
Abstract
DNA methylation is a molecular process that mediates gene-environment interactions. Epigenome-wide association studies (EWAS) using the Illumina Human Methylation BeadChip are powerful tools for quantifying the relationship between DNA methylation and phenotypes. Recently, the Illumina Methylation EPICv2 BeadChip (EPICv2) was released, which includes new features, such as duplicated probes and changed probe names. Several published algorithms have been updated to address these features in EPICv2. However, appropriate EPICv2 preprocessing and integration with previous microarray versions remain complex. Therefore, MethylCallR, an open-source R package designed to provide standard procedures for performing EWAS using Illumina methylation microarrays including EPICv2, was developed. MethylCallR can be used to control duplicated probes in EPICv2, by using pre-set data implemented in MethylCallR or new customized data. MethylCallR includes a straightforward conversion function between different types of Illumina Human Methylation BeadChips. Using MethylCallR, potential outlier sample detection and statistical power estimation were conducted and used to select meaningful probes. Publicly available data was analyzed using MethylCallR and the findings were compared to that of a previous study.
Similar content being viewed by others
Introduction
DNA methylation is one of the most well-studied molecular process that is involved in environmental factors and gene-environment interactions1,2. In recent years, significant associations have been identified between variations in DNA methylation and various phenotypes (e.g., aging, body mass index, smoking status, and various diseases)3,4,5,6. Epigenome-wide association studies (EWAS) are among the most powerful methods used for statistically assessing the associations between epigenetic variations across the genome and various phenotypes7. Microarrays, the most commonly used platform for conducting EWAS, are popular owing to their low resource consumption, high reproducibility, and comprehensive genome coverage8. The Illumina Human Methylation BeadChip is the primary platform chosen by researchers since its reliability. Because of its popularity, the Illumina Methylation EPICv2 BeadChip (EPICv2) has recently been released as a successor to the Illumina Methylation EPIC BeadChip (EPICv1) and Illumina Methylation 450 K BeadChip (450 K). With the powerful advantages of microarrays and improved performance, EPICv2 is expected to become the primary choice for researchers planning to analyze DNA methylation9,10,11.
Unlike EPICv1 and 450 K, EPICv2 requires several new feature considerations for analysis: (i) exactly duplicated probes, (ii) duplicated probes in the probe sequence, (iii) duplicated probes in position, and (iv) probes with different names compared to previous versions. Controlling duplicate probes is important when interpreting results or performing meta-analyses using data from EPICv1 or 450 K. Although detailed descriptions and functional testing of duplicated probes have been reported by Peters et al. fully controlling the replicated probes remains a challenge10. In particular, the reliance on external reference datasets for analysis, such as reference-based cell composition estimation, has become more complicated12,13. Considering that the available algorithms are primarily optimized for EPICv1, it is important to apply them to EPICv2.
To date, well-structured popular pipelines have been widely used to conduct EWAS, which are designed to facilitate the easy utilization of numerous algorithms14,15,16,17,18. Meffil is an EWAS pipeline that includes solutions for efficient analysis of large-scale DNA methylation data18. DMRcate algorithm utilizes Gaussian kernel smoothing to identify significantly differentially methylated regions (DMRs)19. Conumee2.0 is an algorithm that includes useful functions for identifying significant copy number variations, such as tangent normalization, circular binary segmentation, and segmented block bootstrapping20. Although these algorithms have recently been updated to be compatible with EPICv2, only a few EWAS pipelines support the analysis of EPICv2.
This study aims to develop MethylCallR, a pipeline that will consist of modular-designed functions to easily convert array versions (EPICv2, EPICv1, and 450 K) using an address chain file, identify potential outlier samples, and calculate the statistical power for the comprehensive analysis of DNA methylation using Illumina microarrays. MethylCallR pipeline will comprise of standard procedures that include recent algorithms for conducting EWAS and several helpful functions to assist users. MethylCallR can potentially be applied to all epigenetic studies and significantly contributes to enhancing the quality of the results. In this manuscript, we will provide descriptions of several functions included in MethylCallR and a summary of the EWAS results using publicly available data.
