal reference gene β-Actin was utilized for normalization purposes.
Statistical analysis
The statistical calculations and visualizations in this study were conducted using the R software. The KM survival curve, along with the log-rank test, was employed to compare outcomes across different groups. P ≤ 0.05 was considered statistically significant.

The workflow of the study.
Results
Extraction of ADME-related genes with noteworthy prognostic value
The TCGA dataset was utilized to determine the expression levels of 298 genes related to ADME in both normal and breast cancer populations. The differential expression of ADME-related genes across various breast cancer subtypes was observed, as depicted in Supplementary Fig. S1. Within the HER2-enriched subtype, heightened NAT1 expression was significantly correlated with an unfavorable prognosis (P = 0.047) (Supplementary Fig. S2A). Within the Luminal B subtype, elevated ADH1B expression was associated with a poorer prognosis (P = 0.042) (Supplementary Fig. S2B). Furthermore, in the Luminal A subtype, patients exhibiting increased levels of CYP1A1, ABCC9, and CYP46A1 had a worse prognosis, whereas those with heightened CYP21A2 expression demonstrated a more favorable prognosis (Supplementary Fig. S2C). Between normal and breast cancer tissues, 81 DEGs were discovered, containing 26 upregulated and 55 downregulated genes (Fig. 2A,B). The crucial points were subsequently exhibited through the analysis of protein-protein interaction (PPI) of 81 DEGs, as shown in Fig. 2C. We observed that the critical genes were CYP3A4, ABCG2, and CYB2B6 (Fig. 2D).

Identification of candidate ADME-related genes. (A) A heatmap of the DEGs between tumor tissues and normal tissues. (B) A volcano plot of DEGs. (C) The PPI network diagram of DEGs. (D) The number of nodes associated with hub genes in the PPI network was determined. (E) A forest plot was generated to assess the prognostic significance of ADME-related genes.
Creation of a prognostic signature related to ADME
The univariate Cox analysis identified 38 DEGs that were associated with prognosis (Fig. 2E). Following the elimination of overfitting genes through LASSO analysis, an ADME signature consisting of four genes (ABCB5, ATP7B, KCNJ11, and TAP1) was identified (Fig. 3A,B). The risk score is calculated using the following equation:

Construction of the prognostic signature in TCGA dataset. (A,B) The LASSO analysis of the ADME-related genes. (C) Distribution of risk scores and survival time and status for each case. (D) Heatmap showed the expression of risk genes in different risk groups. (E) The PCA analysis. (F) Survival differences between patients in different risk groups. (G) The time-dependent ROC curves of BRCA patients.
Based on the median risk score, breast cancer patients were classified into two different risk subgroups (Fig. 3C). Figure 3D illustrates the diverse manifestation of the four genes among two risk categories. According to results of the PCA, individuals could be effectively categorized into two clusters (Fig. 3E). Figure 3F demonstrates that individuals classified as low-risk exhibit a more favorable prognosis in contrast to those classified as high-risk. Additionally, the ADME signature was assessed through the utilization of the ROC curve, resulting in AUC values of 0.764, 0.708, and 0.710 for the respective 1-, 3-, and 5-year intervals (Fig. 3G).
