
Abstract
Opioid prescription records in existing electronic health record (EHR) databases are a potentially useful, high-fidelity data source for opioid use-related risk phenotyping in genetic analyses. Prescriptions for codeine derived from EHR records were used as targeting traits by screening 16 million patient-level medication records. Genome-wide association analyses were then conducted to identify genomic loci and candidate genes associated with different count patterns of codeine prescriptions. Both low- and high-prescription counts were captured by developing 8 types of phenotypes with selected ranges of prescription numbers to reflect potentially different levels of opioid risk severity. We identified one significant locus associated with low-count codeine prescriptions (1, 2 or 3 prescriptions), while up to 7 loci were identified for higher counts (≥ 4, ≥ 5, ≥6, or ≥ 7 prescriptions), with a strong overlap across different thresholds. We identified 9 significant genomic loci with all-count phenotype. Further, using the polygenic risk approach, we identified a significant correlation (Tau = 0.67, p = 0.01) between an externally derived polygenic risk score for opioid use disorder and numbers of codeine prescriptions. As a proof-of-concept study, our research provides a novel and generalizable phenotyping pipeline for the genomic study of opioid-related risk traits.
Similar content being viewed by others

Introduction
Opioids are among the top 10 most-prescribed prescription medications in the U.S., and about 80% of surgical patients are treated with opioids for acute post-surgical pain1,2.
Opioids are also commonly prescribed for patients with moderate or severe chronic pain that is not managed well by non-opioid drugs3. Starting in the early 1990s, opioid prescriptions increased significantly for pain management, leading to surges in overdoses, opioid use disorder (OUD), and the so-called “opioid crisis”4,5. While opioid drugs are very effective for controlling pain, they are highly addictive6.
Side effects of opioid use include respiratory depression and excessive sedation7. Further, patients who take opioids for longer than 90 days have an increased risk of developing OUD8. In the U.S., up to 3 million people have current or past OUD9. It also has been estimated that 80,816 deaths were related to opioid overdose in the United States in 202110. In recent years, opioid prescription rates have dropped precipitously and most deaths are due to illicit fentanyl, but prescription opioids are still associated with about 12,000 overdose deaths in the U.S. each year11. Additionally, opioid-related adverse drug events (ORADEs) can cause harmful patient outcomes, including inpatient costs, readmissions, and mortality12.
Genome-wide association studies (GWAS) have suggested that both OUD and opioid-related patient responses have strong genetic underpinnings13,14,15,16,17,18,19. GWAS have identified significant genomic loci and related genes that can affect efficacy, metabolism, and adverse effects of opioids, which can in turn cause heterogeneous individual responses to drugs, including both pain levels and development of addiction20,21,22. This is particularly relevant to codeine, in which polymorphism alters the function and expression of the CYP2D6 gene responsible for its metabolism and can vary significantly between individuals23.
With promising studies continuously improving our understanding of the genetic architecture of opioid use disorder24, phenotyping opioid-related conditions in large patient populations remains a significant barrier for exploring the genetics. Collecting OUD-related diagnostic information from patients can be time-consuming, complicating the assembly of large sample sizes for GWAS25. One recent genomic study used medication use as a surrogate phenotype to explore disease etiology26. The results suggest that the genetic signature of taking disease-relevant medication could be used to predict future risk of disease. Electronic health record (EHR) datasets contain a large volume of prescription information with high fidelity, which can serve as a useful source for medication use-based phenotypes27,28,29. This phenotyping method could be particularly useful for diseases like OUD30.
In this proof-of-principle study, we utilized matched EHR and genotyping in the Mass General Brigham (MGB) Biobank, a large clinical data depository with patient records from multiple hospitals, to develop opioid prescription-based phenotypes. We selected codeine, one of the most commonly prescribed opioids worldwide31,32, as a test case with number of prescriptions, an easily generalized trait, for phenotype development. We constructed multiple prescription count-dependent pattern measures for genetic analysis. We then used both GWAS and polygenic risk score methods to investigate the genetic basis of these prescription patterns.
Methods
Data source
The clinical and genetic data in this study were obtained from the MGB Biobank. The MGB Biobank is a large integrated database, including high-quality clinical data from multiple Harvard-affiliated hospitals33. For our genome-phenome association study, we extracted matched genetic and clinical phenotype information from 36,239 European ancestry subjects based on patient self-reported records. The present analysis includes only individuals with European ancestry to minimize the risk of confounding due to ancestry differences. The study’s protocol was reviewed and approved by the Mass General Brigham Human Research Committee (study design summarized in Fig. 1).

Summary of study design.
