
Abstract
Bladder carcinoma (BLCA) represents a common urinary tract malignancy, characterized by aggressive behavior and high recurrence rates. The biological response regulation during tumor proliferation and metastasis is intimately associated with liquid-liquid phase separation (LLPS). For the purpose of enhancing early detection and treatment, this study employed transcriptomic data to examine the prognostic implications of LLPS-associated genes and formulate a predictive model. Clinical and transcriptomic data of bladder cancer patients were sourced from the GEO and TCGA databases. This study applied a clustering algorithm using non-negative matrix factorization (NMF) to classify samples, which were systematically compared based on their liquid-liquid phase separation characteristics. Prognostic models were developed using multivariate Cox regression and the Least Absolute Shrinkage Selection Operator (LASSO) algorithm to establish risk formulas for nine genes. The gene signature’s validity was tested across the entire TCGA cohort (406 cases), the TCGA testing cohort (120 cases), and the external validation dataset GSE13507. The predictive accuracy of the signature system was assessed using receiver operating characteristic (ROC) and Kaplan-Meier curves. Additionally, decision curve analysis incorporating clinicopathological parameters and the genetic signature was employed to predict individual survival. This study identified two distinct molecular subtypes, C1 and C2. Patients with the C1 subtype exhibited significantly better prognoses than those with the C2 subtype. Low-risk group patients consistently showed superior prognoses compared to high-risk groups across the entire TCGA, GEO, and TCGA training cohorts. Furthermore, the LLPS-related gene model demonstrated prognostic value independent of other clinical traits. This study identifies LLPS-associated gene clusters and establishes an independent, accurate prognostic model for BLCA. The model holds potential for clinical application in BLCA prognosis assessment.
Similar content being viewed by others
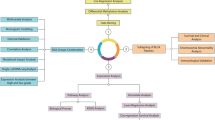


Introduction
Bladder cancer (BLCA) ranks second only to prostate cancer in incidence within the genitourinary system. It is among the top ten most common cancers globally, with a rising annual incidence. BLCA represents one of the most costly malignancies due to lifelong treatment requirements and significantly impacts postoperative quality of life1. Annually, BLCA is responsible for nearly 170,000 deaths worldwide2. It is the fourth most prevalent cancer in men and the most commonly diagnosed malignancy in women3. Approximately 75% of BLCA cases are non-muscle invasive bladder cancers (NMIBCs), characterized by a high recurrence rate of 45% within five years4. Recent years have seen a shift in BLCA treatment from traditional surgery, immunotherapy, and chemotherapy to molecular-level approaches, driven by extensive molecular alterations analysis5.
Cancer is characterized by the disruption of normal homeostatic regulation, leading to uncontrolled cell proliferation. Homeostasis in normal cells is achieved through precise spatiotemporal regulation of life processes6. This regulation is mediated by various subcompartments within living cells. In addition to traditional membrane-bound organelles such as secretory vesicles and the endoplasmic reticulum, many membrane-less organelles exist that maintain coherent structures and compartmentalize specific molecular groups7. Research has revealed that these structures are liquid-phase condensates formed through bioregulatory (liquid-liquid) phase separation (LLPS). Recently, liquid-liquid phase separation (LLPS) has gained prominence as a critical mechanism for spatial and temporal coordination of cellular activities. Studies indicate that essential cellular processes in cancer cells are directly influenced by biomolecular condensates. Consequently, evidence increasingly suggests that LLPS dysregulation may drive carcinogenic activity8.
It is well-established that LLPS induces the formation of condensate-like droplets composed of proteins, RNA, and other biomolecules without intracellular demarcation membranes9. While the protein components of LLPS have been extensively studied, recent research highlights the significant role of RNA in regulating LLPS10. The RPS database (http://rps.renlab.org) offers a comprehensive repository of RNAs associated with LLPS across 20 distinct biomolecular condensates in viruses and eukaryotes. It catalogues 21,613 LLPS-associated RNAs, derived from high-throughput sequencing data, interaction network-based predictions, and literature mining, and provides detailed information on the LLPS process11.
In this study, bladder cancer patients were hierarchically clustered based on LLPS-related gene features using non-negative matrix factorization (NMF), a dimensionality reduction technique that employs high-dimensional mapping12,13. NMF is distinguished by its constraint that both the base components matrix and the mixed coefficients matrix contain only non-negative terms, without imposing orthogonality or independence constraints on the base components14. Subsequently, a risk score (RS) model was developed to predict the prognosis of BLCA patients, correlating clinical features, immune cell infiltration, and staging15. The model’s validity and reliability were evaluated using internal and external validation cohorts.