Results
Running time
MethylCallR comprises wrapper functions that include various algorithms for comprehensive EWAS analysis and helper functions to support them. GSE167202, which consists of 460 samples, was analyzed following standard procedures and outlier sample detection using a single core on an Intel Xeon 3.10 GHz CPU21. By importing data to estimate the cell-type composition, the preprocessing step was 2 h and 32 min. Because the simulations were time-consuming, 20 h and 43 min were required to estimate the statistical power of the EWAS. Additionally, differentially methylated and functional enrichment analyses required 15 min.
Performance of outlier sample detection
It was observed that more probes with actual changes were consistently detected in all cases after removing the detected outlier samples (Fig. 1). This suggests that detecting and removing high-risk outlier samples is important for identifying meaningful DMPs at small effect-size intervals. For each iteration, randomly selected outlier samples were detected using the Mahalanobis distance method (Fig. 2). However, because of the complex interactions between multiple effects in actual data, careful consideration was required to remove outlier samples that were close to the data distribution.

The line plot illustrates the number of detected t-DMPs for each interval of effect size compared to b-DMPs. The x-axis represents the intervals of added effect size, while the y-axis represents the ratio of the number of t-DMPs detected under each condition divided by the number of t-DMPs detected by the b-DMP. The error bar indicates the standard deviation among 60 iterations. Each condition was calculated separately: (A) randomly selected 495 case and 497 control samples with small effect size (mean ± sd, 0.04 ± 0.024; number of outlier samples = 10), (B) randomly selected 495 case and 497 control samples with large effect size (mean ± sd, 0.092 ± 0.054; number of outlier samples = 10), and (C) randomly selected 50 case and 50 control samples with small effect size (mean ± sd, 0.04 ± 0.024; number of outlier samples = 5).

The bar plot shows the proportion of randomly selected outliers detected across all iterations. During the iteration, samples that are identified as outlier in more than 20% of the iterations are designated as “obvious outliers”, while samples identified as outliers in 5–20% of the iterations are designated as “suggestive outliers”. “Non-outlier” is a sample detected as an outlier that is not randomly selected outlier sample. (A) randomly selected 495 case and 497 control samples with small effect size (mean ± sd, 0.04 ± 0.024; number of outlier samples = 10), (B) randomly selected 495 case and 497 control samples with large effect size (mean ± sd, 0.092 ± 0.054; number of outlier samples = 10), (C) randomly selected 50 case and 50 control samples with small effect size (mean ± sd, 0.04 ± 0.024; number of outlier samples = 5).
Differential methylation analysis
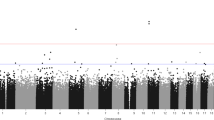
To test MethylCallR, we identified DMPs following standard procedures and outlier sample detection provided by MethylCallR using public data downloaded from GEO (GEO167202), which consisted of EPICv1 data from the peripheral blood of 164 patients with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-positive (Cov+) and 296 SARS-CoV-2-negative (Cov-) participants21. Raw IDAT files of 460 samples were loaded, and quality control was performed. During the quality control step, six samples with low median signal intensity and 106,725 probes, including 990 flagged and 38 mapping failed probes, were removed. BMIQ normalization was performed to control for bias between probe types, and outlier sample detection was performed to identify potential outlier samples22. Two samples were identified as potential outliers and were removed (Fig. 3A). In summary, 759,193 probes from 163 patients with Cov + and 289 Cov- participants were included in the downstream analysis. The known technical batch effects were assessed by correlating the batches and principal components of the DNA methylation data (Fig. 3B), and the array and slide effects were controlled by applying the ComBat algorithm23. The composition of the six types of white blood cells was estimated for each sample based on the reference data implemented in the Flowsorted.Blood.EPIC R package12. Age, sex, race, and white blood cell composition were included as covariates in the differential methylation analysis. Based on statistical power estimation using the pwrEWAS R package (Fig. 3C), 144 significant DMPs were identified (FDR ≤ 0.05, |Δβ| ≥ 0.05) (Supplementary Table S1)24. The top-ranked DMPs were associated with genes that have previously been