
Abstract
Cisplatin-induced injury to renal proximal tubular cells stems from mitochondrial damage-induced apoptosis and inflammation. Dichloroacetate (DCA), a pyruvate dehydrogenase kinase (PDK) inhibitor, a potential generator of ROS and ATP, protects against cisplatin-induced nephrotoxicity by promoting the TCA cycle. However, its effects on apoptotic pathways and ROS production in renal tubular cells remain unclear. Here, we investigated the detailed molecular mechanisms of the DCA’s effects by immunoblot, RT-PCR, RNA-sequencing, and RNA-silencing in a murine renal proximal tubular (mProx) cell line and mouse kidneys. In mProx cells, DCA suppressed cisplatin-induced apoptosis by attenuating the JNK/14-3-3/Bax/caspase-9 and death receptor/ligand/caspase-8 pathways without impeding inflammatory signaling. RNA-sequencing demonstrated that DCA increased the cisplatin-reduced expression of cFLIP, a caspase-8 inactivator, and decreased the expression of almost all oxidative phosphorylation (OXPHOS) genes. DCA also increased NF-kB activation and ROS production, probably enhancing the cFLIP induction and OXPHOS gene reduction, respectively. Furthermore, cFLIP silencing weakened the DCA’s anti-apoptotic effects. Finally, in mouse kidneys, DCA attenuated cisplatin-caused injuries such as functional and histological damages, caspase activation, JNK/14-3-3 activation, and cFLIP reduction. Conclusively, DCA mitigates cisplatin-induced nephrotoxicity by attenuating the JNK/14-3-3/Bax/caspase-9 pathway and inhibiting the caspase-8 pathways via cFLIP induction, probably outweighing the cisplatin plus DCA-derived cytotoxic effects including ROS.
Similar content being viewed by others


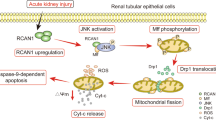
Introduction
Cisplatin is a chemotherapeutic agent widely used to treat solid tumours; however, frequent development of renal injury limits cisplatin-based chemotherapy1,2. Renal proximal tubular damage caused by the activation of cell death and inflammatory pathways is a pathophysiological characteristic of cisplatin-induced nephrotoxicity1,2. Proximal tubular injury arises from apoptosis induced by the intrinsic pathway, depending on mitochondrial injury, and the extrinsic pathway activated by death receptors via inflammatory cytokines such as TNF-α2. In the mitochondrial pathway, cisplatin-stimulated p53 phosphorylation and subsequent upregulation of proapoptotic proteins induce mitochondrial damages, resulting in activation of caspase-9 and caspase-3, leading to apoptosis in cisplatin-induced nephrotoxicity3,4,5. Cisplatin-induced cellular stresses also activate mitogen-activated protein kinases (MAPK), including c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), and p38 kinase (p38), and inflammatory signals, including nuclear transcription factor k-B (NF-κB). These alterations result in the activation of the death receptor pathway via induction of major ligands, such as TNF-α and FasL, causing apoptosis mediated by caspase-8 activation. Moreover, cisplatin stimulates autophagy before apoptosis in cultured renal tubular cells6 and mouse kidneys7. Appropriate augmentation of autophagy protects against cisplatin-induced tubular cell injury8, whereas blockade of autophagy enhances cisplatin-induced nephrotoxicity in vitro6 and in vivo7,9.
Several studies showed that cisplatin-induced nephrotoxicity is characterised by exclusive induction of pyruvate dehydrogenase kinase (PDK) 4, favouring glycolysis and causing considerable renal injury10,11. Dichloroacetate (DCA) inhibits PDK4 and activates pyruvate dehydrogenase (PDH) by reducing its phosphorylation10,11. DCA-induced PDH activation promotes the influx of pyruvate into the tricarboxylic acid (TCA) cycle for the complete oxidation of glucose, leading to increased mitochondrial ATP production10,11. In aerobic glycolysis-favouring cancer cells that have mitochondrial dysfunctions, DCA induces mitochondrial production of reactive oxygen species (ROS) by promoting the TCA cycle and aggravates mitochondrial damages, causing mitochondrial release of cytochrome c and subsequent caspase activation and apoptosis induction12. DCA reportedly diminished cisplatin-induced apoptosis in renal tubular cells and mouse kidneys by reducing mitochondrial damage, oxidative stress, and ATP depletion10,11. The effects on apoptotic pathways and ROS production were unclear. Given that the DCA-promoted TCA cycle may induce ROS production in the cisplatin-damaged mitochondria, the apoptosis-inhibiting effects of DCA could be mediated by mechanisms other than oxidative stress modulation (induction of anti-apoptotic proteins that attenuate activation of caspases-3, -8, or -9). Recently, DCA was reported to modulate autophagy status in cancer cells13,14. p62, a mediator of autophagy, bidirectionally affected the activation of caspase-8 15,16, which may be a fundamental molecule for investigating cross-talk between autophagy and apoptosis during DCA treatment.
DCA may protect against cisplatin-induced nephrotoxicity via various processes, including the modulation of anti-apoptotic proteins and autophagy. The detailed mechanisms underlying these protective effects remain unclear. We investigated the molecular mechanisms underlying the effects of DCA on cisplatin-induced injury, focusing on caspase-9-mediated mitochondrial and caspase-8-mediated death receptor apoptotic pathways, anti-apoptotic proteins, and autophagy, using a murine renal proximal tubular (mProx) cell line and mouse kidneys.
Materials and methods
Materials
Dichloroacetate (DCA) and cisplatin were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan and Sigma-Aldrich, St. Louis, MO, USA), respectively. Rabbit monoclonal antibodies against mouse p53, mouse cleaved caspase-8, human p38, and human NF-κB, mouse MLKL; phosphorylated mouse p53 (p-p53; Ser15), human JNK (p-JNK), human p38 (p-p38), human ERK (p-ERK), human NF-κB (p-NF-κB), human 14-3-3 (p-14-3-3), and mouse MLKL (p-MLKL); and rabbit polyclonal antibodies against human cleaved caspase-3, human JNK, rat ERK, human 14-3-3, mouse cleaved caspase-9, mouse caspase 12, mouse Bid, and human LC3 were purchased from Cell Signaling Technology (Boston, MA, USA). Rabbit polyclonal antibody against human p62, and caspase-3 inhibitor (Z-DEVD-FMK) were purchased from MEDICAL and BIOLOGICAL LABORATORIES Co., Ltd. (Nagano, Japan). Rabbit polyclonal antibodies against β-actin and human cellular FADD-like IL-1β-converting enzyme inhibitory protein (cFLIP); rabbit monoclonal antibodies against human Bax, Cox IV, human pyruvate dehydrogenase (PDH), and phosphorylated PDH (p-PDH); and human recombinant cFLIP were purchased from Abcam Inc. (Cambridge, UK). Rabbit polyclonal antibody against mouse cytokeratin 19 was purchased from Bioss Antibodies, Inc. (Woburn, MA, USA). Horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit immunoglobulin was purchased from Dako (Glostrup, Denmark). Inhibitors of autophagy, bafilomycin A1, and 3-methyladenine (3-MA), were purchased from Adipogen Corp. (San Diego, CA, USA) and Santa Cruz Biotechnology (Dallas, TX, USA), respectively.
Cell cultures
mProx cells were generated as previously described17,18 and confirmed to exhibit proximal tubular phenotype17. mProx cells expressed epithelial cell-specific cytokeratin 19 antigens, whereas K562 cells did not (see Supplementary Fig. S1 online). Cells were grown in modified K-1 medium (50:50 Ham’s F-12/DMEM; Wako Pure Chemical Industries, Ltd.) with 10% FBS (Thermo Fisher Scientific Inc., Waltham, MA, USA) in a humidified 5% CO2 and 95% air atmosphere at 37 °C. mProx cells (passages 15 through 20) were seeded on 12-well plates, and modified K-1 medium was renewed every 2 d until confluence. Human leukaemia cell line K562 was grown in RPMI 1640 medium (Thermo Fisher Scientific Inc.) supplemented with 10% FBS (Thermo Fisher Scientific Inc.) in a humidified 5% CO2 and 95% air atmosphere at 37 °C.