Materials and methods
Data acquisition and collation
Gene expression data for 433 bladder cancer patients, clinical data for 412 patients, and mutation data (calculated by Varscan) for 4 cases were sourced from TCGA (The Cancer Genome Atlas) at https://portal.gdc.cancer.gov/. The GSE13507 dataset was accessed from GEO (Gene Expression Omnibus) at http://www.ncbi.nlm.nih. A total of 7,442 LLPS-associated genes were identified using the RPS database, available at http://rps.renlab.org. Transcriptomic and clinical data were normalized into a visual gene expression matrix using Perl. The GEO dataset was similarly annotated and collated into a matrix. Tumor mutation load was calculated from mutation data. LLPS-related gene expression was extracted from the TCGA gene expression matrix using the limma R package, followed by log2 transformation and p-value correction to compile a list of differential genes.
Sample typing with NMF
Gene expression data from TCGA and GEO were analyzed to identify differential genes linked to liquid-liquid phase separation. These differential expression data were integrated with clinical data to construct the NMF molecular typing model via the NMF R package. NMF hierarchical clustering was applied to the standardized dataset, exploring cluster values (k) from 2 to 10, ultimately determining 2 as the optimal k. Survival differences among subtypes were analyzed using R survival and R survminer packages.
Assessment of immune cell infiltration
The immune cell composition across subtypes (including CD8 + T cells, B cells, monocytes, cytotoxic lymphocytes, endothelial cells, fibroblasts, neutrophils, T cells, NK cells, and myeloid dendritic cells in bladder cancer) was examined. MCPcounter R package quantified immune cell content in each transcriptome sample. Subsequently, ggpubr R package was employed to create violin plots, assessing the variability of immune cell content among patients across subtypes.
Construction of prognostic models
Using the TCGA differential gene expression file and survival data, the merged dataset was obtained through the R package limma. The dataset comprising 406 patients was randomly divided into a testing set (n = 120) and a training set (n= 286) via the R package, ensuring no bias in the distribution of clinical features. Initially, univariate Cox regression analysis was applied to differentially expressed genes associated with LLPS in the training set. This was followed by a comparative assessment of gene expression, survival time, and survival status to derive univariate p-values, which served as a filter for significant gene expressions. Subsequently, the least absolute shrinkage and selection operator (LASSO) regression method was employed for further analysis16. LASSO, a biased estimator for multicollinear data, retains the benefits of subset shrinkage. Based on the LASSO regression results, the ACOX model was developed. The risk score for each sample in the training group was calculated using the model equation, categorizing samples into high-risk (HRG) and low-risk (LRG) groups according to the median risk score. The same calculation method was applied to the testing and GEO patient groups to obtain their risk scores. These steps utilized the glmnet, caret, survival, time ROC, and survminer R packages.
Validation of prognostic models
Predictive accuracy for 1-, 3-, and 5-year survival was evaluated in the training set using survival and receiver operating characteristic (ROC) curves. To assess model stability, identical coefficients were utilized for analyses in the TCGA all-set, TCGA testing set, and the GEO patient group.
GSEA enrichment analysis
Gene Set Enrichment Analysis (GSEA) was performed to analyze and interpret pathway-level changes in transcriptomics experiments17. Active functions or pathways in high-risk and low-risk groups were identified. The c5.go.v7.4.symbols and c2.cp.kegg.v7.4.symbols gene sets were used to generate graphs of high-risk enrichment and highlight the top five pathways. These steps employed the limma, clusterProfiler, org.Hs.eg.db, and enrichplot packages.
Multivariate and univariate cox analyses and model comparison
To establish the independence of genetic features from clinical parameters, both multivariate and univariate Cox regression analyses were executed using the R survival package. This analysis provided hazard ratio (HR), HR fluctuation ranges, and p-values based on TCGA clinical data. A comprehensive analysis of clinical information from TCGA patients included risk scores correlated with age, gender, staging (T-, N-, and M-stage), and cancer grading. The constructed model was validated against existing models.