Verification of the prognostic signature related to ADME and the link between risk scores and clinical features of individuals with BRCA
Moreover, our findings were validated in the test datasets, which stratified individuals diagnosed with BRCA into two distinct risk subgroups. The KM analysis conducted on the datasets TCGA-test, TCGA-all, and GSE20685 demonstrated that individuals classified as low-risk exhibited a more favorable prognosis in comparison to those classified as high-risk (Fig. 4A,C). Furthermore, within the TCGA cohort, high-risk patients exhibited lower rates of recurrence-free survival (RFS) and disease-specific survival (DSS) compared to low-risk patients (Supplementary Fig. S3). Similarly, in the GSE7390, GSE20711, GSE25066, and GSE58812 cohorts, high-risk patients demonstrated a poorer prognosis (Supplementary Fig. S4). In TCGA-test (Fig. 4D), the AUC values for the ROC curve were 0.718, 0.681, and 0.676 for the 1-, 3-, and 5-year periods, respectively. In TCGA-all (Fig. 4E), the corresponding values were 0.737, 0.706, and 0.701. In GSE20685 (Fig. 4F), the AUC values were 0.932, 0.669, and 0.680. In patients diagnosed with Basal-like, HER2-enriched, Luminal A, and Luminal B breast cancer subtypes, those classified as high-risk exhibited a poorer prognosis compared to low-risk patients, with no statistically significant disparity observed in Normal-like subtypes (Supplementary Fig. S5). Furthermore, a survival analysis was performed using clinical characteristics, which demonstrated that individuals with low-risk scores exhibited superior outcomes across multiple categories, encompassing age groups of 65 and below, over 65, absence of metastasis (M0), presence of metastasis (M1), absence of lymph node involvement (N0), presence of lymph node involvement (N1–3), early-stage disease (Stage I + II), advanced-stage disease (Stage III + IV), and smaller tumor size (T1 + 2) (Supplementary Fig. S6). This further demonstrated the reliability of the risk model we developed. Furthermore, we conducted a comparison with the prognostic model of different individuals, and our signature exhibited a higher C-index compared to theirs (Supplementary Fig. S7). The expression of risk genes was validated through the use of RT-qPCR. Supplementary Fig. S8 demonstrated that tumor cells displayed elevated expression of ABCB5 and reduced expression of ATP7B, KCNJ11, and TAP1 in comparison to normal cells.

Validation of the prognostic model. The K-M curves shows the different prognosis in different risk group in TCGA-test (A), TCGA-all (B) and GSE20685 (C). The ROC curves in TCGA-test (D), TCGA-all (E) and GSE20685 (F).
Development of a nomogram for breast carcinoma and assessment of its prognostic predictive capacity
Cox regression analyses yielded compelling evidence supporting the robustness and independence of the risk score as a prognostic indicator (Fig. 5A,B). To further explore the prognostic potential of the ADME signature (Fig. 5C), a novel nomogram was created by incorporating the ADME signature and clinical variables obtained from TCGA. Figure 5D displayed calibration curves that demonstrated a satisfactory capability. Moreover, the nomogram’s ability to predict prognosis was assessed using ROC analysis, comparing it with other factors such as Stage, T, N, M, and age. The AUC value for 1-year survival was 0.954 (nomogram) and 0.789 (risk score) (Fig. 5E). The AUC for 3-year survival was 0.790 (according to the nomogram) and 0.675 (risk score) (Fig. 5F). The AUC value for 5-year survival was 0.718 (nomogram) and 0.700 (risk score) (Fig. 5G). The DCA for 1-, 3-, and 5-year periods demonstrated that this nomogram exhibited a greater overall advantage, as depicted in Fig. 5H,J. These results demonstrated the potential of this innovative nomogram to serve as an excellent prognosis prediction model.

Construction and evaluation of the nomogram. The forest plot for univariate Cox (A) and multivariate Cox regression (B) analysis. (C) Nomogram plot based on risk score and clinicopathological factors. (D) Calibration plot for the validation of the nomogram. The multifactor AUC for 1- (E), 3- (F), and 5-years (G) survival. The DCA curves for 1- (H), 3- (I), and 5-years (J).
Investigation of the TME and its correlation with the efficacy of immunotherapy among patients stratified into different risk score categories
According to the ESTIMATE analysis, it was observed in Fig. 6A that the high-risk group had lower ImmuneScores compared to the low-risk group. Furthermore, the differentiation of immune cell quantities among the two risk categories was investigated using different algorithms. The low-risk group demonstrated increased levels in the majority of immune cells, as depicted in Fig. 6B. According to the ssGSEA analysis, the high-risk patients showed reduced infiltration of B cells, CD8 + T cells, Neutrophils, Mast cells, pDCs, T helper cells, Tfh, Th1 cells, Th2 cells, TIL, and Treg compared to the low-risk patients (Fig. 6C). In the cohort of patients classified as low risk, there was an observed enhancement in specific immune functions, including APC co-inhibition, checkpoint regulation, T cell co-stimulation/co-inhibition (Fig. 6D). This could potentially clarify the reason behind the superior outlook of the low-risk classification. Furthermore, Fig. 6E illustrated the spread of individuals with different risk among various immune subtypes.