To investigate the dose- and time-dependent effects of cisplatin, mProx cells were treated for 24 h with increasing doses of cisplatin (0, 5, 10, and 25 µM) and with 25 µM cisplatin for 0, 3, 6, 16, and 24 h, respectively. To investigate the effects of DCA and a caspase-3 inhibitor on cisplatin-induced changes and their dose-dependency, mProx cells were treated for 24 h with simultaneous administration of cisplatin (25 µM) and DCA (0, 1, 3, 5, 10, or 20 mM) or a caspase-3 inhibitor (0, 5, 10, or 20 µM). The supernatant of mProx cell cultures exposed or not to caspase inhibitors was collected and used to assess cell injury by measuring lactate dehydrogenase (LDH) activity using a LDH assay kit (L-type Wako LD·IF, FUJIFILM Wako Chemical Corporation, Kanagawa, Japan). To investigate the effect of DCA on basal autophagy status, mProx cells were treated for 3 h with or without DCA (10 mM) in the presence or absence of bafilomycin A (200 nM), an inhibitor of autophagic degradation. For starvation experiments, mProx cells were incubated in HBSS medium with Ca2+ and Mg2+ (Wako Pure Chemical Industries, Ltd.) for 3–6 h, as previously reported19,20. To investigate the effects of autophagy inhibition on cisplatin-induced apoptosis, mProx cells were treated with cisplatin (25 µM) for 24 h in the presence or absence of DCA (10 mM) or 3-MA (3 mM), an inhibitor of the late phase of autophagy. Bafilomycin A, cisplatin, DCA, and 3-MA were prepared as stock solutions in dimethyl sulfoxide (DMSO), 0.9% NaCl, and modified K1 medium, respectively, and then further diluted. The final DMSO concentration did not exceed 0.1%.
Annexin V-FITC/Propidium iodide (PI) assay
Phosphatidylserine translocation from the inner to the outer plasma membrane, a marker of early stage apoptosis, was detected using an Annexin V-FITC kit (MBL CO. LTD., Nagoya, Japan) according to the manufacturer’s protocol. Briefly, mProx cells grown in 12-well plates were treated for 24 h with or without cisplatin (25 µM) in the presence or absence of DCA (10 mM). Cells harvested by trypsinization were washed with phosphate-buffered saline and incubated in a binding buffer containing annexin V and a late-phase marker of apoptosis, PI, for 15 min at room temperature in the dark. The stained cells were analyzed by fluorescence-activated cell sorting using a flow cytometer (BD FACSCanto II, BD Biosciences, San Jose, CA, USA) and the FlowJo software version 7.6 (BD Biosciences), with the acquisition of a total of 10,000 events/sample to ensure adequate data.
TaqMan real-time PCR assay
TaqMan real-time PCR was performed as previously described21. Unlabeled specific primers and TaqMan MGB probes (6-FAM dye-labeled) against mouse Bax (Assay ID: Mm00432051_m1), TNF-α (Assay ID: Mm00443258_m1), TNFR1 (Assay ID: Mm00441875_m1), TL1A (Mm00770031_m1), FAS (Mm04206620_m1), FASL (Mm00438864_m1), cFLIP (Mm01255579_m1), PDK4 (Mm01166879_m1), and p62 (Mm00448091_m1) were purchased from Thermo Fisher Scientific Inc. The mRNA levels of each gene were normalized to that of β2-microglobulin mRNA, unless otherwise stated. The average mRNA level of each gene in the untreated cells was set to 1.0.
Immunoblot analysis
Whole cell lysates were prepared using RIPA buffer containing phosphatase inhibitors (Santa Cruz Biotechnology, CA, USA), and mitochondrial and cytosolic fractions of mProx cells were prepared using the EzSubcell Fraction (ATTO Corp., Osaka, Japan) according to the manufacturer’s instructions. Protein lysates (10 µg) were analyzed by immunoblotting, as previously reported17. The membranes to which the proteins were transferred were incubated with anti-p53 (1:1000), anti-p-p53 (1:1000), anti-cleaved caspases-3 (1:1000), anti-cleaved caspase-8 (1:1000), anti-cleaved caspase-9 (1:1000), anti-caspase-12 (1:1000), anti-JNK (1:1000), anti-p-JNK (1:1000), anti-14-3-3 (1:1000), anti-p-14-3-3 (1:1000), anti-p38 (1:1000), anti-p-p38 (1:1000), anti-ERK (1:1000), anti-p-ERK (1:1000), anti-NF-κB (1:1000), anti-p-NF-κB (1:1000), anti-Bid (1:1000), anti-MLKL (1:1000), anti-p-MLKL (1:1000), anti-p62 (1:1000), anti-LC3 (1:500), anti-Cox IV (1:2000), and anti-β-actin (1:6000) antibodies for 20 min at room temperature. The membranes were then incubated with the appropriate horseradish peroxidase-conjugated anti-mouse or anti-rabbit immunoglobulins (1:1000) at room temperature for 1 h. The secondary antibodies were detected using ECL reagents. The amounts of proteins amounts in the cytosol, cell lysates, and mitochondria were normalized to those of β-actin and COX IV, respectively. The average amount of target protein in untreated cells was expressed as arbitrary values, unless otherwise stated. Each target protein and control (β-actin or COX IV) were electrophoresed in different gels, transferred on different membranes, and separately hybridized with the corresponding antibodies, except for cleaved caspase-3, Bid or cellular Bax, and β-actin. Caspase-3, Bid or cellular Bax, and β-actin were electrophoresed in the same gel and transferred on the same membrane, which was cut into two sheets for the analysis of target and control proteins according to the molecular weights before hybridization with the corresponding antibodies. Adequately cropped images of the original blots are represented in the Figures of this manuscript, and the original, full-length images are included in the Supplementary Information.
Measurement of reactive oxygen species (ROS) and ATP
ROS generation by mProx cells was measured using an OxiSelect Intracellular ROS Assay Kit (Cell Biolabs, San Diego, CA, USA) according to the manufacturer’s instructions. After mProx cells seeded in a 96-well black fluorometric plate were incubated in modified K-1 medium for 24 h, they were treated with a cell-permeable probe (DCFH-DA) for 30 min for newly producing DCFH, a detector of ROS, within the cells, and then treated for 1–3 h with increasing doses of DCA (0, 1, 5, 10, or 20 mM) or cisplatin (0, 25, 50, 100, or 500 µM); incubated in the presence or absence of 25 µM cisplatin or 10 mM DCA for 1, 3, 18, or 24 h; or incubated in the presence or absence of 25 µM cisplatin, 10 mM DCA, or 5 mM N-acetyl cysteine (NAC, Wako Pure Chemical Industries, Ltd.) for 24 h. Fluorescence from each well was measured using a standard fluorometric plate reader (Molecular Device SpectraMax iD3, Molecular Devices, LLC., CA, USA) at excitation and emission wavelengths of 480 and 530 nm, respectively. The average ROS level in untreated cells was set to 1.0.
ATP levels were measured using an Intracellular ATP assay kit ver.2 (DOJINDO LABORATORIES, Kumamoto, Japan). mProx cells grown in 96-well plates were incubated for 24–48 h in the presence or absence of DCA (10 mM) or cisplatin (25 µM). Samples were prepared from the cells in each well according to the manufacturer’s instructions, and luminescence was determined using a standard luminescence measurement plate reader (Molecular Device SpectraMax iD3).