Results
Classification of LLPS-related gene samples into two subtypes based on NMF clustering molecular typing
The TCGA-BLCA cohort comprised 433 RNA sequencing samples, including 414 tumor and 19 normal samples. Differential expression analysis identified 579 LLPS-related genes (|log 2 FC|>1, FDR < 0.05) between conventional bladder tissue and BLCA patients. Volcano plots illustrated the differential expression, with 364 upregulated and 215 downregulated genes (Fig. 1A). Clustering of 406 bladder cancer samples was performed using the NMF R package (Fig. 1B,C). Evaluation of cophenetic correlation, silhouette distribution, and residual sum of squares (RSS) determined the optimal number of clusters as two, designated as C1 and C2 subtypes. Progression-free survival (PFS) and overall survival (OS) significantly differed between C1 and C2 subtypes (p < 0.05), with the C2 group exhibiting a poorer prognosis compared to the C1 group (Fig. 1D,E). Molecular subtyping results were also compared with previous studies (Fig. 1F).

(A) Volcano plot illustrating differentially expressed genes in BLCA using TCGA data. (B) Consensus map clustered via the NMF algorithm. (C) Rank (2–10), cophenetic correlation, residual sum of squares (RSS), and silhouette distribution plots. (D) Progression-free survival (PFS) exhibited significant differences between C1 and C2. (E) Overall survival (OS) showed notable disparities between C1 and C2. (F) Alluvial plot depicting the distribution of C2 and C1 across two molecular subtypes.
Analysis of immune infiltration in LLPS-related genes
The prognosis for BLCA patients differed markedly between the two subtypes, reflected in the immune cell infiltration data between C1 and C2. MCPcounter scores indicated substantial differences in cytotoxic lymphocytes, B lineage, monocytic lineage, CD8+ T cells, endothelial cells, neutrophils, fibroblasts, NK cells, T cells, and myeloid dendritic cells across both subtypes (Fig. 2A-J).

Significant variations observed in the Immune scores of cells of the liquid-liquid phase separation (LLPS). (A) Cytotoxic lymphocytes (B) B lineage (C) Monocytic lineage (D) CD8 + T cells (E) Endothelial cells (F) Neutrophils (G) Fibroblasts (H) NK cells (I) T cells (J) Myeloid dendritic cells.
Construction and evaluation of gene signatures in the TCGA training group
The TCGA differential gene expression file was integrated with survival data using the “limma” package, resulting in a risk file comprising 406 cases. Samples were randomly divided into a testing set (120 cases) and a training set (286 cases), ensuring no significant differences between the groups. Univariate Cox proportional hazards regression analysis on the training set identified genes with significant expression (P < 0.05). Subsequently, the LASSO regression model was developed, and cross-validation determined the point of least error (lambda.min) (Fig. 3A,B). Based on LASSO regression results, a multivariate Cox proportional hazards model identified LLPS-related genes with significant differences. Ultimately, a risk score formula incorporating nine genes was derived.
The ROC curve was used to evaluate the accuracy of the RS model in the training set. The model demonstrated an area under the curve (AUC) greater than 0.7 over 1, 3, and 5 years (Fig. 3G). Based on the median RS, the sample was divided into HRG and LRG. The KM survival analysis showed that LRG had a better prognosis compared to HRG (Fig. 3C).

(A) LASSO coefficient profiles were generated using TCGA data. (B) The optimal penalization coefficient (λ) was identified via threefold cross-validation based on partial likelihood deviance. (C–F) Construction and validation of the nine-gene risk score (RS) related to LLPS for BLCA using Kaplan–Meier (KM) curves in different cohorts: (C) TCGA training cohort, (D) GEO cohort, (E) entire TCGA cohort, and (F) TCGA testing cohort. (G–J) Establishment and validation of the nine-gene RS signature associated with LLPS for BLCA with 1, 3, and 5-year ROC analyses across different cohorts: (G) TCGA training cohort, (H) GEO cohort, (I) entire TCGA cohort, and (J) TCGA testing cohort.
Validation of prognostic models in different cohorts
The robustness of the LLPS-related genetic model was assessed by applying the same model and coefficients from the training set to compute risk scores for each sample. BLCA patients were stratified into low-risk and high-risk groups based on median risk values. Survival curves for the GEO cohort (165 cases), the entire TCGA cohort (406 cases), and the TCGA testing cohort (120 cases) were plotted using the survival and survminer packages (Fig. 3D-F). While the survival curves for the GEO and entire TCGA cohorts showed significant differences, the testing cohort did not exhibit notable variations. Consequently, the analysis indicated that HRG had a significantly poorer long-term prognosis compared to the LRG.
