
Abstract
Although the role of low-density granulocytes (LDGs), neutrophils in the peripheral blood mononuclear cell (PBMC) fraction, and neutrophil extracellular traps (NETs) in assessing lupus disease severity is acknowledged, data specific to childhood-onset lupus remains scarce. This study analyzed 46 patients with childhood-onset systemic lupus erythematosus (82.6% females, mean age 14.5 ± 0.3 years), including 26 cases with normal complement levels and 20 with low complement levels, along with 20 healthy adult volunteers. Key parameters that distinguished healthy volunteers from lupus patients and differentiated between lupus patients with low and normal complement were serum interferon (IFN)-α, serum citrullinated histone 3 (CitH3), and extracellular traps (ETs) in LDGs. However, NETs (assessed by nuclear staining morphology), LDG abundance, and other parameters (such as endotoxemia, cytokines, and double-stranded (ds) DNA) did not show such differentiation. When lipopolysaccharide (LPS) was administered to LDGs in the PBMC fraction, it induced ETs in both low and normal complement groups, indicating the inducible nature of ETs. In adult healthy volunteers, activation by recombinant IFN-α or dsDNA in isolated neutrophils induced LDGs and NETs (identified using immunofluorescent staining for CitH3, myeloperoxidase, and neutrophil elastase) at 45 min and 3 h post-stimulation, respectively. Additionally, approximately half of the LDGs underwent late apoptosis at 3 h post-stimulation, as determined by flow cytometry analysis. Activation by IFN-α or dsDNA in LDGs also led to a more pronounced expression of CD66b, an adhesion molecule, compared to regular-density neutrophils, suggesting higher activity in LDGs. In conclusion, IFN-α and/or dsDNA in serum may transform regular-density neutrophils into LDGs before progressing to NETosis and apoptosis, potentially exacerbating lupus severity through cell death-induced self-antigens. Therefore, LDGs and ETs in LDGs could provide deeper insights into the pathophysiology of childhood-onset lupus.
Similar content being viewed by others


Introduction
Systemic lupus erythematosus (SLE or lupus) is a chronic systemic inflammatory disease characterized by the overproduction of autoantibodies against various self-antigens. This condition is caused by multifactorial factors, including genetic, hormonal, and environmental influences1. While the impact of circulating immune complexes in lupus pathogenesis is well-documented, the role of innate immunity is gaining increasing attention2. Neutrophils, the most abundant innate immune cells, are implicated in lupus pathogenesis; upon activation, they rapidly undergo apoptosis3, potentially exacerbating lupus disease activity4. Similarly, neutrophil extracellular traps (NETs), associated with a process known as NETosis, may correlate with autoimmune diseases due to their self-nucleic acid content5,6,7. NETs, comprising nuclear antigens (histone and DNA) and cytoplasmic proteins, are released into the extracellular space for antimicrobial activity8 can enhance exposure to autoantigens and induce chronic inflammation in lupus1.
Low-density granulocytes (LDGs) are a subset of neutrophils found in the peripheral blood mononuclear cell (PBMC) fraction after gradient separation. This procedure is used to differentiate LDGs from regular neutrophils9,10. Type I interferon, a predominant cytokine during active lupus progression, induces the activation of neutrophils, potentially increasing LDG levels and associating them with disease activity11. Although the correlation between LDGs and lupus activity in adult-onset lupus has been established using straightforward measurement methods12, data on LDGs in childhood-onset lupus remain limited13. Childhood-onset lupus often presents with higher prevalence rates of severe clinical manifestations such as lupus nephritis, hematological disorders, and neurological involvement compared to adult-onset SLE14. Thus, understanding LDGs in childhood-onset lupus is crucial for elucidating the role of innate immunity in childhood-onset lupus, potentially leading to novel management strategies.
Despite growing research on the association between neutrophils and lupus disease activity, current lupus activity assessments primarily rely on clinical scores (e.g., SLE Disease Activity Index [SLEDAI], British Isles Lupus Assessment Group [BILAG], and Systemic Lupus Activity Measure [SLAM]) and biomarkers of immune activation, including complement levels (C3, C4, and CH50) and anti-dsDNA levels15,16. Immune complex deposition and complement activation are fundamental to lupus pathogenesis, and low complement levels are often used to assess disease activity17. Consequently, this study categorizes lupus disease severity based on normal and low complement levels.
While NETs and LDGs are potentially correlated with immune activation (including complement, cytokines, and type I interferon), the simultaneous evaluation of both in lupus has been underexplored18,19. Given that both NETs and LDGs represent activated neutrophils and that NETosis in LDGs is a possibility, this study aims to investigate alteration in LDGs, NETs, and extracellular traps (ETs) in the PBMC fraction (potential NETosis in LDGs) in patients with childhood-onset SLE across different disease severities.
Results
Patient characteristics
Among the 46 participants, 38 were females (82.6%) with a mean age of 14.5 ± 0.3 years and a body mass index (BMI) of 23.7 ± 0.8 kg/m2. Participants were divided into groups with normal or low complement levels, reflecting the significance of complement in lupus pathophysiology. Owing to ethical considerations pertaining to the procurement of experimental specimens from healthy children, healthy adults were used as the control group. All patients with lupus were treated with prednisolone and hydroxychloroquine. The control group was older and had a higher percentage of females and hemoglobin levels compared to the lupus groups (Table 1).