reported to be linked to SARS-CoV-2 infection, such as Interferon regulatory factor 7 (IRF7), poly ADP-ribose polymerase 9 (PARP9), and human myxovirus resistance gene 1 (MX1) (Table 1)25,26,27. To determine the gene sets that were enriched with significant DMPs, functional enrichment analysis was performed using the gometh algorithm implemented in the missMethyl R package28. The genes associated with DMPs were significantly enriched in the gene sets related to viral infection and the innate immune response (Table 2). Finally, Manhattan and volcano plots were generated to summarize the EWAS results (Fig. 3D and E). Following the standard procedures of MethylCallR, we identified the DMPs and associated genes reported in the original study by Konigsberg et al., such as oligoadenylate synthetase 1 (OAS1), type I interferon-induced proteins with tetratricopeptide repeats 3 (IFIT3), IRF7, PARP9, and MX121. In particular, DMPs located in genes associated with SARS-CoV-2 infection, which were not identified as significant in the original study, were also identified (adenosine deaminase acting on RNA (ADAR), Filamin B (FLNB), and Scavenger Receptor Class B Member 1 (SCARB1)). ADARis involved in adenosine-to-inosine RNA editing29. This event is closely associated with viral RNA infections and multiple A-to-G substitutions have been observed in the transcriptome of SARS-CoV-230,31. FLNBis a cytoplasmic protein that regulates communication between cytoskeletal networks and connects the actin network to the cell membrane32. The dysregulation of six RNA-binding proteins, including FLNB, has been reported in SARS-CoV-2-infected primary human lung epithelial cells33. SCARB1is involved in the cellular uptake of cholesterol into cells34. It is involved in the intracellular entry of SARS-CoV-2, along with angiotensin-converting enzyme 2 (ACE2)35. These interactions may be influenced by epigenetic alterations in these three genes.

(A) The PCA plot which represents the potential outlier samples and the boundary of the outlier samples for each group. (B) A heatmap illustrating the effect of batches and covariates on DNA methylation data. Blue tiles represent P-values of 0.05 or lower. Darker blue tiles indicate smaller P-values. The Spearman correlation coefficient is displayed in the center of each tile. (C) A line plot shows the statistical power for given sample sizes and effect sizes generated by the pwrEWAS R package24. (D) Manhattan plot. The x-axis represents the chromosomes, the y-axis represents the -log10 scaled P-value, and the significant P-value threshold was indicated by a horizontal red line. Multiple probe sets could be highlighted in different colors. Significant DMPs are highlighted in green, while the top 20 DMPs are highlighted in blue. Among the top 20 DMPs, those annotated to genes associated with SARS-CoV-2 infection are represented in red. (E) Volcano plot. The x-axis represents Δβ, and the y-axis represents -log10 scaled P-value. The red horizontal line indicates the significance threshold for the P-value, while the dark gray vertical line indicates the threshold for Δβ. The genes associated with the top 20 DMPs are labeled. PCA, principal component analysis; DMP, differentially methylated probe; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; Δβ (delta-beta), the average beta value of patients minus the average beta value of controls.
Discussion
MethylCallR is a comprehensive pipeline that provides modular functions for analyzing DNA methylation data generated by Illumina methylation microarrays. Simple and straightforward functions allow non-experts to easily conduct analyses, whereas detailed parameters allow experienced users to customize the analysis according to the study aim. MethylCallR helps users to achieve high-quality analysis by offering various options through the following major features (Supplementary Table S2): (i) MethylCallR provides a guide for controlling duplicated probes in EPICv2 and allow for integration with previous versions of the microarray (EPICv1 and 450 K), (ii) an outlier sample detection algorithm based on Mahalanobis distance can effectively identify and remove potential outlier samples within groups36, (iii) MethylCallR offers wrapper functions that integrate several recent statistical algorithms and enable compatibility with EPICv2 data, and (iv) MethylCallR uses ggplot2 to generate various EWAS summary plots and provides customizable probe highlighting option37. It is expected that MethylCallR will enhance the accessibility of the analysis by offering a user-friendly environment and improving the quality of the results. MethylCallR can be applied to all studies where an Illumina human methylation microarray is available.