Codeine count granularity measurement phenotypes
We screened ~ 16 million medication records from 2010 to 2020 in the study population and identified codeine prescription records by using keyword search. Three categories of codeine prescription count measures were used to develop 8 phenotypes to reflect different levels of information granularity:
-
(1)
Three low-count prescription phenotypes: patients with 1, 2 or 3 codeine prescriptions.
-
(2)
Four high-count prescription phenotypes: patients with 4 or more, 5 or more, 6 or more, or 7 or more codeine prescriptions. For both low- and high-count prescription groups, the control group was defined as patients with no opioid prescriptions.
-
(3)
All-count prescription phenotype: codeine prescription count was coded as integers and winsorized at 8 prescriptions to reduce the influence of outliers.
Genotyping data and quality control
Genotyping was performed by the MGB Biobank team. Prior to imputation, standard GWAS quality control procedures were carried out. These included: (1) sample-level QC. samples with discrepant reported and predicted sex or high missing rates were excluded; (2) Variant-level QC. variants with invalid alleles, allele mismatch with the reference panel, SNPs not found within the reference panel and duplicated, monomorphic variants, indels (insertion and deletions), and variants with low call rate (less than 90%) were excluded. Imputation was performed using the Michigan Imputation Server with 1000 Genomes panel and haplotype phasing was performed using SHAPEIT34,35,36.
Post-imputation quality control was conducted to select high-quality SNPs and control for population stratification and family structure. The relatedness of the cohort was detected by pairwise IBD estimation filtered by pi-hat (1 for 100% identical by descent [IBD], 0.5 for 50%, 0.25 for 25%) using PLINK to estimate the probability of sharing 0, 1, or 2 alleles IBD for any two individuals from the study population. Only autosomal biallelic SNPs with minor allele frequencies (MAF) of at least 1%, an info score above 0.8 and call rates above 98% were retained, which led to ~ 5 million SNPs. A principal components analysis was applied in a linkage-disequilibrium-pruned set of genotyped SNPs to characterize population structure within samples from included individuals.
Genome-wide association and gene-level analysis
We used PLINK 2.0 to conduct the genome-wide association analysis for each codeine prescription phenotype, using linear regression for continuous phenotypes and logistic regression for binary phenotypes37. All association analyses were adjusted for age, sex and the top 5 principal components. We used functional mapping and annotation (FUMA) and multi-marker analysis of genomic annotation (MAGMA) to conduct gene-based tests and pathway analysis38,39. A standard genome-wide significance threshold of p < 5 × 10− 8 was chosen for SNP identification and r2 = 0.6 was set as the cutoff for independent significant SNPs. The maximum distance of linkage disequilibrium (LD) blocks to merge was 250 kb. All Manhattan plots were generated by FUMA.
Disease polygenic risk score and correlation analysis
Summary statistics for multiple disease traits were obtained from two external data resources: (1) the Psychiatric Genomics Consortium (PGC);40 (2) the United Kingdom BioBank using the Pan-UK Biobank developed by team from the Analytical and Translational Genetic Unit (ATGU) of Massachusetts General Hospital and the Broad Institute of the Massachusetts Institute of Technology and Harvard41,42. We selected three categories of phenotypes for PRS development based on clinicians’ suggestions, including: (1) opioid use disorder and alcohol dependence; (2) brain and mental health phenotypes (Alzheimer’s dementia and Attention deficit hyperactivity disorder); and (3) other phenotypes (Hyperhidrosis, Standing height, ECG heart rate, Glaucoma and Diabetic hypoglycemia). Other phenotypes serve as negative controls for PRS. With these external summary statistic datasets, we used PRC-CS43, a python tool that utilizes a Bayesian regression framework to output optimized SNP effect sizes representing these diseases. We then developed patient-level polygenic risk scores among MGB patients for nine conditions, including positive (i.e., OUD) and negative (e.g., hyperhidrosis) controls. The default parameters of PRC-CS were used for the analysis. We used 830,461 SNPs from the 1000 Genomes reference panel for PRS construction. We then calculated Kendall correlations between disease polygenic risk scores and codeine prescription count in MGB patient population.
Results
EHR-derived codeine prescription count phenotypes
Using ~ 16 million medication records in MGB clinical database, we identified 8,639 patients with codeine prescriptions during 2010 to 2020, with approximately 700 to 1500 patients per year (Supplementary Fig. 1). We developed multiple codeine count measures based on the number of separate codeine prescriptions per patient (summarized in Table 1). We then used these measurements to develop 8 phenotypes for genome-wide association analyses. We used linear regression to capture all count distribution patterns with a continuous measure, while logistic regression was applied for either low count or high count patterns. We observed relatively older mean age in high count (patients with four or more codeine prescriptions) group compared with low count group. High count patients also have more incidence of diagnoses and clinical encounters in their EHR records (Supplementary Table 1).