Whole transcriptome sequencing
Confluent mProx cells were treated for 24 h with or without 25 mM cisplatin in the presence or absence of 10 mM DCA (four groups, n = 3 per group). From the cells thus treated, total RNA was extracted using RNeasy mini kit (Qiagen, Venlo, The Netherlands). RNA quality and quantity were assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies). Poly(A) selection, library preparation, and 2 × 150 bp paired-end sequencing were performed, by availing the commercial service of Takara Bio Inc. (Kusatsu, Shiga, Japan) using a SMART-Seq v4 Ultra Low Input RNA Kit (Takara Bio Inc.) and Illumina NovaSeq 6000. A total of 56–81 million reads were sequenced per sample. All raw sequence reads were mapped to the mouse genome (mm10, GRCm38) using the DRAGEN Bio-IT Platform. Demultiplexing and adapter trimming were performed using bcl2fastq v2.20. edgeR v3.20.9 was used to normalize the reads and to detect significant differential expression of genes between mProx cells with different treatments (log2 fold change > 1 or <-1 and q value < 0.05) or among mProx cells with different treatments (P < 0.05, q value < 0.05), where the q value was calculated by correcting the P-value using the Benjamini–Hochberg method. Heatmaps were generated using edgeR or Expression Heatmapper and showed the average of the three replicates scaled across genes. Database for Annotation, Visualization and Integrated Discovery (DAVID, v6.8) was used to detect significantly [P < 0.05 and false discovery rate (FDR) < 0.05] enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways22.
Transfection of small interfering RNA against cFLIP, p62, and PDK4
Silencing of the cFLIP, p62, and PDK4 genes was conducted as previously reported21. Small interfering RNAs (siRNAs) against cFLIP, p62, and PDK4, and control siRNA (a non-targeting siRNA) were purchased from GE Healthcare Dharmacon (Lafayette, CO, USA). Briefly, mProx cells (50% confluence) grown on a 24-well plate were transfected for 40 h with negative control siRNA or siRNA against cFLIP, p62, or PDK4 at a final concentration of 10–50 nmol/L using a transfection reagent (DharmaFECT; Dharmacon). The mProx cells refreshed with the modified K-1 medium were then treated with or without cisplatin (25 µM) in the presence or absence of DCA (10 mM) for an additional 24 h, and the levels of several target proteins (including cleaved caspase-3 and − 8) were analyzed by immunoblotting.
Cisplatin-induced kidney injury animal model
All animal experimental procedures were performed in accordance with appropriate institutional guidelines for animal research, were approved by the Institutional Animal Care and Use Committee of University of Fukui (approval number: 30076 and approval date: 2018/03/30), and followed the recommendations in the ARRIVE guidelines. S129 mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). All mice were raised in standard cages under a controlled 12-h light/dark cycle at 22 ± 2 °C and were allowed free access to water and chow. To evaluate the effect of DCA on cisplatin-induced acute injury, 8–10-week-old male S129 mice were assigned to four groups (control, n = 6; DCA-treated, n = 6; cisplatin-treated, n = 10; and cisplatin- and DCA-treated, n = 11). Two groups were pretreated intra-peritoneally daily with DCA and two groups with normal saline for 2 d. A single nephrotoxic dose of cisplatin (20 mg/kg; Sigma-Aldrich) was administered intra-peritoneally to one DCA- and one saline-pretreated group. Daily DCA or saline treatment was continued for 3 d. The remaining DCA and saline pretreatment groups were administered saline. Daily DCA or saline treatment was continued for 3 d. Mice were euthanized 72 h after the cisplatin injection. Anesthesia or euthanasia were performed using methods consistent with the commonly accepted norms of veterinary best practice.
Renal hematological and histological analyses
Serum creatinine and blood urea nitrogen (BUN) levels were measured using an automatic analyzer, TBA-2000FR (CANON MEDICAL SYSTEMS Corp., Tochigi, Japan). As for histological analysis, the mouse kidneys were fixed in 4% paraformaldehyde, embedded in paraffin wax, and sectioned. The 5-µm-thick sections were stained with hematoxylin and eosin (H&E) and Periodic acid–Schiff (PAS) stain. Tubular damage in PAS-stained kidney sections was examined under a light microscope and scored based on the percentage of cortical tubular necrosis as follows: 0 = normal, 1 = 1–10%, 2 = 11–25%, 3 = 26–45%, 4 = 46–75%, and 5 = 76–100%. Slides were scored in a blinded manner, and individual data were presented as dot plots with the mean ± SEM of 10 representative fields/groups. The criteria for tubular necrosis displaying brush border loss, necrosis, or detachment of the proximal tubular cells and cast formation were defined using previously described methods23,24.
Immunohistochemistry
Immunostaining was performed on 4% paraformaldehyde-fixed paraffin-embedded kidney samples (2 mm sections) from mice treated with saline alone, DCA alone, cisplatin alone, or cisplatin plus DCA. Rabbit polyclonal antibody against cFLIP (1:200) was used as the primary antibody to determine the localization of cFLIP. Positive staining was detected using the EnVision™ FLEX kit/HRP(DAB) (Agilent Technologies Japan, Ltd. Tokyo, Japan) according to the manufacturer’s instructions.
Statistical analyses
All individual data are presented as dot plots with mean ± standard error of the mean (SEM). Student’s t-test or one-way analysis of variance (ANOVA) was used to evaluate the significance of differences between two or among three or more groups, respectively. Statistical significance was set at P < 0.05. All statistical analyses were performed using SPSS (IBM SPSS Statistics 24.0), and graphs were created using GraphPad Prism 9.5.1 (GraphPad Software Inc.).
Results
DCA attenuated cisplatin-induced apoptosis by decreasing caspases-3 and -8, but not -12, activation
Cisplatin (25 µM) time-dependently intensified caspase-3 was activated at 16 and 24 h (Supplementary Fig. S2a online). Cisplatin treatment for 24 h dose-dependently increased cleaved caspase-3, reaching a maximum value over 10-fold at 25 µM (Supplementary Fig. S2b online). DCA attenuated cisplatin-induced caspase-3 activation in a dose-dependent manner, reducing rate plateauing at DCA concentrations of ≥ 5 mM (Fig. 1A and B). DCA (10 mM) significantly decreased cisplatin-induced apoptosis in Q2 (late phase; Fig. 1D) and Q4 (early phase; Fig. 1D). DCA reduced cisplatin-enhanced activation of caspase-8 (Fig. 1E). Thus, treatment with cisplatin (25 µM) and/or DCA (10 mM) for 24 h was used in subsequent experiments unless otherwise stated.

DCA reduced cisplatin-induced apoptosis by suppressing caspase-3 and -8 activation in mProx cells. mProx cells were incubated for 24 h with growth buffer containing cisplatin (0 or 25 µM) in the presence or absence of DCA (A: 1, 3, 5, 10, and 20 mM; B–E: 10 mM). Cleaved caspase-3 (A, B) and caspase-8 (E) levels in cell lysates were determined by immunoblot analyses in mProx cells treated as indicated. A representative blot is shown in the upper panel (A, B, E). (C–D) Harvested cells were stained with Annexin V-FITC and propidium iodide (PI), and analyzed by flow cytometry using fluorescence-activated cell sorting scan. Percentages of Q2 and Q4 were calculated based on the data from each well sample. Individual data are expressed as dot plots with the mean ± SEM of a representative experiment (n = 3). *, P < 0.05 and **, P < 0.01 compared with cisplatin treatment alone (A) or under the indicated conditions (B, D, E), according to one-way ANOVA with Scheffe’s post hoc comparisons.
DCA did not reduce caspase-12, an apoptotic marker of endothelium reticulum stress (Supplementary Fig. S2c online). MLKL phosphorylation was not detected in cisplatin- or cisplatin plus DCA-treated mProx cells (Supplementary Fig. S2d online), suggesting absence of necroptosis. Furthermore, an inhibitor (10 µM) of caspase-3 reduced cisplatin-induced morphological changes (Supplementary Fig. S2e online) and caused a 25% decrease in LDH elevation in the supernatant (Supplementary Fig. S2f), indicating the involvement of caspase-3 activation in cisplatin-derived cytotoxicity of mProx cells.
