
Abstract
In this research, we describe the first aptasensor for the detection of the Rift Valley Fever virus (RVFV). The process involved the selection of aptamers through the systematic evolution of ligands by the exponential enrichment (SELEX) technique. After 12 rounds of selection, 6 aptamers were selected and the corresponding binding affinities were assessed using fluorescence binding assays, revealing dissociation constants ranging from 15.45 to 40.98 nM. Notably, among the aptamers, RV2 and RV3 exhibited the highest binding affinities toward RVFV, with dissociation constants of 15.45 and 18.62 nM, respectively. Thiol-modified aptamers were subsequently immobilized onto screen-printed gold electrodes, facilitating the label-free detection of RVFV through square wave voltammetry. The voltammetric aptasensor demonstrated an excellent sensitivity, with a detection limit of 0.015 ng/mL. In addition, cross-reactivity assessments were conducted, where negligible response was obtained when the aptasensor was exposed to non-specific proteins.
Similar content being viewed by others

Introduction
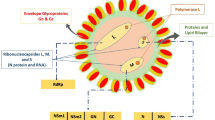
The Rift Valley fever virus (RVFV) is a causative agent of livestock and human severe infections. It is an arthropod-borne virus belonging to the Phlebovirus genus, family of bunyavirus (Bunyaviridae) of the order Bunyavirale1. RVFV is composed of an enclosed spherical structure measuring approximately 80 to 100 nm in diameter, containing an envelope decorated with glycoproteins. It possesses a tripartite single-stranded negative-sense RNA genome; S (small), M (Medium) and L (Large) segments, encoding several structural and non-structural proteins2. RVFV disease was initially characterized after the mortality of around 4,700 lambs and ewes on a farmhouse situated near the shores of Lake Naivasha in the Great Rift Valley of Kenya in 19313. The aetiology of the disease is hypothesized to be associated with ecological and climatic factors4. Over the past decade, there has been an increased occurrence of Rift Valley fever epidemics in West Africa and other nations within the sub-Saharan region. Recent research conducted in Mediterranean countries and the Western Sahara region has revealed the presence of seropositivity in both animals and humans5. In 2000, RVFV was reported for the first time in southwestern Saudi Arabia and northwestern Yemen, where 124 and 166 deaths were respectively, recorded6. Given its high epidemic potential and the potential agricultural and public health impact, RVFV has been listed by the Centres for Disease Control and United States Department of Agriculture as category A pathogen3. In parallel, RVF has been assigned to the list of diseases ‘severe emerging diseases with potential to generate a public health emergency, and for which no, or insufficient, preventive and curative solutions exist’7. It can cause widespread abortion epidemics and high mortality rates of almost 100% in newborn animals, and 10 to 20% in older ruminant animals. RVFV infections are commonly linked to acute diseases and only a minority of patients, may suffer from serious symptoms such as fulminant hepatitis, encephalitis, retinitis, blindness, or a haemorrhagic condition8. RVFV mortality rate in humans is estimated at 0.5–2.0%9.
Various diagnostics strategies have been developed to improve the prevention against RVF disease consequences on health and economic sectors. These methods are based on identifying the viral biomolecules including proteins and nucleic acids. In general, viral proteins are detected by immunoassays based on specific antibodies targeting the antigen, while PCR techniques are used for nucleic acids detection. These latter include real-time quantitative (RT-PCR)10, and reverse transcription loop-mediated isothermal amplification (RT-LAMP)11. Serological tests are also performed for detecting and quantifying the viral antigens as well as anti-RVFV IgGs and IgMs in infected patients or animals12,13. In this context, Hemagglutination inhibition, viral neutralization and immunoassays are usually employed and recognized by the WHO14. However, these classical techniques pose a great health risk for diagnosticians and require sophisticated and expensive facilities. Moreover, RVF diagnosis in naïve countries would not be possible because of the lack to specialized laboratories. To overcome these limitations, integrated systems such lateral flow immunoassays15,16, peptide-assays17 and biosensors have been developed. For instance, optical fiber immunosensors have been described to detect IgG antibody to RVFV18. In 2012, Zhang et al. described a magnetic capture-based Surface Enhanced Raman Scattering (SERS)-based biosensor for targeting DNA derived from RVFV genome. The detection was based on the hybridization of the viral DNA and Raman dye conjugated probe with a complementary sequence immobilized on Au-coated paramagnetic nanoparticles19. Nevertheless, to the best of our knowledge, no electrochemical biosensor has been reported for the detection of RVFV genome or antigens. In the electrochemical biosensing of pathogens, the biorecognition element specific to the target antibody, epitope or DNA is immobilized on a conducting surface known as electrode. The biological interaction between the bioreceptor and its target affect the electron transfer on the surface thus allowing the monitoring of the target concentration though an electrochemical method involving a redox electrolyte20. Electrochemical biosensors have shown highly sensitive detection of pathogens within large dynamic ranges, without the requirement of long sample preparation steps. In addition, the detection can be performed in a label-free mode, where target labelling is not required thus reducing the time and cost of analysis. The pathogen determination is mainly based on the complexation bioreceptor-target, unlike immunoassays that necessitates a secondary binding step21.