Although MethylCallR is expected to be widely used in various studies owing to its multiple strengths, several limitations still need to be addressed. First, the analytical methods for differential methylation analyses are limited. MethylCallR supported differential methylation analysis between the two groups. Other diverse analyses are important because they may offer additional epigenetic clues and significant insights (e.g., multiple group comparisons, differentially variable probes, methylation risk scores, and survival analysis). Given this, there is a plan to include additional functions to perform various analyses using continuous updates. Second, the estimation of cell-type composition in solid tissues can be performed for a limited number of tissue types. However, there is an option to apply the user reference matrix using the “deconvolution” functions implemented in the deconvR R package38. Finally, the criteria for controlling duplicate probes in EPICv2, as provided in the MethylCallR data file, may not always be optimal for all studies. The criteria suggested by MethylCallR prioritize the complete removal of duplicate probes and their compatibility with EPICv1 and 450 K. However, if users have a list of probes they wish to remove, this list can be uploaded to the “MeCall.SetChainFile” function implemented in MethylCallR.
In summary, we have shown that MethylCallR includes straightforward functions for analyzing EPICv2 data and provides an environment to conduct comprehensive EWAS analysis. Users might identify potential outliers and calculate the statistical power of EWAS to select more meaningful epigenetic loci. We believe that MethylCallR could significantly contribute to EWAS studies using the Illumina Human Methylation BeadChips.
Methods
MethylCallR
MethylCallR includes standard procedures from the input of IDAT files to the summary of EWAS, and optional procedures to assist in the interpretation of data (Fig. 4). Standard procedures include data import, quality control, normalization, technical batch effect adjustment, and differential methylation analysis. Optional procedures include outlier sample detection, principal component analysis, generation of EWAS summary plots, and functional enrichment analysis. Owing to its modular design, users can select functions or follow the process recommended by MethylCallR (indicated by the solid lines in Fig. 4) to perform the analysis.

The workflow diagram of MethylCallR.
To develop MethylCallR, four datasets were used: (i) a public dataset downloaded from the Gene Expression Omnibus (GEO) consisting a total of 460 samples (GSE167202)21, (ii) EPICv1 simulation data to evaluate the performance of outlier sample detection39, (iii) demo EPICv2 data downloaded from the Illumina official website (https://support.illumina.com/array/array_kits/infinium-methylationepic-beadchip-kit/downloads.html), and (iv) a public dataset downloaded from GEO (GSE240469) to test the applicability of MethylCallR on EPICv2 data10.