DCA decreased caspase-3 activation by suppressing JNK/14-3-3/Bax/caspase-9 pathways
We examined the effects of cisplatin and/or DCA on mitochondrial apoptotic pathways in mProx cells. Because the mProx cells17 were immortalized by transfection with the SV40 large T antigen which inactivates p536,17,25,26, the phosphorylated p53 (p-p53)/p53 ratio was low in the cytoplasm of mProx cells at baseline (Fig. 2A, control lane), which was consistent with the results of earlier reports25,26. Cisplatin significantly increased protein levels of p-p53 (Fig. 2A), mitochondrial Bax (Fig. 2B), and cleaved caspase-9 (Fig. 2C) and mRNA levels of Bax (Fig. 2D). Cisplatin increased JNK, 14-3-3, and PDH phosphorylation (Fig. 2E–G, respectively). DCA significantly attenuated cisplatin-induced mitochondrial Bax accumulation and caspase-9 activation by 45 and 76%, respectively (Fig. 2C and D). DCA also reduced the cisplatin-stimulated phosphorylation of JNK, 14-3-3, and PDH by 31, 100, and 68%, respectively (Fig. 2E–G).

DCA reduced cisplatin-induced caspase-3 activation by inhibiting the JNK/14-3-3/Bax/caspase-9 pathway. mProx cells were treated for 16–24 h with growth buffer containing cisplatin (0 or 25 µM) in the presence or absence of DCA (10 mM). Whole cell lysates and cytosolic and mitochondrial fractions were prepared as described in the Materials and Methods. Levels of p53 and phosphorylated p53 (p-p53) (A), mitochondrial Bax and Cox IV (B), cleaved caspase-9 and β-actin (C), JNK and phosphorylated JNK (p-JNK) (E), 14-3-3 and phosphorylated 14-3-3 (p-14-3-3) (F), and PDH and phosphorylated PDH (p-PDH) (G) were determined by immunoblotting. The amount of each protein was normalized to that of the indicated antigen. Representative blots are shown in the upper panels (A-C and E–G). Bax mRNA expression (D) was determined by real-time PCR assay and normalized to that of β-microglobulin mRNA. The average Bax mRNA level in untreated cells was set to 1.0. Individual data are expressed as dot plots with the mean ± SEM of a representative experiment (n = 3). NS, not significant; *, P < 0.05; **, P < 0.01 according to one-way ANOVA with Scheffe’s post hoc comparisons.
DCA decreased cisplatin-induced activation of caspase-8 despite sustained activation of pro-inflammatory signalling in extrinsic apoptotic pathways
We investigated the effects of cisplatin and/or DCA on inflammation and death receptor pathways in mProx cells. Cisplatin significantly increased p-p38 (Fig. 3A), p-NF-κB at 3 h, 6 h, 16 h (Supplementary Figs. S3a–c online), and 24 h (Fig. 3B), p-ERK (Fig. 3C), TNF-α, TL1A, FasL, and Fas mRNA levels (Fig. 3D–G). Enhancement of inflammatory signals activated caspase-8 (Fig. 1F), causing Bid cleavage and subsequent decrease in cellular Bid levels (Fig. 3H), indicating activation of the death pathway27,28. DCA did not suppress cisplatin-induced upregulation of p-p38, p-NF-κB, p-ERK, TNF-α mRNA, TL1A mRNA, FasL mRNA, or Fas mRNA (Supplementary Figs. S3a–c online and Fig. 3A–G). DCA significantly reduced cisplatin-induced caspase-8 activation by 66% (Fig. 1F) and increased cisplatin-induced Bid protein reduction by 45% (Fig. 3H).

DCA did not inhibit cisplatin-enhanced inflammation or expression of death receptors and ligands but reduced Bid degradation induced by caspase-8 activation. mProx cells were treated for 16, 18, or 24 h with growth buffer containing cisplatin (0 or 25 µM) in the presence or absence of DCA (10 mM). The levels of p38 and phosphorylated p38 (p-p38) (A), NF-B and phosphorylated NF-B (p-NF-B) (B), ERK and phosphorylated ERK(p-ERK) (C), Bid (h), and β-actin were determined by immunoblotting using whole cell lysates. The amount of each phosphorylated protein was normalized to that of the corresponding protein. Representative blots are shown in the upper panels (A–C, H). The levels of TNF-α (D), TL1A (E), FasL (F), and Fas (G) mRNA were determined by real-time PCR assay and normalized to that of β-microglobulin mRNA. The average mRNA level of each gene in untreated cells was set to 1.0. Individual data are expressed as dot plots with the mean ± SEM of a representative experiment (n = 3). NS, not significant; *, P < 0.05; **, P < 0.01 according to one-way ANOVA with Scheffe’s post hoc comparisons.
DCA, but not cisplatin, increased ROS production in mProx cells
Greater NF-κB activation was observed in DCA-treated mProx cells (Supplementary Figs. S3a–c online and Fig. 3B), possibly caused by oxidative stress. DCA increased ROS production dose-dependently at 1 h and 3 h (Supplementary Figs. S4a and b online); no concentrations of cisplatin increased at 1 h and 3 h (Supplementary Figs. S4c and d online). Treatment with DCA alone and combined with cisplatin significantly increased ROS production from 1 h to 24 h (Supplementary Figs. S5a–d online), associated with NF-κB activation (Supplementary Figs. S3a–c online and Fig. 3B). Although cisplatin alone activated caspase-3 without inducing ROS production (Supplementary Figs. S5d–f online), ROS scavenger NAC considerably reduced this cisplatin-induced caspase-3 activation despite moderate ROS reduction (Supplementary Figs. S5e and f online). Despite elevated ROS production, DCA attenuated cisplatin-induced caspase-3 activation (Supplementary Figs. S5e and f online). It augmented the NAC-mediated suppression of caspase-3 activation (Supplementary Fig. S5e online) despite the higher ROS production than NAC (Supplementary Fig. S5f online). Cisplatin plus DCA treatment produced significantly less ATP at 24 h but more at 48 h (Supplementary Figs. S6a and b online) than cisplatin treatment.
DCA-induced gene expression of anti-apoptotic proteins but downregulated oxidative phosphorylation-related genes in cisplatin-treated mProx cells
DCA reduced cisplatin-induced caspase activation and resultant apoptosis despite ROS induction, ATP reduction, and inflammation activation. Therefore, other factors, including anti-apoptotic proteins, were predicted to be involved in cell-protective effects. RNA sequencing was used to more thoroughly analyze the cisplatin- and/or DCA-induced changes in gene expression in mProx cells treated for 24 h. The KEGG pathway analysis revealed that cisplatin significantly activated inflammatory signalling pathways, including the MAPK, TNF-α, NF-κB, and p53 signalling pathways (Supplementary Tables S1 and S2 online). DCA did not reduce the cisplatin-induced gene expression of inflammatory or p53 signalling pathway-associated proteins (Supplementary Table S3 and Figs. S7 and S8 online). NF-κB-induced anti-apoptotic protein expression, including cellular FADD-like IL-1β-converting enzyme inhibitory protein (cFLIP) and inhibitors of apoptotic proteins (IAPs)29,30; cFLIP and cIAP2 were upregulated by DCA and cisplatin (Supplementary Tables S4a and b online). Furthermore, cFLIP gene upregulation was greater in cisplatin plus DCA-treated cells than in cisplatin alone-treated one, explaining the DCA-induced anti-apoptotic effects (Supplementary Table S4c online, Fig. 4, and Supplementary Table S4d online).

Whole transcriptome sequencing revealed that DCA increases gene expression of the anti-apoptotic protein cFLIP in cisplatin-treated and -untreated mProx cells. mProx cells were treated for 24 h with growth buffer containing cisplatin (0 or 25 µM) in the presence or absence of DCA (10 mM). Total RNA was extracted from the cells and used for whole-transcriptome sequencing. Significant differential expression of anti-apoptotic protein genes (n = 9) was detected in mProx cells subjected to different treatments (P < 0.05 and q value < 0.05, one-way ANOVA; Supplementary Table S4d online). The heat map shows the scaled average expression of the detected genes. Blue and yellow colors indicate low and high expression, respectively.