Analysis of tumor microenvironment of high- and low-risk groups. (A) Differences in Immunescore between the two groups. (B) The examination of disparities in immune cell infiltration between the two cohorts was conducted utilizing multiple algorithms. (C) The analysis of differences in immune cell infiltration between the two risk groups. (D) The examination of variations in immune functions between the two groups was conducted using ssGSEA. (E) The allocation of patients exhibiting high- and low-risk profiles across various immune subtypes.
Next, we analyzed the expression patterns of immune-related genes in different risk patients. The high-risk category showed low expression levels of most immune-related genes (Fig. 7A,D). The TIDE scores played a pivotal role in assessing the efficacy of immunotherapy. Furthermore, an investigation was conducted to analyze the relationship between the TIDE score and risk score. The low-risk category demonstrated significantly lower TIDE scores when compared to the high-risk category (Fig. 7E). As shown in Fig. 7F,I, violin plots showed that a higher IPS in a low-risk category suggested a more robust reaction to PD-1 and CTLA-4 inhibitors, establishing the correlation between IPS and risk groups. Conversely, individuals with a low-risk profile demonstrated a more favorable response to immunotherapy compared to those with a high-risk profile (Fig. 7J).

Evaluation of the immunotherapeutic efficacy in cohorts classified as high- and low-risk. (A–D) The immune-related gene expression levels in different groups. (E) The TIDE score exhibits variations between the two groups. (F–I) The violin plots effectively depicted the association between IPS interventions and different risk groups. (J) Prediction of immunotherapy response.
The functional enrichment analysis of ADME signatures identified numerous pathways that exhibited significant alterations among patients stratified into different risk groups
To enhance comprehension regarding the molecular mechanisms of distinct ADME signature subgroups, we conducted enrichment analyses employing GO, KEGG, and DO. The GO analysis revealed that the DEGs between different risk groups were primarily associated with the development of the epidermis, activity of serine-type endopeptidase, and keratin filament enrichment (Fig. 8A). According to the KEGG findings, these genes were mainly enhanced in the Estrogen, Nitrogen metabolism, and Wnt signaling pathway (Fig. 8B). According to the findings, these genes were mainly found to be abundant in various cancerous tumors, such as gastric carcinoma, cancer of the female reproductive system, and breast cancer (Fig. 8C). Additionally, the GSVA revealed that numerous pathways dramatically altered between breast cancer patients with different risk (Fig. 8D).

Function analysis. (A) GO, (B) KEGG, and (C) DO analysis of DEGs between high and low-risk groups. (D) GSVA enrichment analysis in high- and low-risk groups.
Identification of chemotherapy drug sensitivity on the basis of the prognostic model
In order to obtain additional understanding regarding potential variations in drug responsiveness among the aforementioned risk categories, we conducted an analysis to assess the relationship between the risk scores of breast cancer patients and the IC50 values of chemotherapy and targeted treatment medications. In the high-risk group, the IC50 values of AZ628, CGP-60,474, FTI-277, and GSK429286A were significantly lower compared to the low-risk group. Conversely, the IC50 values of XL-184, KIN001-102, IPA-3, TAE684, and ZSTK474 were higher in high-risk group (Supplementary Fig. S9). The findings indicated that the group at a greater risk showed greater sensitivity to AZ628, CGP-60,474, FTI-277, and GSK429286A, while the group at a lower risk exhibited higher sensitivity to XL-184, KIN001-102, IPA-3, TAE684, and ZSTK474. Furthermore, our analysis of the correlation between risk scores and the sensitivity of frequently utilized breast cancer medications, utilizing data from the GSE130787 dataset, revealed that individuals in the high-risk group exhibited a more favorable response to docetaxel, carboplatin, and trastuzumab treatment (Supplementary Fig. S10).