Lupus activity, inflammatory markers, low-density neutrophils (LDGs), and neutrophil extracellular traps (NETs)
Lupus activity, assessed by the SLEDAI-2 K, was more pronounced in the low complement group compared to the normal complement group (Fig. 1A). Proteinuria levels, indicated by spot urine protein and urine protein to creatinine index (UPCI), were higher in the low complement group, although urine sediments and kidney injury markers (blood urea nitrogen and serum creatinine) showed no significant differences (Fig. 1B-G). Kidney histology revealed a higher activity index in the low complement group, while the chronicity index remained similar between groups (Fig. 1H-I). Crescentic lesions were observed in six out of 26 patients with normal complement (23%) and four out of 20 patients with low complement levels (20%), as depicted in the representative histopathological images (Fig. 1J).

Characteristics of adult healthy volunteers and the patients with normal complement level or low complement (low C3 and/or C4) as indicated by SLEDAI-2 K score (A), kidney injury (blood urea nitrogen and serum creatinine) (B,C), urinary sediments (red blood cells and white blood cells) (D,E), spot urine protein (F), urine protein creatine index (UPCI) (G), and histological index, including activity score, chronicity score, and representative histological picture on Periodic acid–Schiff (PAS) staining (H–J) are demonstrated (n = 14 in adult healthy control and n = 26 and 20 in normal and low-level complement, respectively). *p < 0.05.
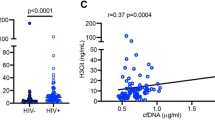
For inflammatory markers, the low complement group exhibited a higher erythrocyte sedimentation rate (ESR) and lower serum albumin levels compared to the control and normal complement groups, although ferritin levels did not differ (Fig. 2A–C). Notably, ESR and serum albumin did not differentiate between the control and normal complement groups (Fig. 2A–C). Serum levels of IL-6, IL-8, endotoxemia, and LDGs in the lupus group were comparable to the control (Fig. 2D–M). However, serum TNF-α, IL-10, IFN-α, and NETs formation, assessed by serum CitH3, dsDNA, and fluorescent-stained nuclear morphology, were elevated in the lupus groups relative to the control (Fig. 2D–M). Among NET biomarkers, only serum CitH3 could distinguish between the normal and low complement groups (Fig. 2K), while other NETs and LDG biomarkers could not (Fig. 2J–N).

Characteristics of adult healthy volunteers and the patients with normal complement level or low complement (low C3 and/or C4) as indicated by routine inflammatory markers, including erythrocyte sedimentation rate (ESR), ferritin, and albumin (A–C), serum cytokines (TNF-α, IL-6, IL-8, IL-10, and IFN-α) (D–H), endotoxemia (I), low-density granulocytes (LDGs) as indicated by percentage of neutrophils in peripheral blood mononuclear cell (PBMC) fraction (J), neutrophil extracellular traps indicators, including serum citrullinated histone 3 (CitH3), serum double strand DNA (dsDNA), and the morphology of nuclei from 4′,6-diamidino-2-phenylindole (DAPI) fluorescent staining with the representative pictures (K–M), and the extracellular traps (ETs) in the PBMC fraction with and without the activation by LPS (N) are demonstrated (n = 14 in adult healthy control and n = 26 and 20 in normal and low complement groups, respectively). *p < 0.05.
Extracellular traps in PBMC Fraction: a potential link to lupus activity
The discrepancy in NET measurements using neutrophil nuclear morphology from the PMNs fraction and serum CitH3 may be attributed to additional CitH3 production from ETs in the PBMC fraction. ET formation was detected in the PBMC fraction of patients with low complements through immunofluorescent staining for MPO and NE, whereas it was not visualized in patients with normal complements or healthy controls (Figs. 2N and 3). Endotoxin (a potent ET inducer) added to the PBMC fraction enhanced ET formation only in samples from patients with normal complement, suggesting possible saturation of ETs in the PBMC fraction of patients with low complement (Figs. 2N and 3).

Representative fluorescent-stained pictures of the extracellular traps (ETs), as determined by the co-presence of neutrophil elastase (green color) with myeloperoxidase (red color), in the peripheral blood mononuclear cell (PBMC) fraction with and without the activation by LPS, are demonstrated. The blue color is from nuclear staining using 4′,6-diamidino-2-phenylindole (DAPI).
Levels of LDGs, NETs formation (serum CitH3, serum dsDNA, and DAPI staining), and ETs in the PBMC fraction did not differ between inactive and active lupus, as classified by SLEDAI scores < 4 and ≥ 4, respectively (Supplementary Fig. 1). Complement activation in lupus may elevate neutrophil activities, partially through activation of various complement component receptors on neutrophils (C3b, C4b, and C5a)20. The specificity of the NE enzyme in activated neutrophils suggests that ETs in the PBMC fraction are primarily derived from LDGs (neutrophils in the PBMC fraction)21,22. Enhanced activity of neutrophils in lupus, indicated by the expression of several genes including ERK-1, HIF-1α, IFI27, ISG15, MX-1, and OAS-1 in the PMN fraction, correlates with elevated serum IFN-α and dsDNA levels, which may be associated with the presence of LDGs and NETs (Fig. 4A–F). Both IFN-α and dsDNA similarly induced LDGs (Fig. 4H–J).