Among the 13 mitochondria-specific genes, DCA or cisplatin significantly upregulated 7 to 9 genes (Supplementary Tables S5a and b online), whereas cisplatin plus DCA downregulated five genes compared to cisplatin alone (Supplementary Table S5c online). Among the oxidative phosphorylation (OXPHOS)-associated genes, cisplatin plus DCA considerably downregulated most genes, whereas cisplatin downregulated or upregulated several different parts of genes (Fig. 5). Among mitochondrial anti-oxidant genes31, DCA plus cisplatin downregulated superoxide dismutase (SOD) 2, peroxiredoxin 3 and 5, and glutathione peroxidase 1 compared to the control or cisplatin alone (Supplementary Tables S6a and b online), explaining DCA-derived effects of decreasing ATP production and increasing ROS production until 24 h.

Whole transcriptome sequencing showed that DCA markedly reduced the expression of oxidative phosphorylation-associated genes in cisplatin-treated mProx cells. mProx cells were treated for 24 h with growth buffer containing cisplatin (0 or 25 µM) in the presence or absence of DCA (10 mM). Total RNA was extracted from the cells used for whole-transcriptome sequencing. Significant differential expression of oxidative phosphorylation-associated genes (n = 114) was detected in mProx cells subjected to different treatments (n = 3 per group) (P < 0.05 and q value < 0.05, one-way ANOVA). The heat map shows the scaled average expression of the detected genes. Blue and yellow colors indicate low and high expression, respectively.
DCA induced cFLIP expression in cisplatin-treated mProx cells
RNA sequencing identified cFLIP as a highly significantly upregulated gene among representative anti-apoptotic proteins in the cisplatin plus DCA-treated mProx cells. We investigated the changes in cFLIP expression. DCA upregulated cFLIP mRNA levels from 6 to 24 h compared with the control (Fig. 6A–D). DCA plus cisplatin increased cFLIP expression from 6 to 24 h compared with cisplatin (Fig. 6A–D). DCA increased cFLIP mRNA level in a dose-dependent manner (Supplementary Fig. S9a online), and a strong positive association was observed between DCA-induced levels of ROS at 1 h and cFLIP mRNA at 6 h (Supplementary Fig. S9b online). In the immunoblot antibody absorption analysis, cFLIP was recognised as a ~ 50-kDa band (Supplementary Fig. S10 online). DCA significantly increased the cisplatin-reduced cFLIP antigen level (Fig. 6E and F). Because ROS-induced NF-κB is a main cFLIP inducer32,33 and cFLIP is the principal inhibitor of caspase-8 activation34, DCA-mediated ROS production and NF-κB activation probably caused cFLIP induction, inhibiting caspase-8 activation in the cisplatin-treated mProx cells.

DCA increased cFLIP expression at mRNA and antigen levels in mProx treated with and without cisplatin. mProx cells were treated for 6 h, 13 h, 16 h, 18 h, or 24 h with growth buffer containing cisplatin (0 or 25 µM) in the presence or absence of DCA (10 mM). The levels of cFLIP mRNA at 6 h (A), 13 h (B), 18 h (C), and 24 h (D) were determined using real-time PCR assay and normalized that of β-microglobulin mRNA. The average cFLIP mRNA level in the untreated cells was set to 1.0. The amounts of cFLIP antigen and β−actin at 16 h (E) and 24 h (F) were determined by immunoblotting using whole cell lysates. The amount of cFLIP was normalized to that of corresponding β-actin. Individual data are expressed as dot plots with the mean ± SEM of a representative experiment (n = 3). NS, not significant; *, P < 0.05; ** P < 0.01 according to one-way ANOVA with Scheffe’s post hoc comparisons. #, P < 0.01 according to Student’s t-test.
cFLIP and PDK4 silencing modulated cisplatin-induced activation of caspase-3 or -8 and affected inhibitory effects of DCA in mProx cells
p62 was recently proposed as a candidate caspase-8 inhibitor in acute kidney injury16. To investigate direct effects of DCA on caspase activation in mProx cells, cFLIP and p62 were silenced using respective siRNA. cFLIP silencing resulted in approximately 90% and 35% reduction in mRNA and protein levels, respectively (Fig. 7A and B). Silencing significantly promoted cisplatin-stimulated activation of caspase-3 and -8 and reduced DCA-derived inhibition rates from 71 to 58% and from 61 to 40% for caspase-3 and caspase-8 activation, respectively (Fig. 7C and D). p62 silencing caused approximately 80% reduction in its mRNA and protein levels (Fig. 7E) but did not affect caspase-8 activation (Fig. 7F). Furthermore, PDK4 silencing decreased its basal mRNA levels by 87% (Supplementary Fig. S11 online), attenuated cisplatin-induced caspase-3 activation, and decreased the DCA-induced inhibition rate (Fig. 7G). Interestingly, PDK4 silencing reduced phosphorylation of PDH, JNK, and 14-3-3 (Fig. 7H–J). This indicated that DCA partially inhibited caspase activation by increasing cFLIP expression and decreasing PDK4 activity.


cFLIP or PDK4 deficiency modulated cisplatin-induced activation of caspase-3 and -8, while p62 deficiency did not influence cisplatin-induced caspase-8 activation. mProx cells were transfected for 40 h with negative control siRNA (A–J), siRNA against cFLIP (A–D), p62 (E, F), or PDK4 (G–J). The cells refreshed with modified K-1 medium were then incubated in K-1 medium containing cisplatin (0 or 25 µM) or DCA (0 or 10 mM) for an additional 24 h. The levels of cFLIP (a) and p62 (E) mRNA were determined using real-time PCR assays and normalized to that of β2-microglobulin mRNA. The average mRNA levels in control siRNA-treated mProx cells was set to 1.0. The amounts cFLIP (B), cleaved caspase-3 (C, G), p62 (E), cleaved caspase-8 (D, F), PDH and p-PDH (H), JNK and p-JNK (I), 14-3-3 and p-14-3-3 (J), and β-actin were determined by immunoblotting. The ratio of cleaved caspase-3 (C) and caspase-8 (D) to β-actin in cFLIP siRNA-transfected mProx cells treated with cisplatin alone and ratio of the cleaved caspase-3 to β-actin in control siRNA-transfected mProx cells treated with cisplatin alone (G) was set to 1.0. Individual data are expressed as dot plots with the mean ± SEM of a representative experiment (n = 3–6). NS, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001 according to Student’s t-test (A, B, E) and one-way ANOVA with Scheffe’s post hoc comparison (C, D, F–J). #, P < 0.05 according to Student’s t-test.
DCA did not affect autophagy status and suppressed caspase activation without autophagy promotion in mProx cells
Bafilomycin A1 inhibited late autophagy (lysosomal degradation). DCA did not increase LC3-II level under basal conditions (incubation in K1 medium) (Supplementary Fig. S12a online); thus, DCA did not promote autophagy in mProx cells under basal conditions. Starvation for 3 h (incubation in HBSS) enhanced autophagy, indicated by a decrease in p62 and increase in LC3 II/I (Supplementary Fig. S12b online). These starvation-induced changes were not augmented by DCA (Supplementary Fig. S12b online). Moreover, DCA did not further stimulate cisplatin-induced autophagy (Supplementary Fig. S12c online). The autophagic inhibitor 3-methyladenine (3-MA; 3 mM) intensified cisplatin-induced caspase-3 and caspase-8 activation by 2.1-fold and 1.6-fold, respectively (Supplementary Fig. S12d online). However, the DCA-mediated inhibition rate for caspase-3 activation did not differ in cisplatin-treated mProx cells with or without 3-MA (60% vs. 55%; Supplementary Fig. S12d online). Conversely, the inhibition rate for caspase-8 was relatively low with 3-MA use compared to that with none (35% vs. 47%; Supplementary Fig. S12d online). These results indicated DCA inhibited cisplatin-induced caspase activation with little involvement of autophagy modulation in mProx cells.