Genome-wide association analysis
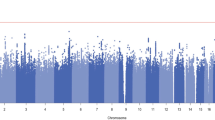
Setting the p-value threshold at 5 × 10− 8, 9 significant genomic risk loci were identified from the all-count phenotype (Fig. 2; Table 2 and Supplementary Fig. 2). The most significant lead SNP was rs2902921 (p = 6.44 × 10− 19), an intergenic SNP on chromosome 4. In addition, two loci (rs709286 and rs11164801-THRAP3, SH3D21, EVA1B, RP11-268J15.5, STK40, LSM10,EVI5, RPL5 and FAM69A) on chromosome 1, one locus on chromosome 2 (rs11680325 – CYP1B1), one locus on chromosome 3 (rs375170584), one locus on chromosome 4 (rs2902921), one locus on chromosome 5 (rs55905691-TSLP, WDR36), one locus on chromosome 11 (rs364139), one locus on chromosome 14 (rs2093210-C14orf39) and one locus on chromosome 17 (rs12453884-TAOK1, ABHD15 and TP53I13) were also identified.

Manhattan plots for GWAS of all prescription count phenotypes.
Various numbers of significant genomic loci were identified from low- and high-count phenotypes (Fig. 2; Table 2, and Supplementary Fig. 3). Two lead significant SNPs identified by three low-count measures (rs13103207 and rs78121242) were in the same LD region with rs2902921 (both R2 > 0.1) identified from the all-count phenotype. High-count phenotypes generally showed more similar genetic associations with the all-count phenotype. Thresholds of 4 or more and 5 or more prescriptions identified 7 and 8 significant genomic loci, respectively. All these loci were shared with the all-count phenotype. Fewer loci were identified with 6 or more and 7 or more prescriptions (5 loci and 3 loci, respectively), although they still overlapped with loci from the all-count phenotype.
Mapped genes and related functions
Using a two-sided distance of +/-10 kb region in proximity to identified genomic loci, we identified genes that could be related with regulatory functions of these variants (summarized in Table 3). Sixteen related genes were found from the all-count phenotype, while 2 to 14 genes were from high-count prescription phenotypes. As more extreme prescription ranges were applied, fewer mapped genes were found, corresponding to fewer significant loci from GWAS. Two genes were remained across all phenotypes: CYP1B1 and C14orf39. CYP1B1 is a member of the cytochrome P450 superfamily of enzymes, one of major enzyme families for drug metabolism44. C14orf39, also known as Six6os1, has been related to primary ovarian insufficiency45.
Comparison between our study and previous opioid genetic studies
We compared our results with previously published opioid-related GWAS (Supplementary Table 2)14,15,16. Of SNPs previously reported, rs9291211 was associated with opioid use in patients of European-ancestry. In our sample, rs9291211 showed various levels of weak associations with different codeine prescription phenotypes, with the relatively stronger signal in 6 or more prescriptions (p = 0.00017677), followed by 7 or more prescriptions (p = 0.00035105). Another three reported SNPs, rs1989903 (opioid use disorder) and rs12130499 (opioid dependence), and rs7188250 (opioid use disorder) also showed weak association p-values in our samples.
Polygenic risk score correlation analysis
We downloaded summary statistics for nine separate conditions from the Psychiatric Genomics Consortium (PGC) and the Pan-UK Biobank and developed patient-level disease polygenic risk scores for these conditions in the MGB study cohort15,46,47,48,49. Among them, codeine prescription count was significantly correlated (Tau = 0.67, p = 0.0127) only with the polygenic risk score for OUD (Table 4).
Discussion
The availability of genomic and clinical data in large data repositories, including Electronic Medical Records and Genomics (eMERGE) and UK Biobank42,50, has enabled researchers to perform more powerful genome-phenome association studies. The All of Us Research Program initiated by the NIH51, with clinical and genomic data expected from 1 million individuals, represents a new era of integrated big data consortia that has the potential to advance precision medicine research to a higher level. Through these studies, information from both the genomic and clinical perspectives can be fully integrated into association models to generate more comprehensive descriptions of disease status. We applied these methods to a large clinical biobank to assess relationships with codeine prescription number, as a test case for opioids, and found 9 loci with strong associations with a high count of codeine prescriptions.