Characteristics of gene expression in neutrophils (PMNs) from adult healthy volunteers and the patients with normal complement level or low complement (low C3 and/or C4) as indicated by ERK-1 (extracellular signal-regulated kinases-1), HIF-1α (hypoxia-inducible factor-1), IFI27 (interferon Alpha Inducible Protein 27), ISG15 (interferon-stimulated gene 15), MX-1 (interferon-induced GTP-binding protein), and OAS-1 (2’-5’-oligoadenylate synthetase 1) (A–F) are demonstrated (n = 8 in adult healthy control and n = 14 and 8 in normal and low complement groups, respectively). The schema of experiments (G) showing neutrophil separation from healthy adults using the gradient separation method that was activated by IFN-α or dsDNA before determination of cell density (percentage of low-density granulocytes; LDGs) using the gradient separation method at 45 min and 3 h post-activation (H,I) and neutrophil extracellular traps in the LDGs portion at 3 h post-activation with representative 4′,6-diamidino-2-phenylindole (DAPI) nuclear stained pictures (J) are demonstrated (the neutrophils in figure H–J were isolated from 8–10 healthy volunteers). *p < 0.05.
A 45-minute activation period was sufficient to induce clear separation between neutrophils into LDGs (low-density cells) and PMNs (regular-density cells) (Fig. 4G,H), with almost all neutrophils transforming into LDGs after 3 h of activation (Fig. 4I). Immunofluorescent analysis showed elevated NETs in LDGs after 3 h of activation by IFN-α or dsDNA compared to the media control, visualized by CitH3 and co-localization of MPO and NE (Fig. 5A–D). Flow cytometry analysis of neutrophils before (PMN_1) and after activation (separated into LDGs and PMN fractions) revealed similar sizes but higher expression of adhesion molecule CD66b in LDGs. The expression of CD66b between IFN-α-induced LDGs and dsDNA-induced LDGs was not different (Fig. 6A,B). Cell death levels (late apoptosis and necrosis) were similar among all groups (PMN_1, LDGs, and PMNs) (Fig. 6C-H). Flow cytometric parameters indicated that the characteristics of normal-density neutrophils before activation (PMN_1) and after stimulation (PMN fraction) were similar (Fig. 6A–H). The separation between LDG and PMN fractions following IFN-α and dsDNA activation took 45 min (Fig. 4I), with almost all cells in the PMN fraction turning into LDGs at 3 h post-incubation and approximately half of these LDGs producing NETs at 3 h post-activation (Fig. 4J).

Neutrophil extracellular traps (NETs) in the low-density granulocytes (LDGs) portion of isolated neutrophils from 8 healthy volunteers after 3 h activation by IFN-α or dsDNA as determined by the fluorescent staining by citrullinated histone 3 (CitH3) and the co presentation of myeloperoxidase (MPO) (red color) and neutrophil elastase (NE) (green color) through the percentage of NETs and the representative pictures (A–D) are demonstrated. The nuclei were stained by 4′,6-diamidino-2-phenylindole (DAPI) (blue color), and the neutrophils in figure A-D were isolated from 8 healthy volunteers. *p < 0.05.

Flow cytometric characteristics of the isolated neutrophils from healthy volunteers before the activation (PMN_1) and after 45 min activation by IFN-α or dsDNA with the separation into low-density granulocytes (LDGs) portion and normal-density neutrophil fraction (PMN) as indicated by cell sizes and an activation marker (CD66b) with the representative flow cytometric patterns (A–C) and cell deaths as determined by late apoptosis as shown by annexin V with propidium iodide (PI), and necrosis (positive for PI) at 45 min and 3 h post-stimulation with the representative flow cytometric patterns (D–H) are demonstrated. The neutrophils in figure A-H were isolated from 8–10 healthy volunteers. MFI, mean fluorescent intensity. *p < 0.05.
Discussion
In this study, serum CitH3 and the abundance of ETs in the PBMC fraction were the only parameters able to differentiate between healthy volunteers, lupus patients with low complement, and those with normal complement.
Despite some similarities in pathogenesis and clinical manifestations between childhood-onset and adult-onset SLE, childhood-onset lupus often presents a more aggressive disease course with higher disease activity23. Current approaches to managing childhood-onset SLE primarily rely on data derived from adult-onset SLE24; however, specific studies on childhood-onset lupus are crucially needed. Regardless of the patient’s age at onset, the classical pathogenesis of SLE involves immune complexes (ICs), which form links between autoantibodies and self-antigens (e.g., ICs between anti-dsDNA and dsDNA), complement activation, and type I IFN (IFN- α)25,26. In SLE, IC deposition activates the classical complement pathway, resulting in reduced blood complement levels and multi-organ damage25,26. Additionally, the internalization of ICs by various antigen-presenting cells, especially dendritic cells, activates endosomal Toll-like receptors (TLRs) by self-DNA (TLR9) and self-RNA (TLR3 and TLR7), leading to the production of IFN-α, a key cytokine associated with SLE27,28.
Recent research has highlighted the significant role of neutrophils—the most abundant innate immune cells in the bloodstream—in lupus pathogenesis. Neutrophils serve as a crucial source of self-antigens through cell death mechanisms such as apoptosis and NETosis1. Activated neutrophils undergo apoptosis within 6–24 h, and NETosis can be triggered by endotoxin, IFN-α, and dsDNA29,30. Complement activation in active lupus may be associated with NETs and ETs in the PBMC fraction, as patients with low complement levels exhibited higher ETs than those with normal complement levels. Interestingly, ET levels in the PBMC fraction did not differ between active and inactive lupus, as determined by the SLEDAI score, suggesting a stronger correlation between ETs and complement activation than with generalized lupus systemic inflammation. While the SLEDAI score provides an acceptable categorization of lupus disease activity based on 24 items from 9 organ systems and a few basic laboratory tests (complete blood count, urine analysis, and complement levels), ET parameters did not vary between active (score ≥ 4) and inactive lupus (score < 4). This indicates a potentially more significant association between ETs and complement activation.