DCA attenuated cisplatin-induced renal injury in mice by suppressing caspase-3 and -8 activation
At 72 h after cisplatin administration, DCA treatment significantly decreased cisplatin-elevated serum creatinine and BUN levels from 0.61 mg/dL to 0.22 mg/dL and 170 mg/dL to 96 mg/dL, respectively (Fig. 8A). DCA treatment significantly ameliorated cisplatin-induced renal tubular damage, characterised by brush border loss, necrosis and detachment of proximal tubular epithelial cells, and tubular casts (Fig. 8B), with decreasing median acute tubular necrosis scores from 3.7 to 2.1 (Fig. 8C). DCA treatment reduced cisplatin-induced activation of caspase-3 and − 8 in the kidneys by 53% and 60%, respectively (Fig. 8D). Cisplatin-induced upregulation of p-p53 and cellular Bax proteins was markedly diminished by DCA (Fig. 8E). DCA considerably suppressed cisplatin-induced phosphorylation of JNK and 14-3-3 (Fig. 8F), consistent with the findings in the mProx cells (Fig. 2E and F). Regarding cisplatin-induced death receptor pathway proteins, DCA reduced TNF-α mRNA levels (Fig. 8G) but increased TNFR1 (Fig. 8H) and Fas mRNA levels (Fig. 8I). In addition, DCA did not reduce cisplatin-induced FasL mRNA levels (Fig. 8J). DCA and cisplatin treatment significantly increased cFLIP mRNA and protein levels by 1.4- and 1.6-fold, respectively, compared to cisplatin treatment for 72 h (Fig. 8K). In kidney tissues of mice treated with saline or DCA, the cFLIP antigen was localized mainly in proximal renal tubules (Supplementary Fig. S13a and 13b online). The staining intensity in proximal renal tubular cells (Supplementary Fig. S13c online) was more significant in mice treated with cisplatin plus DCA than those treated with cisplatin alone (Supplementary Fig. S13d online), where damaged proximal renal tubules showed markedly weak staining for cFLIP (Supplementary Fig. S13c online).


DCA reduced cisplatin-induced acute kidney injury in mice by suppressing caspase-3 and -8 activation. Male S129 mice (8–10 weeks old) were pre-treated with daily intra-peritoneal administration of DCA (250 mg/kg) or saline for two days, intra-peritoneally administered a single dose of cisplatin (20 mg/kg) or saline, followed by daily DCA or saline treatment for three days, and then sacrificed. (A) Serum creatinine and blood urea nitrogen (BUN) levels were measured in the four groups described above. (B) Periodic acid–Schiff (PAS) staining of the kidney tissues of mice. (Original magnification, ×100; *, damaged tubules). (C) Tubular injury score was determined for 10 random fields from each mouse kidney. The amounts of cleaved caspase-3 and caspase-8 (D), p-53 and Bax (E), p-JNK and JNK (F), p-14-3-3 and 14-3-3 (F), cFLIP (K) and β-actin in the kidneys of mice from each group were measured by immunoblotting and normalized to that of the indicated proteins. The levels of TNF-α (G), TNFR1 (H), Fas (I), FasL (J), and cFLIP (K) mRNA were determined using real-time PCR assays and normalized to that of β2-microglobulin mRNA. The average mRNA level of each gene in untreated cells was set to 1.0. Individual data are presented as dot plots with the mean ± SEM (n = 6–9). NS, not significant; *, P < 0.05; **, P < 0.01, ***, P < 0.001 according to one-way ANOVA with Scheffe’s post hoc comparisons. #, P < 0.08 and ##, P < 0.01 according to Student’s t-test (H).
In mouse kidneys, DCA attenuated caspase-3 and -8 activation and suppressed cisplatin-induced nephrotoxicity, probably by inhibiting the JNK/14-3-3/Bax and TNF-α pathways and increasing cFLIP expression in proximal tubular cells without reducing other prominent mediators of death pathways.
Discussion
DCA significantly diminished cisplatin-induced apoptosis in mProx cells by attenuating caspase-3 activation via suppression of the JNK/14-3-3/Bax/caspase-9 pathway and reducing caspase-8 activation via upregulation of cFLIP expression mediated probably by sustained inflammation. Apoptosis suppression was accompanied by no ATP loss salvage or no inflammation inhibition. DCA ameliorated cisplatin-induced acute kidney injury in mice by attenuating caspase-3 and -8 activation. p53, Bax, and TNF-α status differed in mouse kidneys and mProx cells.
A notable finding was that DCA significantly reduced cisplatin-induced caspase-8 activation in mProx cells by inducing cFLIP expression without downregulating the death receptor pathway. DCA-mediated inhibition of Bid degradation confirmed that DCA suppresses caspase-8 activation27,28. Caspase-8 activation involves the interaction between death receptors and their ligands34. DCA did not reduce cisplatin-induced expression or activation of signal molecules related to the death receptor pathways. DCA plus cisplatin upregulated expression of cFLIP, a main inhibitor of caspase-8 activation34,35. RNA sequencing confirmed this. cFLIP silencing enhanced cisplatin-induced caspase activation and weakened inhibitory effects of DCA. cFLIP is a master anti-apoptotic regulator that suppresses the death receptor pathway33 and inhibits stress-induced apoptosis in murine, rat, and human renal tubular cells36,37,38. Thus, the anti-caspase-8 activation effect of DCA likely mediated via cFLIP upregulation. Because NF-κB is the main inducer of cFLIP expression33, DCA-induced NF-κB activation may cause sustained cFLIP upregulation in mProx cells.
DCA-promoted TCA cycle enhanced ROS production in brain tissues39,40 and vascular smooth muscle cells41, as reported for cancer cells12. DCA also induces NF-κB activation in a ROS-independent manner42. Notably, NF-κB activation the increases expression of cFLIP and anti-apoptotic factors, which suppresses the apoptosis pathway29,31,43. In cisplatin-treated mProx cells, DCA induced greater production of ROS, probably leading to greater NF-κB activation as earlier reported39,40,41,44 and consequent induction of cFLIP.
Recent studies showed that DCA reduced cisplatin-induced nephrotoxicity by attenuating cisplatin-induced mitochondrial injury via modulation of glucose oxidation and maintenance of mitochondrial oxidative phosphorylation10,11. How DCA modulates the cisplatin-activated mitochondrial apoptotic pathways remains to be clarified. DCA suppressed the cisplatin-activated mitochondrial apoptotic pathway in mProx cells by reducing mitochondrial Bax accumulation, caspase-9 activation, and caspase-3 activation in our study. It did not mitigate cisplatin-induced phosphorylation of p53 or cellular expression of Bax5. This discrepancy can be explained by involvement of the JNK/14-3-3/Bax pathway21,45. The cytosolic protein 14-3-3 sequesters Bax in the cytosol45. JNK-mediated phosphorylation of 14-3-3 may promote its dissociation from Bax and its translocation to the mitochondria45. In mProx cells, DCA attenuated cisplatin-induced phosphorylation of JNK and 14-3-3, preventing the mitochondrial translocation of Bax. As mitochondrial injury directly activates caspase-9 46, DCA-induced suppression of caspase-9 activation indicates its direct involvement in the mitochondrial apoptotic pathway. Although cisplatin- or inflammation-induced phosphorylation of JNK increased PDK4 mRNA expression11,47, PDK4 inhibition or its gene silencing in mProx cells reduced cisplatin-induced phosphorylation of JNK and 14-3-3 in our study. Therefore, DCA may have attenuated cisplatin-induced apoptosis in mProx cells by reciprocally inhibiting JNK and PDK4.