Preprocessing data
MethylCallR supports the analysis of the EPICv1, EPICv2, and 450 K arrays. MethylCallR imports the IDAT files from the sample directory using the Illuminaio R package40 and generates the objects required for quality control and downstream analysis. Low-quality samples and probes are removed based on the following criteria which is included in the minfi and ChAMP R packages14,15, such as (i) samples with low median log-scaled signal intensity (≤ 10.5), (ii) samples with a high proportion of probes failing the detection P-value (≥ 10%), (iii) probes with a high detection P-value (> 0.01), (iv) probes with fewer than three bead counts in a high proportion of samples (≥ 5%), (v) non-cg probes, (vi) single-nucleotide polymorphism (SNP) associated probes9,41, (vii) ambiguous alignment probes10,42, and (viii) probes not located on the autosomes. MethylCallR includes two additional filtering criteria based on Illumina manifests: (i) flagged and (ii) mapping failed probes. A flagged probe has lower reproducibility, as identified by the manufacturer. The mapping failed probes have unclear genomic positions based on the human reference genome hg38/GRCh38 (hg38). After that, normalization was performed using the 6 available methods (beta-mixture quantile normalization (BMIQ), functional normalization, subset-quantile within array normalization (SWAN), Exponential-Normal mixture signal intensity background correction (ENmix), dasen, and normal-exponential out-of-band normalization (Noob))16,22,43,44,45,46. Normalization can be sequentially performed by specifying one or more algorithms in the method parameter. However, functional normalization, SWAN, ENmix, and Noob can only be utilized as the initial method due to compatibility with the input format. To identify potential outlier samples, outlier sample detection was performed on the dataset based on the user’s decision. To control for technical batch effects, the ComBat algorithm was used to identify known batch effects using a correlation heat map, and Surrogate Variable Analysis was used to identify unknown batch effects23,47. As the last preprocessing step, the composition of six types of white blood cells was estimated based on a previous study (CD8 T-cells, CD4 T-cells, natural killer cells, B-cell, monocytes, and neutrophils)12. MethylCallR was developed to include a wrapper function that estimates cell type proportions for 20 types of solid tissues using references embedded in the EpiSCORE and deconvR R packages (bladder, brain, breast, colon, esophagus, heart, kidney, liver, lung, pancreas, prostate, skin, olfactory epithelium, erythrocytes, adipocytes, vascular, larynx, thyroid, upper gastrointestinal, and cervix)38,48.
For EPICv2 data preprocessing, controlling duplicated probes is vital. To simplify and facilitate it, MethylCallR provides preset data that includes duplicated probes to be removed based on two criteria (Fig. 5): (i) probes that are exactly same with the probes from previous versions (EPICv1 or 450 K), and (ii) superior probes based on previous research (with “superior” and “precision” as a keyword)10. If there are no probes exactly the same as in the previous versions in the duplicated probe set, the second condition is exclusively followed.

This diagram illustrates the method used by MethylCallR to account for duplicated probes in EPICv2. The process involves selecting unique probes (green box) based on the criteria of MethylCallR (red dotted box) from all duplicated probes (gray box). The remaining probes are used to generate the Address_chain_file. A list of the removed probes can be found in Duplicated.Probes.preset (orange box). A summary of the duplicated probes is provided in the gray box ((i) exactly duplicated probes (11,354 probes), (ii) duplicated probes in the probe sequence (1,000 probes), and (iii) duplicated probes in position (268 probes)).
For rapid and accurate array conversion (EPICv2, EPICv1, and 450 K), MethylCallR regenerates the “address chain file” based on the user-provided duplicated probe list. The address chain file is a modified data file in which each row contains the probe addresses and Illumina probe IDs of the matching probes between EPICv2, EPICv1, and 450 K. This file was created based on the Illumina manifest file provided on the official website of Illumina (https://support.illumina.com/?tab=microarrays). To ensure compatibility between EPICv2 and previous versions, functions were included in MethylCallR to easily convert array versions for various object types, including minfi objects, using an address chain file. This function performs the conversion using only the shared probes between different versions of the array.