During active lupus, factors such as endotoxemia (possibly due to IC deposition in the gut)1, IFN-α (secreted by parenchymal cells, fibroblasts, macrophages, neutrophils, and dendritic cells)27,28, and dsDNA31 may induce ETs in both neutrophils and macrophages32,33. In our study, IFN-α activated neutrophils by upregulating several genes and induced NETosis, which supports previous findings34,35. Additionally, dsDNA, a component of cell-free DNA, can induce NETosis through several DNA recognition receptors, including TLR-4, TLR-9, and cytosolic receptors36,37,38. Conversely, NETosis-related cell breakdown increases dsDNA levels, potentially creating a vicious cycle of NETosis induction.
Our in vitro experiments demonstrated that LDGs, derived from immune stimulators such as IFN-α and dsDNA, exhibit elevated cell activities and develop cell death, including NETosis, apoptosis, and necrosis, as early as three hours post-stimulation. This finding aligns with increased CD66b expression, an adhesion molecule, observed 45 min post-stimulation. Consequently, LDGs contribute to lupus exacerbation through cell death mechanisms, supporting the influence of LDGs on childhood-onset lupus as a potential monitoring indicator.
Patients with low complement levels exhibited more severe lupus, as indicated by proteinuria, histological activity index, and other parameters. Serum CitH3, ETs in PBMC, and serum IFN-α effectively differentiated healthy controls from lupus patients with normal complement. For NETosis, serum CitH3 appeared more accurate than parameters derived solely from the neutrophil fraction, such as immunofluorescent staining and PAD4 expression, which are limited to cells from the PMN fraction. Given that NETosis occurs in both the PMN and PBMC fractions, serum CitH3 represents overall ETs, including those from both PMNs and monocytes. In the PBMC fraction, ETs correlate with systemic inflammation and may be saturated in childhood-onset lupus with low complement (indicative of severe systemic inflammation), but not in lupus with normal complement, as LPS addition elevated ETs only in patients with normal complement. This suggests that ETs in patients with normal complement are not saturated by activation, whereas ET levels in PBMC are already high due to lupus inflammatory mediators in patients with low complement.
Although the association between ETs in LDGs and lupus has been previously noted9, data on ETs in the PBMC fraction in childhood-onset lupus remain limited, contrasting with the well-established link between NETosis and lupus severity in adult-onset lupus39. In adult patients with autoimmune diseases, LDGs exhibit a more pronounced ability to generate NETs and synthesize higher amounts of proinflammatory cytokines than normal-density neutrophils40,41,42. Therefore, comparing ET production capabilities in the PBMC fraction between adult-onset and childhood-onset lupus is warranted, encouraging further studies.
For clinical applications, the activation of neutrophils, including LDGs, NETosis, and apoptosis—potentially stimulated by IFN-α or dsDNA—in childhood-onset lupus is an intriguing topic that may correlate with lupus activity. Thus, ETs in the PBMC fraction, serum CitH3, and serum IFN-α might serve as valuable indicators of disease activity in childhood-onset SLE. Evaluating LDGs in PBMC requires only simple centrifugation with a density gradient protocol and ET staining, making it more feasible for resource-limited hospitals compared to other expensive commercial tests (e.g., CitH3 and IFN-α). Further studies on neutrophil activation in childhood-onset lupus are necessary.
Conclusions
In active childhood-onset lupus, immune activators such as IFN-α and dsDNA stimulate neutrophils, resulting in the formation of NETs and LDGs. Consequently, serum CitH3 (a marker of NETs), ETs in the PBMC fraction, and serum IFN-α may serve as valuable indicators of disease activity in childhood-onset SLE.
Materials and methods
Study population and inclusion/ exclusion criteria
A cross-sectional analysis was conducted by enrolling patients with the diagnosis of SLE who were younger than 18 years old and received treatment at the King Chulalongkorn Memorial Hospital (a tertiary care hospital) from 2021 to 2022. All methods and experiments were approved by the Institutional Review Board of the Faculty of Medicine, Chulalongkorn University (IRB No. 753/64) in accordance with the guidelines of the Declaration of Helsinki 1964 and obtained with written informed consent at the time of enrolment using the preserved patient anonymity protocol. All the participants fulfilled the 2019 European League against Rheumatism/American College of Rheumatology classification criteria43. The exclusion criteria were patients with active infection, and the severity stratification of SLE was determined following the Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2 K)44. Body mass index (BMI) was calculated as the ratio of weight in kilograms to height squared in meters. Kidney biopsy results were interpreted based on the ISN/RPS classification and the activity and chronicity indices from the renal histopathology were estimated using a semi-quantitative grading system45. The patients were classified according to the level of complement C3 (83–177 mg/dL) and C4 (19–57 mg/dL), including the normal complement group (C3 and C4 were higher than 83 and 19 mg/dL, respectively) and the low complement group (C3 or C4 were lower than 83 and 19 mg/dL, respectively).