Clinically meaningful phenotypes are critical for disease-oriented genetic research, especially for complex clinical conditions, such chronic diseases or diseases with complicated prescriptions52,53. An accurate and generalizable phenotyping approach could enable a better chance to identify related genetic markers54,55. This is particularly true for disease phenotypes which are challenging to develop, such as phenotypes related to diagnoses of substance dependence and substance use disorder. Due to their sensitive and complex nature, with no simple diagnostic test, this type of diagnostic information will generally be difficult to obtain and hence missing in large numbers of subjects56. Reliance on administrative codes is also problematic; early cases will tend to be missed and diagnoses may be biased by physician factors. Lack of documentation of substance use disorder in patient records also creates a significant limitation for conducting large-scale genetic studies and replications.
Recent studies have suggested that medication use can serve as a useful phenotype method for exploring the genetic basis of medication-related diseases and conditions. Genetic susceptibility of common diseases can be associated with traits of taking relevant medications26. This reverse causality approach provides a useful way to examine disease etiologies by investigating the genetic basis of patients who receive certain medications.
Prescription records can be easily retrieved from EHR databases in large patient populations with high fidelity because prescribing is invariably a core function of EHRs. Using prescription data, related disease traits can be developed. This approach provides phenotypes that can supplement diagnosis-based phenotypes with several unique advantages: (1) for diseases with more difficult (e.g., time consuming or hard to obtain) diagnostic records, relevant medication use can serve as a much easier indirect phenotyping method; (2) for chronic diseases with multiple progression stages, diagnosis-based traits might miss patients with early or subclinical conditions, while medication-based traits could capture a broad range of patients at early stages with less-extreme conditions; (3) medication-based traits can be developed in both a continuous or categorical manner. For example, prescription numbers can serve as a numerical-based measurement, which could potentially provide possibilities to reflect different risk levels in patient populations; (4) large phenotype groups can be created based on prescription records to gain more power for genome-wide association analyses. During phenotyping process, multiple prescription-related variables (medication type, count, dosage, duration etc.) can be used to assemble phenotypes with different levels of granularity.
In this study, we explored the feasibility of conducting opioid-related genetic research using patients’ prescription records. We selected codeine, an opiate with known heterogeneous metabolization between individuals, to capture a patient population with different levels of risk of adverse opioid-related outcomes. We also utilized prescription count to develop targeting phenotypes, requiring no granular prescription information, such as dosage. With this design, we are aiming to test our phenotyping pipeline in a baseline setting with a high generalizability.
Multiple prescription count were used to capture different patterns of codeine exposure. Previous studies have demonstrated that patients with a high count of opioid prescriptions tend to have long-term use and addiction57, suggesting an association between opioid prescription pattern/intensity and levels of future opioid use disorder risk. Considering this finding, we aimed to explore the potential genetic components of this association. Since this association might not be linear, we developed various prescription pattern measures to guide the genetic analysis.
Based on these prescription measures, we observed a count-dependent genotyping-phenotyping pattern, with higher prescription number phenotypes associated with stronger genetic signals. Substantial overlap was also identified across all phenotypes, suggesting a common genetic component among all prescribing counts. In our finding, lower numbers of prescriptions (1, 2, and 3) showed much weaker signals than higher numbers. When patient populations with a greater number of prescriptions (> 6) were selected, we observed a potentially greater specific genetic association relationship with a smaller number of significant SNPs. This pattern is consistent with gene-level analysis with only two genes remaining in the 7 or more phenotype. Both genes were concordant with disease mechanisms from previous studies. CYP1B1, a gene coding for a major enzyme of drug metabolisms, could be particularly relevant to opioid drug responses. Consistent with our finding, a recent study showed the association between CYP1B1 and EHR-derived opioid response58.
We used two approaches to validate our findings. First, we checked previously reported opioid use associated SNPs in our results and identified weak association p-values for several SNPs. Second, we examined the correlations between codeine prescription number with multiple clinical diseases/conditions using polygenic risk scores derived from independent summary statistics. The polygenic risk of opioid use disorder was significantly correlated with the observed number of codeine prescriptions, validating that this risk score is specifically associated with an expected phenotype. Accordingly, a higher mean PRS was observed in high count population (four or more codeine prescriptions) compared with low count patient population (three or less codeine prescriptions). Based on previous literatures, mental disorders are common among patients with opioid use disorder59. Furthermore, there is a positive association between mental disorder and opioid prescriptions60. Opioid use can also be related with other substance use disorders61, suggesting a broad scope of addiction and psychiatric conditions could be also associated opioid prescription with PRS methods in clinical practices.