In the present work, we describe a label-free electrochemical biosensor for Rift Valley Fever Virus diagnostics. Given the numerous advantages of aptamers compared to antibodies, we used DNA aptamers as biorecognition element. Therefore, we report the selection of the first DNA aptamer targeting RVFV and its integration into a label-free voltametric biosensing platform. Herein, the SELEX process (Systematic Evolution of Ligands by EXponential enrichment) was carried out against the recombinant nucleocapsid (N) protein, the most abundant and immunogenic component in the RVF virion22. Six DNA sequences were selected and the aptamer exhibiting the best affinity was integrated into the electrochemical aptasensing platform. The obtained results show that our platform could be an excellent alternative to the existing assays based on nucleic acids and antibodies. The low cost of aptamers and electrodes contributes to an overall reduction in device expenses. The use of DNA aptamers ensures the long-term stability of the biosensor. Furthermore, the analysis requires only small blood sample volumes and reagents.
Materials and methods
Materials and reagents
RVFV Nucleoprotein, Rubella virus-like particles, Zika Virus Envelope Protein and Dengue Virus NS1 Protein Serotypes 1–4 was obtained from The Native Antigen Company (Kidlington, United Kingdom), Pure Cube Ni-NTA (Nickel-Nitrilotriacetic acid) Agarose beads were purchased from Cube Biotech (Monheim, Germany). Magnesium chloride, sodium carbonate anhydrous, sodium azide, acetic acid, boric acid, Ethylenediaminetetraacetic acid (EDTA), sodium bicarbonate, ethanol, Ethidium bromide solution, Tetramethyl ethylenediamine (TEMED), hydrochloric acid, acrylamide/bis-acrylamide, Bromophenol Blue, sodium acetate, ammonium persulfate (APS), Potassium ferrocyanide (K4Fe(CN)6), potassium ferricyanide (K3Fe(CN)6), sodium chloride, mercapto-1-hexanol (MCH), phosphate buffer saline (PBS: pH 7.4), Human serum and bovine serum albumin (BSA) were purchased from Sigma-Aldrich (Ontario, Canada). Tris-base and urea were purchased from Bioshop Inc. (Ontario, Canada). The DNA library (5’-TCCCTACGGCGCTAAC-N40-GCCACCGTGC-TACAAC-3’), labelled forward primer (5’-FAM-TCCCTACGGCGCTAAC-3’), unlabelled reverse primer 5′ -poly dA20-HEG-GTTGTAGCACGGTGG − 3′, M13 primers, and the aptamer sequences were synthesized by Metabion International AG (Planegg, Germany). PCR reagents (10x buffer, dNTPs, MgCl2, Taq polymerase, 6X loading dye, and 100 bp ladder) were obtained from ACE Biotech (Riyadh, Saudi Arabia). DNase/RNase Free Distilled Water, PCR™8/ GW/TOPO™ TA Cloning Kit with One Shot TOP10 and the DH5α T1R chemically competent E. coli cells were obtained from Invitrogen Inc. (New York, USA). Amicon Ultra (0.5 mL) centrifugal filters 3 KDa were purchased from EMD Millipore (Alberta, Canada). SpinX cellulose acetate centrifuge filter tubes with pore size of 0.22 μm were purchased from Corning life sciences (Massachusetts, USA).10X TE (Tris-EDTA) Buffer Solution was purchased from Teknova (USA). Agarose powder and ampicillin were obtained from Bio-Rad (California, USA). X-Gal was obtained from Bio Basic Inc. (Toronto, Ontario, Canada). Luria Bertani Agar was purchased from watin bio-life Advanced Diagnostics mfr (Riyadh, Saudi Arabia).
The binding buffer composed of 20 mM Tris, 1 mM MgCl2 and 150 mM NaCl, pH 8. The washing solution used in the beads coupling step was 30 mM of Imidazole prepared in binding buffer. Elution buffer consists in 300 mM Imidazole prepared in binding buffer. Tris-EDTA buffer is composed of 1 mM EDTA with 10 mM Tris (pH 7.4). The storage solution was 0.05% sodium azide in binding buffer.