Discussion
Breast cancer is a prevalent malignancy that often occurs in women1. While tumor stage and molecular subtype are commonly utilized as prognostic indicators for breast cancer patients, they may not accurately predict prognosis6. The identification of innovative molecular biomarkers is of utmost importance in order to effectively evaluate the prognosis of BRCA patients and deliver precise treatment. This research utilized the LASSO algorithm to develop a novel ADME signature, which was trained on the TCGA cohort, to predict the prognosis of BRCA patients. The reliability of the signature was validated using both the external GEO cohort and the internal TCGA-test cohort. The KM analysis revealed that the group classified as low-risk demonstrated a more favorable prognosis in both the cohort used for training and the cohorts used for validation. Meanwhile, the ADME signature’s predictive ability was demonstrated using ROC analysis. Additionally, we discovered that patients with a lower risk profile exhibited a more favorable response to immunotherapy and were better candidates for ICB treatment. In addition, a novel nomogram including clinical factors was established, which had good accuracy and high AUC value for breast cancer patients. Hence, the ADME pattern proved to be a dependable predictor for predicting the prognosis of breast cancer and assessing the influence of immunotherapy, which could hold considerable importance for the clinical management of breast cancer.
The prognostic signature for breast cancer was comprised of four genes: ABCB5, ATP7B, KCNJ11, and TAP1. These genes have been associated with various cancers and may serve as promising prognostic indicators for breast cancer. ATP binding cassette subfamily B member 5 (ABCB5) has been documented to facilitate the invasion of colorectal cancer (CRC) through the augmentation of AXL signal transduction27. ABCB5 blocking reversed the drug resistance of Merkel Cell Carcinoma (MCC) in vivo xenotransplantation model and inhibited tumor growth, indicating that ABCB5 might be related to the chemical resistance of MCC28. Among individuals with CRC receiving oxaliplatin chemotherapy, Elevated ATPase copper transporting beta (ATP7B) levels were linked to a poor prognosis29. Zhang et al. found KCNJ11 formed a complex with lactate dehydrogenase A and acted as an oncogene in hepatocellular carcinoma (HCC), indicating that it may be possible to design a candidate strategy for preventing HCC that targets KCNJ1130. Ling et al. showed that TAP1 down-regulation was both a poor predictor of outcome for patients with stage I-II CRC and a putative tumor immune escape mechanism31. Segami et al. revealed that better overall survival was correlated with higher TAP1 expression in advanced gastric cancer32. However, more research is still needed to determine how these genes affect breast cancer.
As ADME genes tend to present single-nucleotide polymorphism (SNP). Leung et al. found that SNP rs17143212 was significantly correlated with the expression level of ABCB5 and associated with the risk of liver cancer and the presence of aggressive tumor characteristics33. The variant rs9526814 in ATP7B may potentially enhance resistance to platinum-based chemotherapy in lung cancer patients by influencing the expression of the ATP7B gene, suggesting its potential as a biomarker for predicting platinum chemotherapy sensitivity34. However, the specific role of SNP of ADME-related genes in breast cancer is still unclear, which needs further research.
The TME consists of a diverse array of immune cells, interstitial cells, extracellular matrix components, and tumor blood vessels, all of which collectively contribute significantly to the onset and advancement of cancer35,36,37,38. Throughout the development and expansion of tumors, the composition of infiltrating immune cells within the TME often undergoes changes39,40. Our analysis indicates that patients with high risk scores for BRCA exhibited reduced ImmuneScores. In comparison to the low-risk group, the high-risk group exhibited significant reduction in the majority of immune cells, including T helper cells, Tfh, Th1 cells, Th2 cells, B cells, CD8 + T cells, Neutrophils, Mast cells, pDCs, TIL, and Treg. Furthermore, a substantial proportion of genes linked to the immune system exhibited a trend towards suppression in the high-risk group, whereas the low-risk group displayed notable enhancement in immune function. Studies indicate that crucial components of anti-cancer defense comprise of immune cells41. High-risk individuals have a bleak outlook due to their reduced number of immune cells and weakened immunological function. The findings indicated that individuals with breast cancer who were classified as low-risk had a more favorable response to immunotherapy in comparison to those categorized as high-risk. Precision medicine aims to align drugs with genomic determinants of response23. Identifying the molecular characteristics of tumors that influence responses to specific drug therapies poses a significant challenge due to the heterogeneity of patient populations, the diverse nature of tumors, and the incomplete comprehension of the multitude of molecular factors that influence treatment outcomes23. To address this challenge, we utilized the “pRRophetic” R package to forecast potential drugs that exhibit a strong association with the risk score. Our screening results indicate that various types of chemotherapeutic agents exhibit distinct responses to different risks. The results of this study provide insight into the participation of ADME-associated genes in breast cancer and can be used to guide immunotherapeutic and chemotherapeutic treatments for individuals with breast cancer.