Limitations
This study has several limitations. First, we only selected one type of common opioid drug, codeine, for phenotype development. Other major opioids were not included in the current study, which limits patient population we investigated. Compared with more potent opioids (e.g. hydrocodone and oxycodone), codeine is considered as a relatively weak opioid drug62, with a morphine milligram equivalent 10% of oxycodone. But the population we captured might better reflect early-stage risky population. Another reason to choose codeine is its metabolism. Codeine is one of opioids with clinically actionable gene variants supported by international guideline of drug dosing alterations, making it an interesting research target63. As a proof-of-concept study, we did show the feasibility of our phenotyping pipeline in this population for opioid-related genetic study and validated our finding. Second, the medication use phenotype, compared with diagnosis-based traits, is an indirect approach to reflect the at-risk population. The patient population we captured using this approach could be more heterogeneous with a broader spectrum of disease progression status, which can create heterogeneity in associated genetic signals. In the meantime, the specificity and sensitivity of this phenotyping system can be adjusted by using different cut-off thresholds. In our study, by testing different stringent phenotyping criteria, i.e., the number of prescriptions, we did observe a codeine count-dependent pattern for genetic hits. This provides the potential to calibrate optimal phenotyping thresholds to serve genetic studies with different purposes. For example, researchers can use this method to investigate phenotypes with different sensitivities or specificities for targeted diseases or conditions. Third, the phenotypes we created in the current study only focused on prescription count (numbers of records in EHR database). We did not include dosage or quantity information, which is another important component of prescription decision-making, and we were unable to incorporate prescriptions received outside of the MGB hospital system or prescriptions that were written but not filled. Further, the length of time for codeine prescriptions was not incorporated in current phenotyping pipeline due to lack of high-granular prescription time/duration information and more complete medical history records. Considering these prescription variables require more complete EHR dataset, which could be lacking in many current biobank data depositories, prescription count may be a more generalizable phenotyping method across databases. As a next step, we will incorporate other opioids and standardized opioid dosage and prescription duration information in future studies for a more advanced phenotyping pipeline. We will further incorporate other medical records, including prescription records for outpatient setting (drug monitoring program), patient medical history (e.g. psychiatric comorbidities) and co-prescription records (e.g. stimulant prescriptions). We will explore and identify optimal risk threshold, uncertainties or confidence intervals of PRS. With that, we will develop PRS tool to predict patient-level or population-level risk of opioid use disorder.
Conclusion
We utilized patient-level medication data from a large clinical biobank to develop codeine prescription number phenotypes for genetic research. We observed an interesting pattern of prescription-count dependent genomic signals, suggesting that medication prescription-based phenotypes could be used to capture various levels of opioid-related risk populations in genetic study. Our results provided a novel and generalizable phenotyping framework for opioid-related genetic research.
Data availability
The clinical/genetic datasets generated and analyzed during the current study are not publicly available due to hospital IRB regulation and patient privacy. The genetic summary statistics are available from the corresponding author upon request.
References
Rocha, V. et al. Geographic Variation in Top-10 prescribed Medicines and potentially inappropriate medication in Portugal: An ecological study of 2.2 million older adults. Int. J. Environ. Res. Public. Health. https://doi.org/10.3390/ijerph191912938 (2022).
Ladha, K. S. et al. Opioid prescribing after surgery in the United States, Canada, and Sweden. JAMA Netw. Open 2, e1910734. https://doi.org/10.1001/jamanetworkopen.2019.10734 (2019).
Martell, B. A. et al. Systematic review: Opioid treatment for chronic back pain: Prevalence, efficacy, and association with addiction. Ann. Intern. Med. 146, 116–127. https://doi.org/10.7326/0003-4819-146-2-200701160-00006 (2007).
Gardner, E. A., McGrath, S. A., Dowling, D. & Bai, D. The Opioid Crisis: Prevalence and markets of opioids. Forensic Sci. Rev. 34, 43–70 (2022).
Jani, M. et al. Opioid prescribing among new users for non-cancer pain in the USA, Canada, UK, and Taiwan: A population-based cohort study. PLoS Med. 18, e1003829. https://doi.org/10.1371/journal.pmed.1003829 (2021).
Volkow, N. D., Jones, E. B., Einstein, E. B. & Wargo, E. M. Prevention and treatment of opioid misuse and addiction: A review. JAMA Psychiatry 76, 208–216. https://doi.org/10.1001/jamapsychiatry.2018.3126 (2019).
Paul, A. K. et al. Opioid Analgesia and Opioid-Induced adverse effects: A review. Pharmaceuticals (Basel). https://doi.org/10.3390/ph14111091 (2021).
Banerjee, G. et al. High-dose prescribed opioids are associated with increased risk of heroin use among United States military veterans. Pain 160, 2126–2135. https://doi.org/10.1097/j.pain.0000000000001606 (2019).