Instrumentation
Bio-Rad T100 Thermal cycler (Hercules, California, USA) was used for the PCR amplification. The concentration of DNA and protein was determined using Nanodrop 2000 UV–Vis spectrophotometer (Thermo Scientific, Ottawa, Canada). The fluorescence intensity of the DNA was measured by using Nanodrop ND3300 fluorospectrometric (Thermo Scientific, Ottawa, Canada). The electrophoresis was run through Powerpack-current Power supply and supplied by Bio-Rad (California, United States). UVP BioDoc-It Imaging system (UK) was used in the DNA band analyzing. All electrochemical measurements, including cyclic voltammetry (CV) and square wave voltammetry (SWV), were conducted using a Metrohm (Switzerland) Autolab potentiostat, specifically, the PGSTAT302N model. The experimental setup was managed by Nova 1.11 software. Screen-Printed Gold Electrodes were purchased from Metrohm DropSens, Inc (Asturias, Spain). They are composed of Ceramic substrate (3.4 × 1.0 × 0.05 cm) comprising a gold auxilliary electrode and silver as reference. The working electrode diameter is 4 mm.
SELEX process
Immobilization of RVFV Nucleoprotein (N) on Ni-NTA Agarose beads
The mechanism of binding was based on the interaction between Histidine tag of the nucleoprotein and the Ni-NTA on the agarose beads (Figure S.1). The process of conjugating the RVFV nucleoprotein to the Ni-NTA Agarose beads started by applying the beads to many washes using a binding buffer to remove any residual preservative solution. Then, 100 µL of RVFV were added to tube containing 2 mL of the binding buffer. The washed beads were mixed with the protein solution at a 1:1 ratio and incubated at 4 ◦C overnight with end-over-end rotation. Subsequently, the protein-conjugated beads underwent a washing process using 2 mL of washing solution. Following that, the beads were subjected to incubation in the washing buffer for 1 h, during which rotation was performed to block the activity of unbound sites present on the beads surface. Following the blocking of the conjugated beads, a total of five washes were performed using binding buffer. Then, the conjugated beads were stored at 4 ◦C in TE buffer mixed with 0.05% sodium azide for further use in the SELEX process.
In vitro selection of DNA aptamers against RVFV protein
First, the DNA library and primers were designed as it was previously described by our group23. In the first cycle of selection 100 µL of the RVFV protein-conjugated beads are incubated with 3 nmol of DNA library while the amount of 150 pmol is used in the subsequent SELEX cycles. Prior to the incubation of the beads, these latter were washed five times with 300 µL of binding buffer to remove the unbound protein and eliminate the sodium azide. In parallel, the ssDNA was subjected to a heating process at a temperature of 90 ◦C for 5 min. Subsequently, it was cooled down to 4 °C for a period of 10 min, followed by a further 10 min incubation at room temperature. The ssDNA mixture was poured into the washed beads into the Spin-X filter tube. Following that, the filter tube was incubated at room temperature for a duration of 2 h with continuous rotation in an end-over-end method. After that, the beads were washed five times using binding buffer until the total absence of any observable fluorescence. This step was conducted to confirm the elimination of the non-bound DNA. In parallel, the bound DNA was separated using 300 µL of elution buffer and treated with heating process at approximately 90 °C for 10 min. This step was repeated until the last elution showed no fluorescence signal. Next, a centrifugal filter with a molecular weight cutoff of 3 kDa was used to concentrate and desalt the DNA that had been collected. Following the end of the seventh cycle, a negative-selection step was performed, involving the incubation of a DNA pool obtained from the previous positive cycle with blank beads; non conjugated to the RVFV nucleoprotein. After each cycle DNA was amplified by PCR generating double-stranded DNA (dsDNA) with around 72 bp (bp). Then, the dsDNA was separated into ssDNA using denaturing Urea PAGE (Polyacrylamide Gel Electrophoresis). The SELEX cycles were stopped at the 12th cycle, then unlabelled primers were used to amplify the eluted ssDNA after purification, and the final product was used in the cloning step. Forty reactions were used for the PCR amplification step, each containing 50 µL of the master mixture: 10x buffer, 10 mM dNTP, 25 mM MgCl2, 0.2 µM primers (forward and reverse), and 2 units of Taq polymerase. The PCR thermal cycle started at 95 °C for 5 min, then 15 cycles of 95 °C for 30 s/54°C for 30 s/72°C for 45 s, and a 10 min extension at 72 °C as a final step. The amplified product was confirmed by running 2% agarose gel electrophoresis with ethidium bromide at 110 V for 30 min. Subsequently, the amplified dsDNA underwent ethanol precipitation, wherein three volumes of pure cold ethanol and 0.1 volume of 3 M sodium acetate (pH 5.5) were added. The solution was incubated for 1 h at -80 °C and then centrifuged at 4 °C for 30 min. Next, the supernatant was removed, and the pellet washed by 75% ethanol and dried at 37 °C to eliminate any residual ethanol. The pellet was reconstituted in a water and formamide loading dye mixture with a volume ratio of 1:2 and this mixture was incubated at 90 °C. Then, the fluorescence-labelled ssDNA was separated by running the mixture through a 12% denaturing urea polyacrylamide gel at a voltage of 300 V for 1 h. The fluorescence-labelled ssDNA band was visualized using a gel imager. The band was subsequently cut from the gel and the DNA was extracted by adding TE buffer, freeze/thaw cycle, followed by incubating the DNA mixture in the rotator at 37 °C overnight. Finally, the eluted ssDNA solution was desalted and concentrated by a 3 KDa filter, then used to start a new SELEX cycle.