Serum analysis for renal injury, systemic inflammation, and NETs
All participants underwent clinical and laboratory evaluation using pre-prandial morning specimen collections according to a standardized unit protocol, and the samples were measured from the central laboratory of the King Chulalongkorn Memorial Hospital. The routine laboratory results, including complete blood count, blood urea nitrogen, serum creatinine, serum albumin, serum complements (C3 and C4), ferritin, and erythrocyte sedimentation rate (ESR), were measured in the central laboratory of the King Chulalongkorn Memorial Hospital using the Sysmex XN9203 Analyzer (Kobe, Hyogo, Japan) and Cobas c502 (Roche Diagnostics, Basel, Switzerland). Serum cytokines (TNF-α, IL-6, IL-8, IL-10, and IFN-α) and serum lipopolysaccharide (LPS), referred to as endotoxemia, were measured by enzyme-linked immunosorbent assay (ELISA) (Invitrogen, Waltham, MA, USA) and the HEK-Blue LPS Detection Kit (InvivoGen™, San Diego, CA, USA), respectively. Serum CitH3 and dsDNA were measured by ELISA assay (Cayman Chemical, Ann Arbor, MI, USA) and Quant-iT™ PicoGreen reagents (Thermo Fisher Scientific, Waltham, MA, USA), respectively.
Low-density granulocytes (LDGs) and extracellular traps in the PBMC fraction
With density gradient cell separation8, the low-density granulocytes (LDGs), the polymorphonuclear cells (PMNs) in the fraction of peripheral blood mononuclear cells (PBMCs), and NETs formation, neutrophils from the PMN fraction with altered nuclear morphology were analyzed. Briefly, blood samples were mixed with Ficoll-Paque (Robbins Scientific Corporation, Sunnyvale, CA, USA) and Polymorphprep™ (Axis-Shield, Oslo, Norway) in a 1:1:1 ratio in a 50 mL-sterile tube and centrifuged at 1000 ×g for 30 min at room temperature without brake. Then, the PBMC and PMN fractions were identified at the upper and lower parts of the Ficoll-Paque, respectively, and LDGs were determined as percentages of neutrophils in the PBMC fraction after staining by Wright’s Giemsa color. All cells, including neutrophils (cells with multilobe nuclei) and lymphoid cells (cells with a single lobe-nucleus), in the PBMC fraction were counted by light microscopy. With the PMN layer, which is largely neutrophils (> 98%), the cells were collected, washed with RPMI 1640 (Thermo Fisher Scientific, Logan, Utah, USA), and resuspended with RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (FBS). The contaminating erythrocytes were removed by ammonium chloride lysis (ACK) buffer. Neutrophil preparations were at least 98% of the total cells as evaluated by trypan blue exclusion (Sigma-Aldrich, Singapore) and Wright’s Giemsa staining (Biotech, Bangkok, Thailand). Moreover, the cells with extracellular traps in the PBMC fraction (ETs in PBMC) were also detected by the co-presence of immunofluorescent color using anti-myeloperoxidase (anti-MPO; ab25989) together with anti-neutrophil elastase (anti-NE; ab68672) (Abcam, Cambridge, MA, USA). The specific secondary antibodies coupled with Alexa Fluor 647 (red color) and Alexa Fluor 488 (green color) were used for fluorescent visualization, and the fluorescence intensity was analyzed by a confocal microscope (ZEISS LSM 980, Carl Zeiss, Germany). Although NE and MPO are mostly released by activated neutrophils, both enzymes are also possibly expressed by other cells (monocytes and macrophages)21,46,47. The ETs in the PBMC portion are likely derived from both neutrophils and monocytes. To determine the capacity of ETs formation in the PBMC fraction, the PBMC cells were further activated by lipopolysaccharide (LPS) (Escherichia coli 026: B6) (Sigma-Aldrich) at 100 ng/mL for 3 h before ETs detection using immunofluorescent staining.
Extracellular traps in the neutrophil fraction and the expression of several genes
Neutrophils isolated from the density centrifugation as mentioned above were used to determine NETs and the expression of several genes. For NETs formation, NETs were detected by nuclear morphology staining with a blue-fluorescent DNA-staining color of 4′,6-diamidino2-phenylindole (DAPI)6,8,48. The expression of several genes in neutrophils was demonstrated in the relative quantitation of the comparative threshold method (2−ΔΔCt) as normalized by β-actin using real time polymerase chain reaction (RT-PCR). The PCR samples were prepared using a TRIzol RNA extraction (Invitrogen), a nanodrop 100 spectrophotometer, high-capacity cDNA reverse transcription, and SYBR Green PCR Master Mix for quantitative RT-PCR with the QuantStudio6 Flex Real-time PCR System (Thermo Scientific). The list of primers is shown in Table 2.