Schuckit, M. A. Treatment of Opioid-Use disorders. N. Engl. J. Med. 375, 357–368. https://doi.org/10.1056/NEJMra1604339 (2016).
Statistics, N. C. f. H. U.S. Overdose Deaths In. https://www.cdc.gov/nchs/pressroom/nchs_press_releases/2022/202205.htm (2021).
Ahmad, F. B., Rossen, C. J. & Sutton, L. M. P. Provisional Drug Overdose Death Counts. National Center for Health Statistics. https://www.cdc.gov/nchs/nvss/vsrr/drug-overdose-data.htm (2023).
Urman, R. D. et al. The Burden of Opioid-related adverse drug events on hospitalized previously opioid-free Surgical patients. J. Patient Saf. 17, e76–e83. https://doi.org/10.1097/PTS.0000000000000566 (2021).
Song, W. et al. Genome-wide association analysis of opioid use disorder: A novel approach using clinical data. Drug Alcohol Depend. 217, 108276. https://doi.org/10.1016/j.drugalcdep.2020.108276 (2020).
Nelson, E. C. et al. Evidence of CNIH3 involvement in opioid dependence. Mol. Psychiatry 21, 608–614. https://doi.org/10.1038/mp.2015.102 (2016).
Polimanti, R. et al. Leveraging genome-wide data to investigate differences between opioid use vs. opioid dependence in 41,176 individuals from the Psychiatric Genomics Consortium. Mol. Psychiatry 25, 1673–1687. https://doi.org/10.1038/s41380-020-0677-9 (2020).
Deak, J. D. et al. Genome-wide association study in individuals of European and African ancestry and multi-trait analysis of opioid use disorder identifies 19 independent genome-wide significant risk loci. Mol. Psychiatry 27, 3970–3979. https://doi.org/10.1038/s41380-022-01709-1 (2022).
Cheng, Z. et al. Genome-wide association study identifies a regulatory variant of RGMA associated with opioid dependence in European Americans. Biol. Psychiatry 84, 762–770. https://doi.org/10.1016/j.biopsych.2017.12.016 (2018).
Gelernter, J. et al. Genome-wide association study of opioid dependence: Multiple associations mapped to calcium and potassium pathways. Biol. Psychiatry 76, 66–74. https://doi.org/10.1016/j.biopsych.2013.08.034 (2014).
Hancock, D. B. et al. Cis-expression quantitative trait loci mapping reveals replicable associations with Heroin Addiction in OPRM1. Biol. Psychiatry 78, 474–484. https://doi.org/10.1016/j.biopsych.2015.01.003 (2015).
Singh, A., Zai, C., Mohiuddin, A. G. & Kennedy, J. L. The pharmacogenetics of opioid treatment for pain management. J. Psychopharmacol. 34, 1200–1209. https://doi.org/10.1177/0269881120944162 (2020).
Hwang, I. C. et al. OPRM1 A118G gene variant and postoperative opioid requirement: A systematic review and meta-analysis. Anesthesiology 121, 825–834. https://doi.org/10.1097/ALN.0000000000000405 (2014).
Nishizawa, D. et al. Genome-wide association study identifies a potent locus associated with human opioid sensitivity. Mol. Psychiatry 19, 55–62. https://doi.org/10.1038/mp.2012.164 (2014).
Virbalas, J., Morrow, B. E., Reynolds, D., Bent, J. P. & Ow, T. J. The prevalence of Ultrarapid Metabolizers of Codeine in a Diverse Urban Population. Otolaryngol. Head Neck Surg. 160, 420–425. https://doi.org/10.1177/0194599818804780 (2019).
Chawar, C. et al. A systematic review of GWAS identified SNPs associated with outcomes of medications for opioid use disorder. Addict. Sci. Clin. Pract.. https://doi.org/10.1186/s13722-021-00278-y (2021).
Hu, L. L., Sparenborg, S. & Tai, B. Privacy protection for patients with substance use problems. Subst. Abuse Rehabil. 2, 227–233. https://doi.org/10.2147/SAR.S27237 (2011).
Wu, Y. et al. Genome-wide association study of medication-use and associated disease in the UK Biobank. Nat. Commun. 10, 1891. https://doi.org/10.1038/s41467-019-09572-5 (2019).
Jennings, M. V. et al. Identifying high-risk comorbidities associated with opioid use patterns using Electronic Health record prescription data. Complex. Psychiatry 8, 47–55. https://doi.org/10.1159/000525313 (2022).