Cloning and sequencing of the aptamers selected against RVFV nucleoprotein
After the last round of SELEX, symmetric PCR was run, then the bands were checked using 2% agarose gel. The ligation was carried out by incubating the ligation mixture: 2 µL of PCR product, 1 µL of salt solution, 2 µL of H2O, and 1 µL of the vector at room temperature for 30 min. After that, E. coli competent cells were transferred into ice and 2 µL of the ligated product were immediately added. Then, the cells were incubated at the ice for 30 min and transferred at 42℃ for 30 s and the tube was placed on ice for 1–2 min. 250 µl of Super Optimal broth with Catabolite repression (SOC) media was subsequently added to the cells and rotated at 37 ℃ for 1 h. Next, 50 µl of ampicillin and 40 µl of X-gal were added to the plates and spread. After 1 h rotation, the cells were added on three different plates with three different volumes (25 µl, 50 µl, and 75 µl) and kept upside down at 37 ℃ for 24 h. The plates were examined to identify colonies with visible growth, and those displaying a positive white or light blue appearance were selected. The ssDNA aptamer inserts were subjected to amplification through colony PCR, using M13 forward and reverse primers. Verification of the successful incorporation of the aptamer sequences was achieved through the detection of a distinct 300 bp band via 2% agarose gel electrophoresis. Ultimately, the PCR products from the colonies were subjected to sequencing and alignment using the PRALINE program, accessible at https://www.ibi.vu.nl/programs/pralinewww/.
Binding affinity study of the aptamers to RVFV and determination of dissociation constants (Kd)
A series of fluorescein-labelled aptamers were diluted using the binding buffer (0-400 nM). Each solution was then treated with the heating/cooling cycle. Following that, the sample was incubated with 50 µL of beads and transferred into spin X filter tubes. The solution was subjected to incubation at ambient temperature for 60 min with continuous rotation. The beads underwent a washing process using 300 µL of binding buffer. This process was repeated five times. For the elution step, 300 µL of elution buffer was added to the sample. The tubes were then incubated at 90 °C for 10 min, after which centrifugation was performed at 13,000 rpm for 1 min. The eluates were collected, and the fluorescence of each sample was determined using a nanodrop fluorometer at an emission wavelength of 515 nm. Subsequently, the concentration of ssDNA input (from 0 to 100 nM) was plotted against the measured fluorescence intensity. Saturation curves have been created for each aptamer sequence. The Kd values were determined through nonlinear regression analysis using Prism software.
Fabrication of the electrochemical aptasensor
The selected aptamer was first conjugated to a thiol group to enable its immobilization on the gold electrodes. Then, 10 µL of the RVFV-aptamer (1 µM) was dropped onto the surface resulting in the spontaneous formation of a self-assembled monolayer (SAM). The incubation of the Au electrode with the aptamer was carried out overnight in a water-saturated environment at 4 °C. Subsequently, the electrode underwent a washing process using a PBS buffer solution with a pH of 7.4. The blocking of the remaining sites was accomplished by incubating the electrodes with 1 mM MCH prepared in PBS buffer (pH 7.4) for 30 min at room temperature. After washing with PBS, the electrode was ready to use for the electrochemical experiments. The label-free detection of the nucleoprotein was performed by SWV conducted in a solution containing 5 mM ferro/ferricyanide prepared in a 10 mM PBS buffer with a pH of 7.4. The SWV scan potential range was from 0.4 to -0.4 V with a step potential of -5 mV, an amplitude of 20 mV, and a frequency of 25 Hz.
The selectivity of the RVFV aptasensor toward its the targeted nucleoprotein was studied by incubating the functionalized gold surface with Rubella virus-like particles, Zika Virus Envelope Protein, Dengue Virus NS1 Protein, BSA, and human globulin. The test was performed by incubating 10 µL of the potential interferents (50 pg/mL) on the RVFV electrode. Then, the sensor response was evaluated as described above. To demonstrate the clinical applicability of the aptasensor, human serum was diluted 100 folds in 10 mM PBS buffer. Then, it was spiked with increasing concentrations of RVFV protein (50 pg/ml to 1000 pg/ml). 10 µL of each spiked serum was incubated with the RVFV aptasensor. Then, the corresponding electrochemical response was recorded and compared to that obtained in the buffer solution.