Subsequent ROC analysis evaluated the prognostic classification of risk scores and assessed the classification efficiency for 1, 3, and 5-year survival. These analyses aligned with the performance observed in the TCGA training cohort. Utilizing the timeROC package, ROC curves (Fig. 3H-J) revealed that the areas under the 1, 3, and 5-year ROC curves for the GEO and entire TCGA cohorts exceeded 0.6, while the TCGA testing cohort surpassed 0.5. This suggested the constructed model predicted 1, 3, and 5-year survival with high accuracy.
The relationship between OS and potential variables was examined using univariate and multivariate Cox regression models, incorporating gender, age, grade, pathological grade, TNM stage, and RS (Table 1). The results demonstrated that age (HR: 1.029, 95% CI: 1.012–1.965, p < 0.0001), TNM stage (HR: 1.613, 95% CI: 1.323–1.965, p < 0.0001), and RS (HR: 1.339, 95% CI: 1.257–1.426, p < 0.0001) significantly impacted OS.
Risk model assessment for prognosis prediction
The risk model was evaluated against previously reported BLCA feature models by comparing survival and ROC curves to validate its prognostic accuracy. Three reports were selected through a targeted literature review18,19,20 (Fig. 4). Comparative analysis of prognostic models for bladder cancer indicated that the current model exhibited the largest area under the ROC curve (Fig. 4A-D) and the smallest p-value (Fig. 4E-H). The C-index and RMS for each model were calculated, revealing that the developed risk model had the highest values in both metrics (Fig. 4I-J), highlighting its superior performance in predicting long-term prognosis.

(A) ROC curves of the constructed model. (B–D) ROC curves of three previously published gene signatures. (E) KM curves of the constructed model. (F–H) KM curves of three previously published gene signatures. (I) C-index of all four prognostic risk models, with the LLPS model exhibiting the highest value. (J) RMS time curve of all four prognostic risk models, indicating a 60-month overlap.
Enrichment analysis between subtypes
A correlation analysis was performed between typing results from the TCGA cohort and published immunotyping results. GO functions and KEGG pathway enrichment annotations were applied to 406 sample risk files. This analysis identified the top five pathways enriched in the HRG, based on NES (normalized enrichment score) and q-value (adjusted p-value) (Fig. 5A,B). The pathways were listed below;

(A) The top 5 pathways gene signature assessment in the HRG using KEGG analysis. (B) The top 5 pathways gene signature assessment in the HRG using GO analysis.
Clinical, genetic, and immune cell correlation analysis
Box plots for clinical correlation analysis were generated using the ggpubr R package. RS varied across clinical subgroups of BLCA patients, showing higher values in patients over 65 years of age, with high grade, stage III-IV, and high TNM staging (Fig. 6A-D). Kaplan–Meier (KM) survival curves for stage I-II and III-IV subtypes further validated the model (Fig. 6E,F), indicating that risk characteristics were reliable predictors across clinical subgroups.
To examine the correlation between RS and target genes, expression levels of key pathway target genes were extracted and analyzed. A positive correlation was observed between RS and LLPS-related genes, including PHGDH, MMP9, ADIPOQ, and NAMPT (Fig. 7A).
In the TCGA-BLCA cohort, immune cell fractions were calculated for each sample and their correlation with RS was evaluated. The analysis revealed a positive correlation between RS and both endothelial cells (p < 0.05) and fibroblasts (p < 0.05) (Fig. 7B).

(A–D) RS comparison of BLCA samples with their corresponding clinical data of BLCA: (A) Age (B) Grade (C) Stage (D) T1-T4. (E, F), Assessment of the nine-gene risk model in various stage groups in LRG and HRG using OS KM curves.

(A) Association between RS of BLCA samples and expression levels of representative genes for generic pathway targets in oncology. (B) Association between RS of BLCA samples and immune scores.
Discussion
Bladder cancer pathogenesis is a complex, multigene, and multi-step process. Recent studies have identified numerous membrane-free compartments, termed biomacromolecular condensates, which participate in various biological activities through liquid-liquid phase separation assemblies of phase-separated condensates21. These condensates can affect tumor initiation and progression.
This study identified two molecular subtypes of BLCA, C1 and C2, through NMF clustering analysis of BLCA cases from the GEO and TCGA databases based on LLPS-associated genes. Immune infiltration levels exhibited significant differences between these molecular subtypes, including CD8 + T cells, B lineage, cytotoxic lymphocytes, endothelial cells, fibroblasts, neutrophils, monocytic lineage, NK cells, T cells, and myeloid dendritic cells. Additionally, OS and PFS correlation analyses indicated differences between C1 and C2.