Furthermore, this investigation also examined the variances in physiological mechanisms, signaling pathways, and immune responses among individuals at different risk. Enrichment analysis was conducted on the genes that exhibited differential expression in high-risk and low-risk patients, indicating their involvement in various tumor-related signaling pathways such as the Estrogen, ErbB, Ras, and Wnt signaling pathway. The findings indicate that genes associated with ADME may be involved in controlling the progression of BRCA through the regulation of signal pathways related to tumors.
Nevertheless, our study is subject to several limitations. Selection bias may be present due to the use of publicly available datasets for the majority of analyses, as well as the retrospective collection of all samples. Furthermore, additional in vitro and in vivo experiments are required to validate our findings.
Conclusions
In conclusion, we developed a prognostic signature associated with ADME genes that can assist in predicting survival, guiding immunotherapy, and determining clinical results. The study has the potential to offer greater understanding of the role of ADME-associated genes in BRCA and aid in the advancement of more efficient treatments for this condition.
Data availability
The study’s GEO database (GSE20685, GSE7390, GSE20711, GSE25066, GSE58812) can be downloaded from https://www.ncbi.nlm.nih.gov/gds/. The TCGA-BRCA gene matrix presented in the study could be obtained from the TCGA database(https://portal.gdc.cancer.gov/).
Change history
31 December 2024
A Correction to this paper has been published: https://doi.org/10.1038/s41598-024-83823-4
References
Harbeck, N. et al. Breast cancer. Nat. Rev. Dis. Primers. 5 (1), 66. https://doi.org/10.1038/s41572-019-0111-2 (2019).
Loibl, S., Poortmans, P., Morrow, M., Denkert, C. & Curigliano, G. Breast cancer. Lancet. 397 (10286), 1750–1769. https://doi.org/10.1016/s0140-6736(20)32381-3 (2021).
Wang, S. et al. Clinical significance and immunogenomic landscape analyses of the immune cell signature based prognostic model for patients with breast cancer. Brief. Bioinform. 22(4). https://doi.org/10.1093/bib/bbaa311 (2021).
Tang, L. et al. DCST1-AS1 promotes TGF-β-induced epithelial-mesenchymal transition and enhances chemoresistance in triple-negative breast cancer cells via ANXA1. Front. Oncol.10, 280. https://doi.org/10.3389/fonc.2020.00280 (2020).
Zhang, R. et al. EZH2 inhibitors-mediated Epigenetic Reactivation of FOSB Inhibits triple-negative Breast cancer progress. Cancer Cell. Int.20, 175. https://doi.org/10.1186/s12935-020-01260-5 (2020).
Cao, R. J. et al. Disabled-1 is down-regulated in clinical breast cancer and regulates cell apoptosis through NF-κB/Bcl-2/caspase-9. J. Cell. Mol. Med.23(2), 1622–1627. https://doi.org/10.1111/jcmm.14047 (2019).
Pak, L. M. & Morrow, M. Addressing the problem of overtreatment in breast cancer. Expert Rev. Anticancer Ther.22(5), 535–548. https://doi.org/10.1080/14737140.2022.2064277 (2022).
Klein, K. et al. A new panel-based next-generation sequencing method for ADME genes reveals novel associations of common and rare variants with expression in a human liver cohort. Front. Genet.10, 7. https://doi.org/10.3389/fgene.2019.00007 (2019).
Tang, X. et al. Development and validation of an ADME-Related gene signature for survival, treatment outcome and immune cell infiltration in head and neck squamous cell carcinoma. Front. Immunol.13, 905635. https://doi.org/10.3389/fimmu.2022.905635 (2022).
Tao, L. et al. Recent progresses in the exploration of machine learning methods as in-silico ADME Prediction tools. Adv. Drug Deliv Rev.86, 83–100. https://doi.org/10.1016/j.addr.2015.03.014 (2015).