Results and discussion
In vitro selection of the DNA aptamers against RVFV
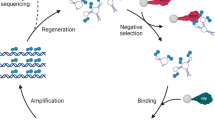
As it was mentioned above, Ni-NTA agarose beads were used as immobilization support for the RVFV nucleoprotein. Ni-NTA, nickel (II) ions bound to nitrilotriacetic acid (NTA), forms a complex in which NTA acts as a chelator that securely coordinates divalent nickel at four binding sites, preventing metal leaching from the resin24. Nickel exhibits superior binding capacity when compared to other divalent metals like cobalt, particularly with histidine residues in His-Tag recombinant proteins25. The RVFV protein was immobilized onto Ni-NTA agarose beads by linking the His-Tag at the protein’s C-terminal to the Ni-NTA groups on the beads. To minimize nonspecific binding between the beads and DNA, the remaining free Ni-NTA groups on the beads were blocked using a low concentration of imidazole. The coupling between the beads and the RVFV nucleoprotein were confirmed by measuring the protein concentration at 280 nm before and after the coupling. The measurements carried out in the washes after coupling, revealed a significant reduction in protein concentration, indicating successful coupling of most of the protein amount to the beads. To select a specific aptamer for the RVFV protein, 12 selection cycles were performed. The initial cycle involved a pool of ssDNA with 1.8 × 1015 random sequences comprising 40-nucleotides. Each cycle consists of eight consecutive steps (Fig. 1A). These steps included incubating the beads with the pool of ssDNA, followed by washing to remove unbound sequences. The elution of the bound sequences was accomplished using EB (300 mM Imidazole), wherein Imidazole served as a competitive agent to displace histidine-tagged proteins from the beads26. The eluted DNA was amplified through symmetric PCR and analysed via 2% agarose gel electrophoresis to confirm product size. Next, asymmetric PCR amplification was employed in combination with denaturing urea polyacrylamide gel electrophoresis to obtain ssDNA. Asymmetric PCR used dsDNA as a template with a 10 µM fluorescently labelled Forward primer to generate a substantial quantity of Forward DNA aptamer. The used reverse primer was extended with Poly A to enlarge the size of and a HEG (Hexaethylene glycol) spacer to block the PCR amplification27.

(A) SELEX steps for Aptamer selection. (B) Negative selection step.
This aptamer was purified through ethanol precipitation based on positively charged sodium ions neutralizing the negative charges on the DNA’s PO4− groups, making it less soluble in water due to reduced hydrophilicity in ethanol28. Given that asymmetric PCR products contain both ssDNA and ds DNA, the dsDNA can interfere with the selection process. For that, the strands of different lengths were separated using denaturing urea-polyacrylamide gel electrophoresis, as urea disrupted the hydrogen bonds between DNA strands, causing differential migration29. This process enriched the DNA pool with potentially high-affinity sequences for the target.
The fluorescence intensity of the eluted sequences in each cycle was monitored to ensure the enrichment. Figure 2A shows the obtained fluorescence revealing an initial increase in DNA recovery within the first four cycles, indicating the enrichment of RVFV-binding DNA. We noted that the enrichment plateaued by the seventh cycle. Then, a negative-selection step was performed to remove the non-specifically bound DNA where we observed a remarkable

(A) SELEX enrichment of RVFV aptamers, the bar graph demonstrates the quantity of ssDNA eluted from the beads during each cycle of selection (B) Analysis of the selected sequences by multiple sequence alignment using the PRALINE software. (C) Dissociation constants (Kd) determination (C).
decrease in fluorescence intensity (Fig. 1.B). After that, four additional SELEX rounds were conducted before reaching a plateau in the fluorescence intensity, indicating the saturation of the target binding sites.
Sequence alignment and determination of the dissociation constants of RVFV aptamers
To analyse the selected sequences, a multiple sequence alignment was performed using the PRALINE program (https://www.ibi.vu.nl/programs/pralinewww/). The sequences were classified into six distinct groups based on their similarities. Notably, each group displayed significant consensus regions shared by the sequences within that group, with some sequences being identical. Consequently, one sequence from each group was chosen for further studies on the binding affinity with RVFV, as illustrated in Fig. 2B.
Afterwards, the binding affinity of each sequence was determined by using a fluorescence binding assay. For that, the primer sites were truncated, and the aptamers were labelled with a carboxyfluorescein (FAM). The affinity study was conducted under the same SELEX conditions. The process involved incubating RVFV nucleoprotein-conjugated beads with varying concentrations of fluorescent aptamers, followed by a washing step to remove unbound aptamers. The aptamers that had bound to the beads were then eluted, and their fluorescence signal were measured and plotted against the aptamer concentration. Non-linear regression analysis was used to determine the Kd values for the selected aptamers (Fig. 2.C). Of the six selected aptamers, all demonstrated significant affinity for RVFV, with low Kd values ranging from 15.45 nM to 40.98 nM. As shown in table S1 (Supporting information), aptamers RVF 2 and RVF 3 exhibited the highest affinity for the RVFV protein, with the Kd values of 15.45 nM and 18.62 nM, respectively. Therefore, these two aptamers were employed in subsequent experiments.