Breitenstein, M. K., Liu, H., Maxwell, K. N., Pathak, J. & Zhang, R. Electronic health record phenotypes for precision medicine: Perspectives and caveats from treatment of breast Cancer at a single Institution. Clin. Transl Sci. 11, 85–92. https://doi.org/10.1111/cts.12514 (2018).
Wei, W. Q. & Denny, J. C. Extracting research-quality phenotypes from electronic health records to support precision medicine. Genome Med. https://doi.org/10.1186/s13073-015-0166-y (2015).
Kember, R. L. et al. Cross-ancestry meta-analysis of opioid use disorder uncovers novel loci with predominant effects in brain regions associated with addiction. Nat. Neurosci. 25, 1279–1287. https://doi.org/10.1038/s41593-022-01160-z (2022).
Mishriky, J., Stupans, I. & Chan, V. The views of Australian adults experiencing pain on the upscheduling of codeine-containing analgesics to ‘prescription only’. Int. J. Clin. Pharm. 43, 386–393. https://doi.org/10.1007/s11096-020-01026-z (2021).
Robert, M., Jouanjus, E., Khouri, C., Sam-Lai, F., Revol, B. & N. & The opioid epidemic: A worldwide exploratory study using the WHO pharmacovigilance database. Addiction 118, 771–775. https://doi.org/10.1111/add.16081 (2023).
Castro, V. M. et al. The Mass General Brigham Biobank Portal: An i2b2-based data repository linking disparate and high-dimensional patient data to support multimodal analytics. J. Am. Med. Inf. Assoc. 29, 643–651. https://doi.org/10.1093/jamia/ocab264 (2022).
Fairley, S., Lowy-Gallego, E., Perry, E. & Flicek, P. The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 48, D941–D947. https://doi.org/10.1093/nar/gkz836 (2020).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287. https://doi.org/10.1038/ng.3656 (2016).
Delaneau, O., Marchini, J. & Zagury, J. F. A linear complexity phasing method for thousands of genomes. Nat. Methods 9, 179–181. https://doi.org/10.1038/nmeth.1785 (2011).
Chang, C. C. et al. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience. https://doi.org/10.1186/s13742-015-0047-8 (2015).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826. https://doi.org/10.1038/s41467-017-01261-5 (2017).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219. https://doi.org/10.1371/journal.pcbi.1004219 (2015).
Sullivan, P. F. et al. Psychiatric Genomics: An update and an agenda. Am. J. Psychiatry 175, 15–27. https://doi.org/10.1176/appi.ajp.2017.17030283 (2018).
Team, P. U. Pan-Ancestry Genetic Analysis of the UK Biobank. https://pan.ukbb.broadinstitute.org (2020).
Sudlow, C. et al. UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779. https://doi.org/10.1371/journal.pmed.1001779 (2015).
Ge, T., Chen, C. Y., Ni, Y., Feng, Y. A. & Smoller, J. W. Polygenic prediction via bayesian regression and continuous shrinkage priors. Nat. Commun. 10, 1776. https://doi.org/10.1038/s41467-019-09718-5 (2019).
Li, F., Zhu, W. & Gonzalez, F. J. Potential role of CYP1B1 in the development and treatment of metabolic diseases. Pharmacol. Ther. 178, 18–30. https://doi.org/10.1016/j.pharmthera.2017.03.007 (2017).
Fan, S. et al. Homozygous mutations in C14orf39/SIX6OS1 cause non-obstructive azoospermia and premature ovarian insufficiency in humans. Am. J. Hum. Genet. 108, 324–336. https://doi.org/10.1016/j.ajhg.2021.01.010 (2021).
Martin, J. et al. A genetic investigation of Sex Bias in the prevalence of Attention-Deficit/Hyperactivity disorder. Biol. Psychiatry 83, 1044–1053. https://doi.org/10.1016/j.biopsych.2017.11.026 (2018).
Walters, R. K. et al. Transancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nat. Neurosci. 21, 1656–1669. https://doi.org/10.1038/s41593-018-0275-1 (2018).
Jansen, I. E. et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413. https://doi.org/10.1038/s41588-018-0311-9 (2019).
Karczewski, K. et al. Pan-UK Biobank GWAS improves discovery, analysis of genetic architecture, and resolution into ancestry-enriched effects. medRxiv https://doi.org/10.1101/2024.03.13.24303864 (2024).
McCarty, C. A. et al. The eMERGE Network: A consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med. Genomics 4, 13. https://doi.org/10.1186/1755-8794-4-13 (2011).
All of Us Research Program. The all of Us Research Program. N. Engl. J. Med. 381, 668–676. https://doi.org/10.1056/NEJMsr1809937 (2019).