Hovelson, D. H. et al. Characterization of ADME gene variation in 21 Populations by exome sequencing. Pharmacogenet. Genom.27(3), 89–100. https://doi.org/10.1097/fpc.0000000000000260 (2017).
Hu, D. G., Marri, S., McKinnon, R. A., Mackenzie, P. I. & Meech, R. Deregulation of the genes that are involved in drug absorption, distribution, metabolism, and excretion in hepatocellular carcinoma. J. Pharmacol. Exp. Ther.368(3), 363–381. https://doi.org/10.1124/jpet.118.255018 (2019).
Jittikoon, J. et al. Comparison of genetic variation in drug ADME-related genes in thais with Caucasian, African and Asian HapMap populations. J. Hum. Genet.61(2), 119–127. https://doi.org/10.1038/jhg.2015.115 (2016).
Arbitrio, M. et al. Pharmacogenomic profiling of ADME gene variants: Current challenges and validation perspectives. High. Throughput. 7(4). https://doi.org/10.3390/ht7040040 (2018).
Hu, D. G., Mackenzie, P. I., McKinnon, R. A. & Meech, R. Genetic polymorphisms of human UDP-glucuronosyltransferase (UGT) genes and cancer risk. Drug Metab. Rev.48(1), 47–69. https://doi.org/10.3109/03602532.2015.1131292 (2016).
Drozdzik, M. & Oswald, S. Expression and regulation of drug transporters and metabolizing enzymes in the human gastrointestinal tract. Curr. Med. Chem.23(39), 4468–4489. https://doi.org/10.2174/0929867323666161024154457 (2016).
Fisel, P., Schaeffeler, E. & Schwab, M. DNA methylation of ADME genes. Clin. Pharmacol. Ther.99(5), 512–527. https://doi.org/10.1002/cpt.343 (2016).
Cai, Y. et al. The downregulated drug-metabolism related ALDH6A1 serves as predictor for prognosis and therapeutic immune response in gastric cancer. Aging (Albany NY)14(17), 7038–7051. https://doi.org/10.18632/aging.204270 (2022).
Xu, Z. et al. The roles of drug metabolism-related ADH1B in immune regulation and therapeutic response of ovarian cancer. Front. Cell. Dev. Biol.10, 877254. https://doi.org/10.3389/fcell.2022.877254 (2022).
Nan, Y. et al. DLGAP1-AS2-mediated phosphatidic acid synthesis activates YAP signaling and confers chemoresistance in squamous cell carcinoma. Cancer Res.82(16), 2887–2903. https://doi.org/10.1158/0008-5472.Can-22-0717 (2022).
Hong, K. et al. Identification and validation of a novel senescence-related biomarker for thyroid cancer to predict the prognosis and immunotherapy. Front. Immunol.14, 1128390. https://doi.org/10.3389/fimmu.2023.1128390 (2023).
Cao, J. et al. Development of anoikis-related genes signature to predict the prognosis in gastric cancer patients. Front. Oncol.12, 1096608. https://doi.org/10.3389/fonc.2022.1096608 (2022).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res.28(1), 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci.28(11), 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res.51(D1), D587–D592. https://doi.org/10.1093/nar/gkac963 (2023).
Xu, R. et al. Cancer-associated Fibroblast Related gene Signature in Helicobacter pylori-based Subtypes of Gastric Carcinoma for Prognosis and Tumor Microenvironment Estimation in silico Analysis. Front. Med. (Lausanne). 10, 1079470. https://doi.org/10.3389/fmed.2023.1079470 (2023).
Guo, Q. et al. ATP-binding cassette member B5 (ABCB5) promotes tumor cell invasiveness in human colorectal cancer. J. Biol. Chem.293(28), 11166–11178. https://doi.org/10.1074/jbc.RA118.003187 (2018).
Kleffel, S. et al. ABCB5-targeted chemoresistance reversal inhibits merkel cell carcinoma growth. J. Invest. Dermatol.136(4), 838–846. https://doi.org/10.1016/j.jid.2015.12.038 (2016).