Autophagy is upregulated in cisplatin-induced injury to renal tubular cells and kidney tissues6,7, whereas autophagy inhibition amplifies caspase activation and apoptosis6,7,9. These alterations were also observed in mProx cells in our study. Autophagy augmentation protects against injury to renal proximal tubular cells8,16; DCA induces protective autophagy in cancer cells13,14. DCA did not affect autophagy status under basal, starvation, or cisplatin-treated conditions in mProx cells.
It is essential to consider the influence of ROS on the status of mProx cells. ROS is generally considered cytotoxic, and DCA-induced ROS was shown to damage nerve39,40 and cancer cells12. In this study, DCA and DCA plus cisplatin increased ROS production to the same extent in the mProx cells (Supplementary Figure S5a-S5d online). DCA alone decreased ATP production (Supplementary Figure S6 online) and OXPHOS-associated gene expression (Fig. 5) without MAPK or caspase activation, whereas cisplatin considerably reduced ATP production (Supplementary Figure S6 online) and activated the MAPK pathways and caspases with negligible ROS induction (Supplementary Figure S5a-S5d online). DCA plus cisplatin further reduced ATP production (Supplementary Figure S6 online) and its associated gene expression (Fig. 5) without suppressing inflammation but attenuating caspase activation, compared to cisplatin. Moreover, NAC-derived ROS reduction weakened caspase-3 activation in the mProx cells treated with cisplatin alone and cisplatin plus DCA (Supplementary Figure S5f online). These results indicate that ROS had a varying degree of harmful effect on any of the mRox cells treated with DCA, cisplatin, or DCA plus cisplatin. DCA was presumed to induce ROS production by promoting the TCA cycle due to increased PDH activity. Conceivably, in mProx cells, the ROS production enhanced the cisplatin-induced reduction of OXPHOS gene expression and ATP production and sustained the cisplatin-induced inflammation. On the contrary, DCA suppressed PDK4 activity and the JNK/14-3-3/Bax/caspase-9 pathway. It also increased the expression of anti-apoptotic proteins, mainly cFLIP, probably via sustained cisplatin-derived inflammation, attenuating the cisplatin-induced caspase-8 activation. Intriguingly, low to intermediate amounts of ROS were reported to activate NF-kB without inducing apoptosis44, potentially corresponding to ROS produced by DCA. Furthermore, there should be additional protective effects of DCA that are yet to be identified. Thus, it seems plausible that the DCA’s protective effects comprehensively outweighed the cisplatin plus DCA-derived cytotoxic effects, including enhanced ROS production.
Our study had a few limitations. First, the subcellular site of DCA-induced ROS production was unclear, as total intracellular ROS levels were measured. Second, the mechanism by which DCA reduced the expression of OXPHOS genes in cisplatin-treated mProx cells was not investigated, although ROS production was considered a cause. Third, it was unclear why a significant ROS production was not detected in cisplatin-treated mProx cells using standard assay, contradicting early studies on cisplatin-induced nephropathy. Therefore, we should have also measured ROS levels in mProx cells using alternative methods, although NAC considerably reduced cisplatin-induced caspase-3 activation. Fourth, the cell-protective effects of a caspase-8 inhibitor remain to be clarified. Fifth, to further clarify the mechanisms of DCA’s protective effects, other possible pathways in non-tumour cells should have been investigated. Sixth, the presence of apoptosis in the kidneys of cisplatin-treated mice also remained to be investigated.
In summary, DCA attenuated cisplatin-induced injury in mProx cells and mouse kidneys by inhibiting caspase-3 activation via suppression of the JNK/14-3-3/Bax/caspase-9 pathway and inhibiting caspase-8 activation via induction of cFLIP expression, despite ROS elevation and no ATP loss salvage or inflammation inhibition. These findings indicate new mechanisms of action for DCA as a candidate agent to mitigate cisplatin-induced nephrotoxicity, which probably outweigh the deleterious effects of ROS.
Data availability
The data underlying this article will be shared on reasonable request to the corresponding authors.
References
Ozkok, A. & Edelstein, C. L. Pathophysiology of cisplatin-induced acute kidney injury. BioMed research international 967826. https://doi.org/10.1155/2014/967826 (2014).
Oh, G. S. et al. Cisplatin-induced kidney dysfunction and perspectives on improving treatment strategies. Electrolyte Blood Pressure: E BP. 12, 55–65. https://doi.org/10.5049/EBP.2014.12.2.55 (2014).
Park, M. S., De Leon, M. & Devarajan, P. Cisplatin induces apoptosis in LLC-PK1 cells via activation of mitochondrial pathways. J. Am. Soc. Nephrology: JASN. 13, 858–865. https://doi.org/10.1681/ASN.V134858 (2002).
Wei, Q., Dong, G., Franklin, J. & Dong, Z. The pathological role of bax in cisplatin nephrotoxicity. Kidney Int. 72, 53–62. https://doi.org/10.1038/sj.ki.5002256 (2007).
Zhang, D. et al. Tubular p53 regulates multiple genes to mediate AKI. J. Am. Soc. Nephrology: JASN. 25, 2278–2289. https://doi.org/10.1681/ASN.2013080902 (2014).
Yang, C., Kaushal, V., Shah, S. V. & Kaushal, G. P. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am. J. Physiol. Renal. Physiol. 294, F777–787. https://doi.org/10.1152/ajprenal.00590.2007 (2008).
Takahashi, A. et al. Autophagy guards against cisplatin-induced acute kidney injury. Am. J. Pathol. 180, 517–525. https://doi.org/10.1016/j.ajpath.2011.11.001 (2012).
Li, J. et al. Metformin protects against Cisplatin-Induced Tubular Cell apoptosis and acute kidney Injury via AMPKalpha-regulated autophagy induction. Sci. Rep. 6, 23975. https://doi.org/10.1038/srep23975 (2016).
Jiang, M. et al. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 82, 1271–1283. https://doi.org/10.1038/ki.2012.261 (2012).
Galgamuwa, R. et al. Dichloroacetate prevents cisplatin-Induced nephrotoxicity without compromising cisplatin anticancer properties. J. Am. Soc. Nephrology: JASN 27, 3331–3344. https://doi.org/10.1681/ASN.2015070827 (2016).
Oh, C. J. et al. Pyruvate dehydrogenase kinase 4 deficiency attenuates cisplatin-induced acute kidney injury. Kidney Int. 91, 880–895. https://doi.org/10.1016/j.kint.2016.10.011 (2017).
Bhat, T. A., Kumar, S., Chaudhary, A. K., Yadav, N. & Chandra, D. Restoration of mitochondria function as a target for cancer therapy. Drug Discovery Today. 20, 635–643. https://doi.org/10.1016/j.drudis.2015.03.001 (2015).
Gong, F. et al. Dichloroacetate induces protective autophagy in LoVo cells: involvement of cathepsin D/thioredoxin-like protein 1 and Akt-mTOR-mediated signaling. Cell Death Dis. 4, e913. https://doi.org/10.1038/cddis.2013.438 (2013).
Jia, H. Y. et al. Dichloroacetate induces protective autophagy in esophageal squamous carcinoma cells. Oncol. Lett. 14, 2765–2770. https://doi.org/10.3892/ol.2017.6562 (2017).
Hou, W., Han, J., Lu, C., Goldstein, L. A. & Rabinowich, H. Autophagic degradation of active caspase-8: a crosstalk mechanism between autophagy and apoptosis. Autophagy. 6, 891–900. https://doi.org/10.4161/auto.6.7.13038 (2010).
Kaushal, G. P. & Shah, S. V. Autophagy in acute kidney injury. Kidney Int. 89, 779–791. https://doi.org/10.1016/j.kint.2015.11.021 (2016).
Takaya, K. et al. Involvement of ERK pathway in albumin-induced MCP-1 expression in mouse proximal tubular cells. Am. J. Physiol. Renal. Physiol. 284, F1037–1045. https://doi.org/10.1152/ajprenal.00230.2002 (2003).