Song, W., Huang, H., Zhang, C. Z., Bates, D. W. & Wright, A. Using whole genome scores to compare three clinical phenotyping methods in complex diseases. Sci. Rep. 8, 11360. https://doi.org/10.1038/s41598-018-29634-w (2018).
Mosley, J. D. et al. Identifying genetically driven clinical phenotypes using linear mixed models. Nat. Commun. 7, 11433. https://doi.org/10.1038/ncomms11433 (2016).
DeBoever, C. et al. Assessing digital phenotyping to enhance genetic studies of human diseases. Am. J. Hum. Genet. 106, 611–622. https://doi.org/10.1016/j.ajhg.2020.03.007 (2020).
Sinnott, J. A. et al. Improving the power of genetic association tests with imperfect phenotype derived from electronic medical records. Hum. Genet. 133, 1369–1382. https://doi.org/10.1007/s00439-014-1466-9 (2014).
King, C., Englander, H., Priest, K. C., Korthuis, P. T. & McPherson, S. Addressing missing data in substance use research: A review and data justice-based approach. J. Addict. Med. 14, 454–456. https://doi.org/10.1097/ADM.0000000000000644 (2020).
Deyo, R. A. et al. Association between initial opioid prescribing patterns and subsequent long-term use among opioid-naive patients: A statewide retrospective cohort study. J. Gen. Intern. Med. 32, 21–27. https://doi.org/10.1007/s11606-016-3810-3 (2017).
Lopes, G. S. et al. Identification of sex-specific genetic associations in response to opioid analgesics in a White, non-hispanic cohort from Southeast Minnesota. Pharmacogenom. J. 22, 117–123. https://doi.org/10.1038/s41397-022-00265-9 (2022).
Santo, T. Jr. et al. Prevalence of mental disorders among people with opioid use disorder: A systematic review and meta-analysis. Drug Alcohol Depend. 238, 109551. https://doi.org/10.1016/j.drugalcdep.2022.109551 (2022).
Sullivan, M. D., Edlund, M. J., Zhang, L., Unutzer, J. & Wells, K. B. Association between mental health disorders, problem drug use, and regular prescription opioid use. Arch. Intern. Med. 166, 2087–2093. https://doi.org/10.1001/archinte.166.19.2087 (2006).
Compton, W. M., Valentino, R. J. & DuPont, R. L. Polysubstance use in the U.S. opioid crisis. Mol. Psychiatry 26, 41–50. https://doi.org/10.1038/s41380-020-00949-3 (2021).
Von Korff, M. et al. De facto long-term opioid therapy for noncancer pain. Clin. J. Pain 24, 521–527. https://doi.org/10.1097/AJP.0b013e318169d03b (2008).
Wong, A. K., Somogyi, A. A., Rubio, J. & Philip, J. The role of pharmacogenomics in opioid prescribing. Curr. Treat. Options Oncol. 23, 1353–1369. https://doi.org/10.1007/s11864-022-01010-x (2022).
Acknowledgements
This study was supported by National Institute on Drug Abuse (NIDA) 1K01DA059572-01.The authors would like to acknowledge contributions of The Mass General Brigham (MGB) Biobank for providing genomic data and health information data, and The MGB Biobank Team for providing all the technique support.We gratefully acknowledge all the studies and databases that made GWAS summary data available: Psychiatric Genomics Consortium, the Pan-UKB project and UK Biobank.Psychiatric Genomics Consortium: https://pgc.unc.edu/for-researchers/download-results/Pan-UKB team: https://pan.ukbb.broadinstitute.org. 2020.UK Biobank: https://biobank.ctsu.ox.ac.uk/crystal/exinfo.cgi? src=accessing_data_guide.
Author information
Authors and Affiliations
Contributions
WS initiated the study and developed the study cohort. WS, ML and RL designed and conducted data analysis. DB, KM, SW, RU and AS provided important clinical opinions. DB and AW were involved in study supervision. All authors are participated in manuscript development and are accountable for integrity of this work.
Corresponding author
Ethics declarations
Competing interests
Richard D. Urman received consulting fees from AcelRx and has received funding from National Institutes of Health.
Ethics approval
This project was reviewed and approved by the Mass General Brigham (MGB) Human Research Committee. Due to the retrospective nature of the study, the MGB Institutional Review Board waived the need of obtaining informed consent. All methods were carried out in accordance with relevant guidelines and regulations.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Song, W., Lam, M., Liu, R. et al. A genome-wide Association study of the Count of Codeine prescriptions. Sci Rep 14, 22780 (2024). https://doi.org/10.1038/s41598-024-73925-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-73925-4