Martinez-Balibrea, E. et al. Increased levels of copper efflux transporter ATP7B are associated with poor outcome in colorectal cancer patients receiving oxaliplatin-based chemotherapy. Int. J. Cancer124(12), 2905–2910. https://doi.org/10.1002/ijc.24273 (2009).
Zhang, K. et al. NFκB mediated elevation of KCNJ11 promotes tumor progression of hepatocellular carcinoma through interaction of lactate dehydrogenase A. Biochem. Biophys. Res. Commun.495(1), 246–253. https://doi.org/10.1016/j.bbrc.2017.11.011 (2018).
Ling, A. et al. TAP1 down-regulation elicits immune escape and poor prognosis in colorectal cancer. Oncoimmunology6(11), e1356143. https://doi.org/10.1080/2162402x.2017.1356143 (2017).
Segami, K. et al. Clinical significance of TAP1 and DLL4 expression in patients with locally advanced gastric cancer. Vivo35(5), 2771–2777. https://doi.org/10.21873/invivo.12562 (2021).
Leung, I. C. et al. Genetic variation in ABCB5 associates with risk of hepatocellular carcinoma. J. Cell. Mol. Med.24(18), 10705–10713. https://doi.org/10.1111/jcmm.15691 (2020).
Li, Y. Q., Chen, J., Yin, J. Y., Liu, Z. Q. & Li, X. P. Gene expression and single nucleotide polymorphism of ATP7B are associated with platinum-based chemotherapy response in non-small cell lung cancer patients. J. Cancer9(19), 3532–3539. https://doi.org/10.7150/jca.26286 (2018).
Li, M. et al. Predicting response to neoadjuvant chemoradiotherapy in rectal cancer: From biomarkers to tumor models. Ther. Adv. Med. Oncol.14, 17588359221077972. https://doi.org/10.1177/17588359221077972 (2022).
Li, W. N., Zhang, S. J., Feng, J. Q. & Jin, W. L. Repurposing vitamin C for cancer treatment: focus on targeting the tumor microenvironment. Cancers (Basel). 14(11). https://doi.org/10.3390/cancers14112608 (2022).
Nierves, L. et al. Multi-omic profiling of the leukemic microenvironment shows bone marrow interstitial fluid is distinct from peripheral blood plasma. Exp. Hematol. Oncol.11(1), 56. https://doi.org/10.1186/s40164-022-00310-0 (2022).
Wang, S. et al. Immune cell infiltration-based signature for prognosis and immunogenomic analysis in breast cancer. Brief. Bioinform22(2), 2020–2031. https://doi.org/10.1093/bib/bbaa026 (2021).
Chen, Z. et al. Flow cytometry-assessed PD1/PDL1 status in tumor-infiltrating lymphocytes: a link with the prognosis of diffuse large B-cell lymphoma. Front. Oncol.11, 687911. https://doi.org/10.3389/fonc.2021.687911 (2021).
Yang, L. et al. Clinical significance of the immune microenvironment in ovarian cancer patients. Mol. Omics14(5), 341–351. https://doi.org/10.1039/c8mo00128f (2018).
Yajuk, O. et al. The PD-L1/PD-1 axis blocks neutrophil cytotoxicity in cancer. Cells. 10(6). https://doi.org/10.3390/cells10061510 (2021).
Acknowledgements
We thank HOME for Researchers (www.home-for-researchers.com) platform.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation were performed by [Zhixuan Wu], [Yangyang Guo] and [Liangchen Qu]. Data collection and analysis were performed by [Zhixuan Wu], [Hewei Zhang] and [Xiaowu Wang]. The first draft of the manuscript was written by [Zhixuan Wu] and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: In the original version of this Article, Zhixuan Wu was incorrectly affiliated with ‘The Dingli Clinical College of Wenzhou Medical University, Wenzhou, Zhejiang Province 325000, China’. The correct affiliation is listed in the correction notice.
Electronic supplementary material
Below is the link to the electronic supplementary material.










Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, Z., Guo, Y., Qu, L. et al. Development and validation of a prognostic signature of breast cancer based on drug absorption, distribution, metabolism and excretion (ADME)-related genes. Sci Rep 14, 21619 (2024). https://doi.org/10.1038/s41598-024-72635-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-72635-1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}