Electrochemical aptasensing of RVFV
Characterization of the RVFV aptasensor fabrication steps
The aptamer RVF3 was chosen for the electrochemical detection because it has shown better analytical performance when compared to RVF2 aptamer. As it is depicted in Fig. 3, the thiol-modified aptamer was immobilized on a screen-printed gold electrode via self-assembly. Upon exposure to the gold, the disulfide bonds broke, resulting in the formation of semi-covalent bonds on the gold surface30. After the aptamer immobilization, the electrodes were blocked with MCH, forming a mixed monolayer. This MCH blocking step is crucial not only for reducing non-specific adsorption on the surface but also for preserving the anchored ssDNA conformation and regulating the DNA film formation31.

Fabrication steps and application of the aptasensor for the electrochemical detection of RVFV protein using SWV.
The fabrication steps of the aptasensor were characterized through cyclic voltammetry (CV) analysis using the ferro/ferricyanide redox couple, as depicted in Fig. 4. As it can be seen from the voltammograms, the bare Au electrode (black curve) displays reversible peaks with anodic/cathodic peak separation (ΔE) of approximately 0.12 V, indicating a clean gold surface. The immobilization of aptamers (red curve) resulted in a decrease in the cathodic and anodic peak currents due to the introduction of negatively charged DNA, which repelled the redox anions. These results confirm the successful assembly of aptamers on the Au surface. Moreover, a slight peak-to-peak separation was seen, corresponding to the aptamer voltammogram. Finally, the MCH blocking (blue curve) caused a reduction in the peak current and an increase in ΔE (0.43 V) due to the blocking of the remaining free gold surface.

(A) Cyclic voltammetry (CV) characterizations of the aptasensor fabrication steps (Au-) (black curve), aptamer-Au (Au/Apt) (red curve) and MCH (Au/ APT/MCH) (blue curve). cyclic voltammetry was performed with a scan rate of 50 mV s⁻¹, scanning over a potential range from + 0.6 V to -0.6 V.
Electrochemical detection of the RVFV nucleoprotein
To demonstrate that the selected aptamer can be successfully applied for determination of RVFV, the aptamer modified-SPGSs were separately incubated with increasing concentrations of the viral nucleoprotein for 35 min at room temperature. This incubation time was selected after an optimization step where we tested different duration varying from 5 to 45 min (Figure S.2). Square wave voltammetry measurements were first carried out on the aptasensor before addition of the analyte. The measurements were conducted in a 5 mM [Fe (CN)6] 4−/3− solution within PBS buffer at pH 7.4 The SWV scan potential range was from 0.4 to -0.4 V with a step potential of 5 mV, an amplitude of 20 mV, and a frequency of 25 Hz. Figure 5A shows the square wave voltammograms recorded in the range of concentration from 0 to 5000 ng/mL. A gradual reduction in the peak currents was noted on the RVFV sensor by increasing the targeted protein amount. The decreasing signal can be explained by the specific binding between the aptamer and RVFV protein which inhibits the electron transfer to the transducing surface. The obtained peak currents were used for calculating the sensor responses ((i0 − i)/i0%) and plotting the calibration curve. As it can be seen from Fig. 5B, the curve displays a linear response within the wide working range of 0.075ng/mL to 5000 ng/mL, with a regression equation of ((i0 − i)/i0%) = 56.060944 + 4.69291 log RVFV [ng/mL] and an R2 value of 0.95994. The detection limit (LOD) for RVFV was determined to be 0.015 ng/mL, it was calculated using the formula 3 σ/b, where σ represents the standard deviation of the blank aptasensor, and b is the slope of the calibration curve32. To assess the reproducibility, RVFV aptasensor underwent three independent measurements for each set of data.

(A) Square Wave Voltammograms of the RVFV aptasensor before and after incubation with varied RVFV nucleoprotein concentrations prepared in Binding Buffer. The measurements were conducted in a 5 mM [Fe (CN)6] 4−/3− solution within PBS buffer at pH 7.4 The SWV scan potential range was from 0.4 to -0.4 V with a step potential of 5 mV, an amplitude of 20 mV, and a frequency of 25 Hz. (B) Calibration curve for the RVFV aptasensor, depicting a plot of the logarithmically transformed concentrations of RVFV protein against the aptasensor response ((i0-i)/ i0%). The error bars represent standard deviations derived from triplicate measurements.