Okada, H. et al. Dexamethasone induces connective tissue growth factor expression in renal tubular epithelial cells in a mouse strain-specific manner. Am. J. Pathol. 168, 737–747. https://doi.org/10.2353/ajpath.2006.050656 (2006).
Wei, Y., Pattingre, S., Sinha, S., Bassik, M. & Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 30, 678–688. https://doi.org/10.1016/j.molcel.2008.06.001 (2008).
Lee, J. M. et al. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 516, 112–115. https://doi.org/10.1038/nature13961 (2014).
Shan, J. et al. PPAR-delta activation reduces cisplatin-induced apoptosis via inhibiting p53/Bax/caspase-3 pathway without modulating autophagy in murine renal proximal tubular cells. Clin. Exp. Nephrol. 25, 598–607. https://doi.org/10.1007/s10157-021-02039-2 (2021).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592. https://doi.org/10.1093/nar/gkac963 (2023).
Oh, G. S. et al. Pharmacological activation of NQO1 increases NAD(+) levels and attenuates cisplatin-mediated acute kidney injury in mice. Kidney Int. 85, 547–560. https://doi.org/10.1038/ki.2013.330 (2014).
Tavares, M. B. et al. Acute tubular necrosis and renal failure in patients with glomerular disease. Ren. Fail. 34, 1252–1257. https://doi.org/10.3109/0886022X.2012.723582 (2012).
Dimri, G., Band, H. & Band, V. Mammary epithelial cell transformation: insights from cell culture and mouse models. Breast cancer Res.: BCR. 7, 171–179. https://doi.org/10.1186/bcr1275 (2005).
Kim, C. W. et al. Immortalization of human corneal epithelial cells using simian virus 40 large T antigen and cell characterization. J. Pharmacol. Toxicol. Methods 78, 52–57. https://doi.org/10.1016/j.vascn.2015.11.005 (2016).
Kim, W. S. et al. The caspase-8/Bid/cytochrome c axis links signals from death receptors to mitochondrial reactive oxygen species production. Free Radic. Biol. Med. 112, 567–577. https://doi.org/10.1016/j.freeradbiomed.2017.09.001 (2017).
Huang, K. et al. Cleavage by caspase 8 and mitochondrial membrane association activate the BH3-only protein bid during TRAIL-induced apoptosis. J. Biol. Chem. 291, 11843–11851. https://doi.org/10.1074/jbc.M115.711051 (2016).
Karin, M. & Lin, A. NF-kappaB at the crossroads of life and death. Nat. Immunol. 3, 221–227. https://doi.org/10.1038/ni0302-221 (2002).
Gullulu, O., Hehlgans, S., Rodel, C., Fokas, E. & Rodel, F. Tumor suppressor protein p53 and inhibitor of apoptosis proteins in Colorectal Cancer-A Promising Signaling Network for therapeutic interventions. Cancers. 13 https://doi.org/10.3390/cancers13040624 (2021).
Nakano, H. et al. Reactive oxygen species mediate crosstalk between NF-kappaB and JNK. Cell Death Differ. 13, 730–737. https://doi.org/10.1038/sj.cdd.4401830 (2006).
Zeng, Y. et al. Edaravone attenuated Particulate Matter-Induced Lung inflammation by inhibiting ROS-NF-kappaB signaling pathway. Oxidative Med. Cell. Longev. 2022 (6908884). https://doi.org/10.1155/2022/6908884 (2022).
Safa, A. R. c-FLIP, a master anti-apoptotic regulator. Exp. Oncol. 34, 176–184 (2012).
Tsuchiya, Y., Nakabayashi, O. & Nakano, H. FLIP the switch: regulation of apoptosis and necroptosis by cFLIP. Int. J. Mol. Sci. 16, 30321–30341. https://doi.org/10.3390/ijms161226232 (2015).
Hughes, M. A. et al. Co-operative and hierarchical binding of c-FLIP and Caspase-8: a unified model defines how c-FLIP isoforms differentially control cell fate. Mol. Cell. 61, 834–849. https://doi.org/10.1016/j.molcel.2016.02.023 (2016).
Du, C., Guan, Q., Yin, Z., Zhong, R. & Jevnikar, A. M. IL-2-mediated apoptosis of kidney tubular epithelial cells is regulated by the caspase-8 inhibitor c-FLIP. Kidney Int. 67, 1397–1409. https://doi.org/10.1111/j.1523-1755.2005.00217.x (2005).
Xu, D. et al. c-Myc promotes tubular cell apoptosis in ischemia-reperfusion-induced renal injury by negatively regulating c-FLIP and enhancing FasL/Fas-mediated apoptosis pathway. Acta Pharmacol. Sin. 40, 1058–1066. https://doi.org/10.1038/s41401-018-0201-9 (2019).
Wang, S. et al. Anti-IL-2 receptor antibody decreases cytokine-induced apoptosis of human renal tubular epithelial cells (TEC). Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association -. Eur. Ren. Assoc. 26, 2144–2153. https://doi.org/10.1093/ndt/gfq714 (2011).
Gao, X. et al. PDK4 decrease neuronal apoptosis via inhibiting ROS-ASK1/P38 pathway in early brain Injury after subarachnoid hemorrhage. Antioxid. Redox. Signal. https://doi.org/10.1089/ars.2021.0083 (2022).
Wei, W. et al. Dichloroacetic acid-induced dysfunction in rat hippocampus and the protective effect of curcumin. Metab. Brain Dis. 36, 545–556. https://doi.org/10.1007/s11011-020-00657-5 (2021).
Yang, Y. et al. AKT-independent activation of p38 MAP kinase promotes vascular calcification. Redox Biol. 16, 97–103. https://doi.org/10.1016/j.redox.2018.02.009 (2018).
Pan, Y. et al. The critical role for TAK1 in trichloroethylene-induced contact hypersensitivity in vivo and in CD4(+) T cell function alteration by trichloroethylene and its metabolites in vitro. Toxicol. Appl. Pharmcol. 380, 114705. https://doi.org/10.1016/j.taap.2019.114705 (2019).
Lalaoui, N. & Vaux, D. L. Recent advances in understanding inhibitor of apoptosis proteins. F1000Research 7 (2018). https://doi.org/10.12688/f1000research.16439.1
Gloire, G., Legrand-Poels, S. & Piette, J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem. Pharmacol. 72, 1493–1505. https://doi.org/10.1016/j.bcp.2006.04.011 (2006).
Tsuruta, F. et al. JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. EMBO J. 23, 1889–1899. https://doi.org/10.1038/sj.emboj.7600194 (2004).
Kuida, K. Caspase-9. Int. J. Biochem. Cell Biol. 32, 121–124. https://doi.org/10.1016/s1357-2725(99)00024-2 (2000).
Park, H. & Jeoung, N. H. Inflammation increases pyruvate dehydrogenase kinase 4 (PDK4) expression via the Jun N-Terminal kinase (JNK) pathway in C2C12 cells. Biochem. Biophys. Res. Commun. 469, 1049–1054. https://doi.org/10.1016/j.bbrc.2015.12.113 (2016).
Acknowledgements
The authors thank all the participants of this study. The authors also thank Miki Takezawa and Ai Yoshimura for their valuable assistance during the study.
Author information
Authors and Affiliations
Contributions
HK and K Kamiyama designed the experiments. K Kamiyama, TI, IT, MK, and TS performed in-vitro and animal experiments. HK and NT analyzed the RNA-seq data. HK and K Kasumo interpreted the data. HK and MI wrote and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kimura, H., Kamiyama, K., Imamoto, T. et al. Dichloroacetate reduces cisplatin-induced apoptosis by inhibiting the JNK/14-3-3/Bax/caspase-9 pathway and suppressing caspase-8 activation via cFLIP in murine tubular cells. Sci Rep 14, 24307 (2024). https://doi.org/10.1038/s41598-024-75229-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-75229-z
Keywords
This article is cited by
-
Large-sized polystyrene microplastics induce oxidative stress in AML12 cells
Scientific Reports (2025)