Neutrophil responses to IFN-α and dsDNA, the in vitro experiments
Because the increased LDGs in the blood of patients with lupus might be due to an induction by type I interferon or free dsDNA in the blood, IFN-α, and dsDNA were tested with isolated neutrophils from healthy volunteers. The dsDNA was isolated from blood sample using 0.5 mL of whole blood mixed with 1 mL of lysis buffer (Erythrocyte lysis buffer, Qiagen, Hilden, Germany) and centrifuged at 400 ×g for 5 min at 4 °C. After that, the pellet was mixed with 100 µL of STE buffer (NaCl-Tris-EDTA), 60 µL of 10% sodium dodecyl sulfate (SDS), and 50 µg/mL of proteinase K at 56 °C for 3 h. Then, chloroform (200 µL) and phenol (200 µL) were used to separate the nucleic acid, and 6 M NaCl was used to precipitate protein. Then, the supernatant containing nucleic acid was added by ethanol to precipitate DNA, and the DNA pellet was dried at room temperature and dissolved in 50 µL of sterile water. Subsequently, the isolated neutrophils using a gradient separation protocol as mentioned above at 1 × 107 cells were incubated with recombinant human IFN-α2 (BioLegend, San Diego, CA, USA) at 25 ng/mL or dsDNA at 200 ng/mL before the determination of cell density again by gradient separation to differentiate neutrophils with low density (LDGs) and regular density at the upper and lower parts of the Ficoll-Paque, respectively. In the LDGs portion, NETs were measured using nuclear morphology by DAPI staining and immunofluorescent analysis. For immunofluorescence, the antibodies against neutrophil elastase, myeloperoxidase, and citrullinated histone H3 with DAPI. The secondary antibodies, including goat anti-rabbit IgG (ab150077) (green color), goat anti-mouse IgG (ab150115), and donkey anti-rabbit IgG (ab150075) (red color) (Abcam, Cambridge, MA, USA) were used. Then, the images were analyzed with the ZEISS LSM 980 (Carl Zeiss, Germany).
Flow cytometry analysis of untreated PMNs versus stimulated cells (PMNs and LDGs)
To determine the differences between the pre-stimulated polymorphonuclear cells (PMNs) in the PMN fraction and the PMN after stimulation by IFN-α and dsDNA, which were turned into LDGs or maintained as the PMNs, flow cytometry analysis was performed. Then, the pre-stimulated PMN (PMN_1) and post-stimulated cells from the LDGs portion (the low-density cells) and from the neutrophil fraction (the regular-density cells) were washed with staining buffer (0.5% bovine serum albumin in PBS) and resuspended cells in Annexin V binding buffer. Cells were stained with fluorochrome-conjugated antibodies against anti-CD66b (an adhesion molecule) and APC Annexin V apoptosis detection with propidium iodide (PI) (Cat640932; BioLegend, San Diego, CA, USA). All stained cells were examined by flow cytometry using CytoFLEX (Beckman Coulter Life Sciences, USA), and the data were analyzed by the CytExpert software (Beckman Coulter Life Sciences, USA).
Statistical analysis
Data analysis was done using Stata v14 (StataCorp, College Station, TX, USA), and p values of < 0.05 were considered statistically significant, while GraphPad Prism version 10.0 software (La Jolla, CA, USA) was used for figure generation. Descriptive data are reported as the median ± range for quantitative variables and the number (%) for categorical variables. The normality test was performed by the two-sample Kolmogorov-Smirnov test using p ≥ 0.05 in both control and disease groups (the combination between normal and low complement lupus) as a criterion for passing the test. The Kruskal-Wallis test followed by multiple comparison (the Dunn’s test) and the Mann-Whitney U test were used for multiple group comparison and 2-group analysis, respectively.
Data availability
The datasets analysed during the current study are not publicly available due to ethics in human research but are available from the corresponding author on reasonable request.
References
Charoensappakit, A., Sae-khow, K. & Leelahavanichkul, A. Gut barrier damage and gut translocation of pathogen molecules in lupus, an impact of Innate Immunity (macrophages and neutrophils) in autoimmune disease. Int. J. Mol. Sci. 23, 8223 (2022).
Bhunyakarnjanarat, T. et al. Prominent indomethacin-induced enteropathy in Fcgriib deficient lupus mice: an impact of macrophage responses and immune deposition in gut. Int. J. Mol. Sci. 22, 1377 (2021).
Fox, S., Leitch, A. E., Duffin, R., Haslett, C. & Rossi, A. G. Neutrophil apoptosis: relevance to the innate immune response and inflammatory disease. J. Innate Immun. 2, 216–227. https://doi.org/10.1159/000284367 (2010).
Mahajan, A., Herrmann, M. & Muñoz, L. E. Clearance deficiency and cell death pathways: a model for the pathogenesis of SLE. Front. Immunol. 7, 35. https://doi.org/10.3389/fimmu.2016.00035 (2016).
Lee, K. H. et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: a comprehensive review. Autoimmun. Rev. 16, 1160–1173. https://doi.org/10.1016/j.autrev.2017.09.012 (2017).
Saisorn, W. et al. Acute kidney injury induced lupus exacerbation through the enhanced neutrophil extracellular traps (and apoptosis) in Fcgr2b deficient lupus mice with renal ischemia reperfusion Injury. Front. Immunol. 12, 669162. https://doi.org/10.3389/fimmu.2021.669162 (2021).
Saithong, S. et al. A synergy between endotoxin and (1→3)-beta-D-glucan enhanced neutrophil extracellular traps in Candida administered dextran sulfate solution induced colitis in FcGRIIB-/- lupus mice, an impact of intestinal fungi in lupus. J. Inflamm. Res. 14, 2333–2352. https://doi.org/10.2147/jir.S305225 (2021).
Saithong, S. et al. Neutrophil extracellular traps in severe SARS-CoV-2 infection: a possible impact of LPS and (1→3)-β-D-glucan in blood from Gut translocation. Cells 11. https://doi.org/10.3390/cells11071103 (2022).