Selectivity studies
The aptasensor’s selectivity was tested with some non-specific proteins, including Rubella virus-like particles, Zika Virus Envelope Protein, Dengue Virus NS1 Protein, BSA, and human globulin. For that, the aptasensor was incubated with the potential interferents, separately, on different electrodes. This step is crucial to demonstrate that our aptamer is specific to RVFV nucleoprotein without cross-reactivity to any potential interferents. For that, the RV-3-modified SPGEs were incubated with a fixed amount of the different proteins, separately. The voltammetric responses were recorded by SWV and used to calculate the percentages ((i0 − i)/i0%) to build the histogram shown in Fig. 6. We note that the aptasensor’s responses to all the non-specific proteins were notably 12 times lower than the response obtained for RVFV nucleoprotein. These findings affirm the high selectivity of the RVFV aptasensor and establish its suitability for accurately quantifying RVFV without any interference from other proteins.

Comparative Analysis of the RVFV Aptasensor’s Response to 50 ng/mL RVFV, ZIKA, Dengue, Rubella, BSA, and Globulin, with Error Bars Indicating Standard Deviation from Three Replicate Measurements.
Applicability of the aptasensor in human serum samples
The effectiveness of the developed aptasensor for real applications was also verified by testing it with serum samples spiked with different amounts RVFV nucleoprotein. By comparing the calculated values with the added concentrations, we obtained excellent recovery rates ranging from 96 to 104.67% as shown in Table 1. These findings validate the strong selectivity and minimal interference from the surrounding matrix. This suggests that our aptasensor could be applied successfully in biological fluids. Additionally, the method’s reproducibility was assessed by calculating relative standard deviations from triplicate measurements, all of which were found to be less than 3%. This underscores the high level of consistency and reliability of the RVFV aptasensor.
Conclusion
In summary, we have successfully identified and characterized ssDNA aptamers designed for Rift Valley fever diagnostics. Among the identified aptamers, the aptamers RV-2 and RV-3 with the lowest dissociation constants (Kd) of 15.45 nM and 18.62 nM displayed the highest binding affinity for RVFV. The aptamers applicability was validated by employing RV-3 in the construction of a label-free electrochemical sensing platform. Dose to dose experiments were conducted, revealing that apt-RV3 provide an excellent analytical performance within a wide linear range and a high sensitivity. In addition, the proposed strategy has shown a good selectivity towards different virus particles and serum proteins. The real sample applicability was also demonstrated in real serum samples spiked with the viral nucleoprotein. Our aptasensor offer numerous advantages over existing methods, including enhanced sensitivity, stability, low-cost, besides to the great potential for miniaturization. In parallel, the detection is based on the interaction aptamer-pathogen epitope without involving the antibodies presence. This method is very useful in diagnosing infections that do not generate significant amounts of antibody in the organism. Finally, the selected aptamer could be applied to detect the RVFV nucleoprotein by using other detection techniques including colorimetry and fluorescent assays.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Maes, P. et al. Taxonomy of the family Arenaviridae and the order Bunyavirales: Update 2018. Arch. Virol. 163, 2295–2310 (2018).
Nair, N. et al. Rift Valley fever virus—Infection, pathogenesis and host immune responses. Pathogens 12(9), 1174 (2023).
Bird, B. H. et al. Rift Valley fever virus. J. Am. Vet. Med. Assoc. 234(7), 883–893 (2009).
Muturi, M. et al. Ecological and subject-level drivers of interepidemic Rift Valley fever virus exposure in humans and livestock in Northern Kenya. Sci. Rep. 13(1), 15342 (2023).
Versteirt, V. et al. Systematic literature review on the geographic distribution of Rift Valley fever vectors in Europe and the neighbouring countries of the Mediterranean Basin. EFSA Support. Publ. 10(4), 412E (2013).
Al-Afaleq, A.I., M.F.J.V.-B. Hussein, and Z. Diseases, The status of Rift Valley fever in animals in Saudi Arabia: A mini review. Vector-Borne Zoonotic Dis. 2011. 11(12): p. 1513–1520.
WHO. 2018 Annual review of diseases prioritized under the Research and Development Blueprint (World Health Organization Geneva, 2018).
Bird, B. H. et al. Rift Valley fever virus lacking the NSs and NSm genes is highly attenuated, confers protective immunity from virulent virus challenge, and allows for differential identification of infected and vaccinated animals. J. Virol. 82(6), 2681–2691 (2008).
Archer, B.N., et al. Outbreak of Rift Valley fever affecting veterinarians and farmers in South Africa. S. Afr. Med. J.101(4), 263–266 (2008).
Mwaengo, D. et al. Detection and identification of Rift Valley fever virus in mosquito vectors by quantitative real-time PCR. Virus Res. 169(1), 137–143 (2012).
Han, Q. et al. Development of a visible reverse transcription-loop-mediated isothermal amplification assay for the detection of Rift Valley fever virus. Front. Microbiol. 11, 590732 (2020).