The TCGA dataset (406 samples) was divided into testing and training cohorts. LASSO-Cox regression and univariate Cox analysis were employed to construct a prediction model based on nine LLPS-related genes (MTHFD1L, P4HB, PDGFRA, OAS3, AHNAK, PDXK, IGSF8, FASN, NT5DC3), which accurately predicted BLCA patient survival. Previous research has identified MTHFD1L (Methylenetetrahydrofolate dehydrogenase 1-like), a mitochondrial enzyme, as a key factor in bladder cancer cell proliferation, colony formation, and invasion, with overexpression linked to overall survival in bladder cancer patients22. The expression level of P4HB (Prolyl 4-hydroxylase, beta polypeptide) allows for early-stage BLCA patient identification and prognosis prediction23. OAS3 (2’, 5’-oligoadenylate synthase) serves as a biomarker for disease staging, treatment response, prognosis, and tumor microenvironment across various cancer types24. Both NT5DC3 (5’-nucleotidase domain-containing 3) and AHNAK (Neuroblast differentiation-associated protein) influence the growth of hepatocellular carcinoma25,26.
Data were categorized into HRG and LRG using survival curves, and 1-, 3-, and 5-year survival rates were predicted via ROC curves. The prognostic model was validated across different sets, with the scoring model applied similarly in both the training and GEO sets, yielding favorable AUC values and significant prognostic differences between the two subgroups. Notably, the high-risk group exhibited a poorer prognosis compared to the low-risk group.
Additionally, the constructed model outperformed previous models in predicting long-term prognosis. It also demonstrated that the risk score could serve as an independent risk factor. Subgroup analysis confirmed the model’s effectiveness in identifying and predicting BLCA patients with varying clinical features. Patient risk scores, calculated based on characteristics, correlated with age (≤ 65 years or > 65 years), grade (high or low), stage (I-II or III-IV), and T stage (T1-T2 or T3-T4), indicating the model’s broad applicability.
This study demonstrates that the prognostic model based on liquid-liquid phase separation (LLPS) offers a potential method for BLCA typing. This model facilitates the early identification of BLCA patients and predicts their prognosis. However, the study’s limitation lies in examining the role of LLPS in bladder cancer only at the molecular level, necessitating further experimental validation of these model genes’ functions.
Data availability
Data supporting this study’s results are publicly available in [RNAs involved in liquid-liquid phase separation (RPS)] at [http://rps.renlab.org], [The Cancer Genome Atlas (TCGA)] at [https://portal.gdc.cancer.gov/], and [Gene Expression Omnibus (GEO)] at [http://www.ncbi.nlm.nih].
References
Panebianco, V. et al. VI-RADS for bladder cancer: Current applications and future developments. J. Magn. Reson. Imaging JMRI. 55, 23–36 (2022).
Patel, V. G., Oh, W. K. & Galsky, M. D. Treatment of muscle-invasive and advanced bladder cancer in 2020. CA Cancer J. Clin.70, 404–423 (2020).
Lenis, A. T., Lec, P. M., Chamie, K. & Mshs, M. D. Bladder cancer: A review. JAMA324, 1980–1991 (2020).
Identification and Quantification of Iron Metabolism Landscape on Therapy and Prognosis in Bladder Cancer. - PubMed, accessed 25 Apr 2023; https://pubmed.ncbi.nlm.nih.gov/35265613/.
Siracusano, S., Rizzetto, R. & Porcaro, A. B. Bladder cancer genomics. Urologia. 87, 49–56 (2020).
Mehta, S. & Zhang, J. Liquid-liquid phase separation drives cellular function and dysfunction in cancer. Nat. Rev. Cancer. 22, 239–252 (2022).
Liquid-Liquid Phase Separation (LLPS) in Cellular Physiology and Tumor Biology - PubMed, accessed 25 Apr 2023; https://pubmed.ncbi.nlm.nih.gov/34522448/.
Alberti, S. & Dormann, D. Liquid-liquid phase separation in disease. Annu. Rev. Genet.53, 171–194 (2019).
Liquid Phase Condensation in Cell Physiology and Disease - PubMed, accessed 25 Apr 2023; https://pubmed.ncbi.nlm.nih.gov/28935776/.
RNA and Liquid-Liquid Phase Separation - PubMed, accessed 25 Apr 2023; https://pubmed.ncbi.nlm.nih.gov/33997539/.
RPS: A Comprehensive Database of RNAs Involved in Liquid-Liquid Phase Separation - PubMed, accessed 25 Apr 2023; https://pubmed.ncbi.nlm.nih.gov/34718734/.