Ning, X., Wang, W. M. & Jin, H. Z. Low-density granulocytes in Immune-mediated inflammatory diseases. J. Immunol. Res. 2022, 1622160. https://doi.org/10.1155/2022/1622160 (2022).
Blanco-Camarillo, C., Alemán, O. R. & Rosales, C. Low-density neutrophils in healthy individuals display a mature primed phenotype. Front. Immunol. 12, 672520. https://doi.org/10.3389/fimmu.2021.672520 (2021).
Tay, S. H., Celhar, T. & Fairhurst, A. M. Low-density neutrophils in systemic lupus erythematosus. Arthritis Rheumatol. 72, 1587–1595. https://doi.org/10.1002/art.41395 (2020).
van den Hoogen, L. L. et al. Neutrophil extracellular traps and low-density granulocytes are associated with the interferon signature in systemic lupus erythematosus, but not in antiphospholipid syndrome. Ann. Rheum. Dis. 79, e135. https://doi.org/10.1136/annrheumdis-2019-215781 (2020).
Midgley, A. & Beresford, M. W. Increased expression of low density granulocytes in juvenile-onset systemic lupus erythematosus patients correlates with disease activity. Lupus 25, 407–411. https://doi.org/10.1177/0961203315608959 (2016).
Rianthavorn, P. & Prurapark, P. Risk factors of infection-associated mortality in children with lupus nephritis in under-resourced areas. Lupus 28, 1727–1734. https://doi.org/10.1177/0961203319882498 (2019).
Jesus, D. et al. Performance of SLEDAI-2K to detect a clinically meaningful change in SLE disease activity: a 36–month prospective cohort study of 334 patients. Lupus 28, 607–612 (2019).
Fernando, M. M. & Isenberg, D. A. How to monitor SLE in routine clinical practice. Ann. Rheum. Dis. 64, 524–527. https://doi.org/10.1136/ard.2003.015248 (2005).
Ekdahl, K. N. et al. Interpretation of serological complement biomarkers in disease. Front. Immunol. 9, 2237 (2018).
Torres-Ruiz, J. et al. Low-density granulocytes and neutrophil extracellular traps as biomarkers of disease activity in adult inflammatory myopathies. J. Clin. Rheumatol. 28, e480–e487. https://doi.org/10.1097/rhu.0000000000001772 (2022).
Torres-Ruiz, J. et al. The role of low density granulocytes and NETosis in the pathogenesis of adult-onset still’s disease. Clin. Exp. Rheumatol. 37 (Suppl 121), 74–82 (2019).
Akk, A. et al. Complement activation on neutrophils initiates endothelial adhesion and extravasation. Mol. Immunol. 114, 629–642. https://doi.org/10.1016/j.molimm.2019.09.011 (2019).
Fouret, P. et al. Expression of the neutrophil elastase gene during human bone marrow cell differentiation. J. Exp. Med. 169, 833–845. https://doi.org/10.1084/jem.169.3.833 (1989).
Marinaccio, L. et al. Peptide human neutrophil elastase inhibitors from natural sources: an overview. Int. J. Mol. Sci. 23, 2924 (2022).
Levy, D. M. & Kamphuis, S. Systemic lupus erythematosus in children and adolescents. Pediatr. Clin. North Am. 59, 345–364. https://doi.org/10.1016/j.pcl.2012.03.007 (2012).
Pennesi, M. & Benvenuto, S. Lupus nephritis in children: novel perspectives. Medicine (Kaunas) 59. https://doi.org/10.3390/medicina59101841 (2023).
Bolouri, N. et al. Role of the innate and adaptive immune responses in the pathogenesis of systemic lupus erythematosus. Inflamm. Res. 71, 537–554. https://doi.org/10.1007/s00011-022-01554-6 (2022).
Weinstein, A., Alexander, R. V. & Zack, D. J. A review of complement activation in SLE. Curr. Rheumatol. Rep. 23, 16. https://doi.org/10.1007/s11926-021-00984-1 (2021).
Ivashkiv, L. B. & Donlin, L. T. Regulation of type I interferon responses. Nat. Rev. Immunol. 14, 36–49. https://doi.org/10.1038/nri3581 (2014).
Kaplan, M. J. Neutrophils in the pathogenesis and manifestations of SLE. Nat. Rev. Rheumatol. 7, 691–699. https://doi.org/10.1038/nrrheum.2011.132 (2011).
Greenlee-Wacker, M. C. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol. Rev. 273, 357–370. https://doi.org/10.1111/imr.12453 (2016).
Sadeghi, M., Dehnavi, S., Jamialahmadi, T., Johnston, T. P. & Sahebkar, A. Neutrophil extracellular trap: a key player in the pathogenesis of autoimmune diseases. Int. Immunopharmacol. 116, 109843. https://doi.org/10.1016/j.intimp.2023.109843 (2023).
Postal, M. et al. Type I interferon in the pathogenesis of systemic lupus erythematosus. Curr. Opin. Immunol. 67, 87–94. https://doi.org/10.1016/j.coi.2020.10.014 (2020).
Liu, S. et al. Neutrophil extracellular traps are indirectly triggered by lipopolysaccharide and contribute to acute lung injury. Sci. Rep. 6, 37252. https://doi.org/10.1038/srep37252 (2016).
Weng, W., Hu, Z. & Pan, Y. Macrophage extracellular traps: current opinions and the state of research regarding various diseases. J. Immunol. Res. 2022, 7050807. https://doi.org/10.1155/2022/7050807 (2022).