Paweska, J. T. et al. IgG-sandwich and IgM-capture enzyme-linked immunosorbent assay for the detection of antibody to Rift Valley fever virus in domestic ruminants. J. Virol. Methods 113(2), 103–112 (2003).
Paweska, J. T., van Vuren, P. J. & Swanepoel, R. J. Validation of an indirect ELISA based on a recombinant nucleocapsid protein of Rift Valley fever virus for the detection of IgG antibody in humans. J. Virol. Methods 146(1–2), 119–124 (2007).
Wilson, W., et al., Diagnostic approaches for Rift Valley fever. In Vaccines and Diagnostics for Transboundary Animal Diseases 73–78. (Karger Publishers, 2013).
Sayed, R. H. et al. Development of a lateral flow kit for detection of IgG and IgM antibodies against Rift Valley fever virus in sheep. Indian J. Vet. Sci. Biotechnol. 15(2), 63–68 (2019).
Domfe, T. et al. Development of a versatile half-strip lateral flow assay toward the detection of Rift Valley fever virus antibodies. Diagnostics 12(11), 2664 (2022).
Duburcq, X. et al. Peptide–protein microarrays for the simultaneous detection of pathogen infections. Bioconjug. Chem. 15(2), 307–316 (2004).
Sobarzo, A. et al. Optical fiber immunosensor for the detection of IgG antibody to Rift Valley fever virus in humans. J. Virol. Methods 146(1–2), 327–334 (2007).
Zhang, H., et al., Surface-enhanced Raman scattering detection of DNAs derived from virus genomes using Au-coated paramagnetic nanoparticles. Langmuir 22012. 28(8): p. 4030–4037.
Cesewski, E. & Johnson, B. N. Electrochemical biosensors for pathogen detection. Biosens. Bioelectron. 159, 112214 (2020).
Cooper, M.A. Label-Free Biosensors: Techniques and Applications (Cambridge University Press, 2009).
Williams, R. et al. Validation of an IgM antibody capture ELISA based on a recombinant nucleoprotein for identification of domestic ruminants infected with Rift Valley fever virus. J. Virol. Methods 177(2), 140–146 (2011).
Chinnappan, R. et al. In vitro selection of DNA aptamers and their integration in a competitive voltammetric biosensor for azlocillin determination in waste water. Analytica Chimica Acta 1101, 149–156 (2020).
Gaberc-Porekar, V. & Menart, V. Perspectives of immobilized-metal affinity chromatography. J. Biochem. Biophys. Methods 49(1–3), 335–360 (2001).
Freitas, A. I., Domingues, L. & Aguiar, T. Q. Tag-mediated single-step purification and immobilization of recombinant proteins toward protein-engineered advanced materials. J. Adv. Res. 36, 249–264 (2022).
Mahanta, S. et al. A minimal fragment of MUC1 mediates growth of cancer cells. PLoS ONE 3(4), e2054 (2008).
Chinnappan, R. et al. Aptamer selection and aptasensor construction for bone density biomarkers. Talanta 224, 121818 (2021).
Kohlberger, M. & Gadermaier, G. SELEX: Critical factors and optimization strategies for successful aptamer selection. Biotechnol. Appl. Biochem. 69(5), 1771–1792 (2022).
Marimuthu, C., Tang, T. H., Tominaga, J., Tan, S. C. & Gopinath, S. C. Single-stranded DNA (ssDNA) production in DNA aptamer generation. Analyst 137(6), 1307–1315 (2012).
Eissa, S. & Zourob, M. Aptamer-based label-free electrochemical biosensor array for the detection of total and glycated hemoglobin in human whole blood. Sci. Rep. 7(1), 1016 (2017).
Eissa, S. & Zourob, M. Selection and characterization of DNA aptamers for electrochemical biosensing of carbendazim. Anal. Chem. 89(5), 3138–3145 (2017).
Eissa, S., Almusharraf, A. Y. & Zourob, M. A comparison of the performance of voltammetric aptasensors for glycated haemoglobin on different carbon nanomaterials-modified screen printed electrodes. Mate. Sci. Eng. C. 101, 423–430 (2019).
Acknowledgements
Authors would like to acknowledge the generous support from the Research, Development and Innovation Agency (RDIA) for the support (Project No. 12813-alfaisal-2023-FU-R-2-1-HW-) .
Author information
Authors and Affiliations
Contributions
M.A and A. R. performed all the experimental work and wrote the first draft. M.Z inititaed the idea and work, supervised the work and secured the funding. All authors contributed to the writing and prooof reading.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Alharbi, M.A., Rhouati, A. & Zourob, M. Development of a label-free electrochemical aptasensor for Rift Valley fever virus detection. Sci Rep 14, 23892 (2024). https://doi.org/10.1038/s41598-024-74314-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-74314-7