Wang, C.-Y. et al. Dual hyper-graph regularized supervised NMF for selecting differentially expressed genes and tumor classification. IEEE/ACM Trans. Comput. Biol. Bioinform.18, 2375–2383 (2021).
Wang, C-Y., Liu, J-X., Yu, N. & Zheng, C-H. Sparse graph regularization non-negative matrix factorization based on Huber loss model for cancer data analysis. Front. Genet.10, 1054 (2019).
A Flexible R Package for Nonnegative Matrix Factorization - PubMed, accessed 25 Apr 2023; https://pubmed.ncbi.nlm.nih.gov/20598126/.
Identification of Molecular Subtypes and Prognostic Signature for Hepatocellular Carcinoma Based on Genes Associated with Homologous Recombination Deficiency - PubMed, accessed 25 Apr 2023; https://pubmed.ncbi.nlm.nih.gov/34912005/.
Head and Neck Squamous Cell Carcinoma Subtypes Based on Immunologic and Hallmark Gene Sets in Tumor and Non.-tumor Tissues - PubMed, accessed 25 Apr 2023; https://pubmed.ncbi.nlm.nih.gov/35187059/.
GSEA-InContext: Identifying Novel and Common Patterns in Expression Experiments - PubMed, accessed 25 Apr 2023; https://pubmed.ncbi.nlm.nih.gov/29950010/.
Cao, R. et al. An EMT-related gene signature for the prognosis of human bladder cancer. J. Cell. Mol. Med.24, 605–617 (2020).
Lv, J., Zhu, Y., Ji, A., Zhang, Q. & Liao, G. Mining TCGA database for tumor mutation burden and their clinical significance in bladder cancer. Biosci. Rep.40, BSR20194337 (2020).
Robertson, A. G. et al. Comprehensive molecular characterization of muscle-invasive bladder Cancer. Cell. 174, 1033 (2018).
Liquid-Liquid Phase Separation in Biology: Mechanisms, Physiological Functions and Human Diseases - PubMed, accessed 25 Apr 2023; https://pubmed.ncbi.nlm.nih.gov/32548680/.
Expression and Role of Methylenetetrahydrofolate Dehydrogenase 1 Like (MTHFD1L) in Bladder Cancer - PubMed, accessed 25 Apr 2023; https://pubmed.ncbi.nlm.nih.gov/31401334/.
P4HB: A Novel Diagnostic and Prognostic Biomarker for Bladder Carcinoma - PubMed, accessed 25 Apr 2023; https://pubmed.ncbi.nlm.nih.gov/33376528/.
OAS3 is a Co-immune Biomarker Associated with Tumour Microenvironment Disease Staging, Prognosis, and Treatment Response in Multiple Cancer Types - PubMed, accessed 25 Apr 2023; https://pubmed.ncbi.nlm.nih.gov/35592250/.
AHNAK Contributes to Hepatocellular Carcinoma Growth by Interacting with IGF-1R - PubMed, accessed 25 Apr 2023; https://pubmed.ncbi.nlm.nih.gov/36557813/.
Comprehensive Analysis of Prognostic Value and Immune Infiltration of the NT5DC Family in Hepatocellular Carcinoma. - PubMed, accessed 25 Apr 2023; https://pubmed.ncbi.nlm.nih.gov/35047040/.
Acknowledgements
Gratitude is extended to the TCGA database for providing the platform and to the contributors for uploading their valuable datasets.
Funding
The project for enhancing the basic research capabilities of young and middle-aged teachers in Guangxi universities (No: 2022KY0083). Self-funded scientific research of the Guangxi Zhuang Autonomous Region Health Committee (No: Z20190841). Self-funded scientific research of the Guangxi Zhuang Autonomous Region Health Committee (No: 20210968). Self-funded scientific research of the Guangxi Zhuang Autonomous Region Health Committee (No: 20210487).
Author information
Authors and Affiliations
Contributions
W.M. designed and conceived the study. Q.S. analyzed data. S.G. completed, drafted and revised the manuscript. Z.Y. provided advice and technical assistance. All authors have contributed to and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
All data are sourced from public databases, with ethics approval obtained by the data providers.
Consent for publication
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guo, S., Su, Q., Yang, Z. et al. Development and validation of a novel liquid-liquid phase separation gene signature for bladder cancer. Sci Rep 14, 22552 (2024). https://doi.org/10.1038/s41598-024-73422-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-73422-8