Peng, Y. et al. The potential roles of type I interferon activated neutrophils and neutrophil extracellular traps (NETs) in the pathogenesis of primary Sjögren’s syndrome. Arthritis Res. Ther. 24, 170. https://doi.org/10.1186/s13075-022-02860-4 (2022).
Gul, E. et al. Type I IFN-related NETosis in ataxia telangiectasia and Artemis deficiency. J. Allergy Clin. Immunol. 142, 246–257. https://doi.org/10.1016/j.jaci.2017.10.030 (2018).
Suksawad, N. et al. Cyclic GMP-AMP synthase (cGAS) deletion reduces severity in bilateral nephrectomy mice through changes in neutrophil extracellular traps and mitochondrial respiration. Biomedicines 11. https://doi.org/10.3390/biomedicines11041208 (2023).
Charoensappakit, A. et al. Cell-free DNA as diagnostic and prognostic biomarkers for adult sepsis: a systematic review and meta-analysis. Sci. Rep. 13, 19624. https://doi.org/10.1038/s41598-023-46663-2 (2023).
Chen, T. et al. Receptor-mediated NETosis on neutrophils. Front. Immunol. 12, 775267. https://doi.org/10.3389/fimmu.2021.775267 (2021).
Yu, Y. & Su, K. Neutrophil Extracellular traps and systemic Lupus Erythematosus. J. Clin. Cell. Immunol. 4https://doi.org/10.4172/2155-9899.1000139 (2013).
Villanueva, E. et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 187, 538–552 (2011).
Denny, M. F. et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J. Immunol. 184, 3284–3297 (2010).
Skrzeczynska-Moncznik, J. et al. Differences in staining for neutrophil elastase and its controlling inhibitor SLPI reveal heterogeneity among neutrophils in psoriasis. J. Investig. Dermatol. 140, 1371–1378.e1373 (2020).
Aringer, M. et al. European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. 71, 1400–1412. https://doi.org/10.1002/art.40930 (2019).
Tselios, K., Gladman, D. D. & Urowitz, M. B. How can we define low disease activity in systemic lupus erythematosus? Semin Arthritis Rheum. 48, 1035–1040. https://doi.org/10.1016/j.semarthrit.2018.10.013 (2019).
Bajema, I. M. et al. Revision of the International Society of Nephrology/Renal Pathology Society classification for lupus nephritis: clarification of definitions, and modified National Institutes of Health activity and chronicity indices. Kidney Int. 93, 789–796. https://doi.org/10.1016/j.kint.2017.11.023 (2018).
Yang, M. et al. Neutrophil elastase promotes neointimal hyperplasia by targeting toll-like receptor 4 (TLR4)-NF-κB signalling. Br. J. Pharmacol. 178, 4048–4068. https://doi.org/10.1111/bph.15583 (2021).
Siraki, A. G. The many roles of myeloperoxidase: from inflammation and immunity to biomarkers, drug metabolism and drug discovery. Redox Biol. 46, 102109. https://doi.org/10.1016/j.redox.2021.102109 (2021).
Saithong, S. et al. Candida administration worsens neutrophil extracellular traps in renal ischemia reperfusion injury mice: an impact of gut fungi on acute kidney injury. J. Innate Immun. 14, 502–517. https://doi.org/10.1159/000521633 (2022).
Acknowledgements
This research was funded by the Program Management Unit for Human Resources and Institutional Development, Research and Innovation (B16F640175 and B48G660112) with Rachadapisek Sompote Matching Fund (RA-MF-22/65, RA-MF-13/66, and RA-MF-eAsia), and Thailand Science research and Innovation Fund Chulalongkorn University (HEAF67300087 and HEA663000039). W.S. was funded by Second Century Fund (C2F) for Ph.D. candidates from Chulalongkorn University.
Author information
Authors and Affiliations
Contributions
W.S. designed and coordinated all the experiments, performed the experiments, analyzed data, and wrote and approved the manuscript. P.P. performed the experiments and approved the manuscript. C.S. and N.S. performed clinical data collection and analyzed data. P.R. supervised to conception and study design and approved the manuscript. A.L. designed and coordinated all the experiments, analyzed the data, and wrote and approved the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
The Human Research Ethics Committee of the Chulalongkorn University Faculty of Medicine accepted this study (approval number 753/64). This study involving human participants was conducted in accordance with the ethical standards of the institutional and national research committee and with the Declaration of Helsinki of 1964 and its later amendments.
Consent to participate
Written informed consent was obtained at the time of enrollment, and the data were kept anonymous.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Saisorn, W., Santiworakul, C., Phuengmaung, P. et al. Extracellular traps in peripheral blood mononuclear cell fraction in childhood-onset systemic lupus erythematosus. Sci Rep 14, 23177 (2024). https://doi.org/10.1038/s41598-024-74705-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-74705-w
Keywords
This article is cited by
-
Low-density granulocytes and neutrophil extracellular traps in dengue, impacts of interferon alpha and cell-free DNA
Scientific Reports (2025)
-
Lacticaseibacillus rhamnosus attenuates uremic toxins in patients with nondialysis chronic kidney disease through the anti-inflammatory molecules
Scientific Reports (2025)
-
Accelerating and protective effects toward cancer growth in cGAS and FcgRIIb deficient mice, respectively, an impact of macrophage polarization
Inflammation Research (2025)