Outlier sample detection
Quality control of samples primarily consider the median of the log-scaled signal intensity or the proportion of low-quality probes14,15. Because these approaches do not consider the distribution of samples within a group, potential outlier samples of good quality may be missed. In MethylCallR, potential outlier samples were identified using Mahalanobis distance from the subsamples within each group36. In each iteration, a sample that fell outside the confidence region of the chi-squared distribution was considered an outlier sample. Using the EPICv1 simulation data, the performance of the proposed method was evaluated. We used the EPICv1 simulation data provided in Tristan Zindler’s previous study to test the effectiveness of Mahalanobis distance method39. Using data generator R code, we generated virtual samples of n = 992 and n = 100 separately with technical variability of EPICv1. We simulated the following conditions: (i) randomly selected 495 case and 497 control samples with small effect size (mean ± sd, 0.04 ± 0.024; number of outlier samples = 10), (ii) randomly selected 495 case and 497 control samples with large effect size (mean ± sd, 0.092 ± 0.054; number of outlier samples = 10), and (iii) Randomly selected 50 case and 50 control samples with small effect size (mean ± sd, 0.04 ± 0.024; number of outlier samples = 5). No significant DMPs by chance was identified in all conditions (FDR = 1 at all probes). Each condition was tested in the following steps: Step1) random effect sizes were assigned to randomly selected probes (approximately 10% of all probes) of case samples. These probes are defined as True-DMPs. Then, significant DMPs were re-calculated and defined as Baseline-DMPs (FDR ≤ 0.05, |Δβ| ≥ 0.02), Step2) in order to generate outlier samples, random effect sizes were assigned to original DNA methylation level of randomly selected probes from randomly selected samples. Then, significant DMPs were identified as DMPs-with-outlier (FDR ≤ 0.05, |Δβ| ≥ 0.02), Step3) after outliers were identified and removed using Mahalanobis distance, DMPs were re-calculated and defined as DMPs-without-outlier (FDR ≤ 0.05, |Δβ| ≥ 0.02), and Step4) we compare the number of True-DMPs (t-DMP) included in Baseline-DMP (b-DMP), DMPs-with-outlier (ow-DMP), and DMPs-without-outlier (ow/o-DMP) within each delta-beta interval and count the number of correctly detected outliers during all iterations. To assist users, MethylCallR returns ggplot2 objects that visualize the distribution of samples within each group and boundaries for outliers37.
Differentially methylated analysis
Based on preprocessed data, differential methylation analysis at both the probe and regional levels can provide valuable epigenetic insights. The limma R package in MethylCallR was used to identify differentially methylated probes (DMPs)49. The genomic positions of all probes were based on hg38 (1-based). Importantly, a statistical power estimation can be conducted to set appropriate delta-beta (Δβ) thresholds and allow users to select more reliable epigenetic loci24. To identify DMRs, five algorithms were implemented in MethylCallR (Bumphunter, DMRcate, seqlm, dmrff, and combined-pvalues)19,50,51,52,53.
In addition, the user can perform functional enrichment analysis to identify gene sets enriched with significant DMPs. MethylCallR can be performed using the gometh algorithm, which considers complex relationships between CpG sites and genes through Wallenius non-central hypergeometric distribution28. This function conducts an analysis based on the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases, and consolidates significant results54,55.
Copy number variation analysis
MethylCallR provides a unified function that starts with the preprocessing method provided by the ChAMP and Conumee R packages, and includes copy number segmentation using the circular binary segmentation algorithm15,20,56. This function returns the segmentation results and marker data required to use GISTIC 2.0, which is useful for identifying recurrent focal alterations57.
Plot generations
MethylCallR generates high-quality plots using the ggplot2 R package to summarize EWAS results37. Manhattan, volcano, and stacked bar plots were generated to represent the ratios of genic and CpG annotations for the DMPs. In a Manhattan plot, the user can provide a list of objects to simultaneously highlight multiple probe sets in different colors. Additionally, gene names can be labeled to annotate specific CpG sites.
Data availability
MethylCallR is publicly available on Github (https://github.com/Yang9704/MethylCallR). The manuals for testing MethylCallR are publicly available on Github (https://github.com/Yang9704/MethylCallR/tree/main/doc). Test datasets for validating MethylCallR can be downloaded from the NCBI GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE167202; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE240469). The essential data for simulating performance of outlier sample detection are available at https://doi.org/10.1186/s12859-020-03559-6.
References
Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21 (2002).
Mattei, A. L., Bailly, N. & Meissner, A. DNA methylation: a historical perspective. Trends Genet. 38, 676–707 (2022).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, 3156 (2013).
Wahl, S. et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 541, 81–86 (2017).
Zeilinger, S. et al. Tobacco Smoking Leads to Extensive Genome-Wide Changes in DNA Methylation. PLOS ONE. 8, e63812 (2013).
Jin, Z. & Liu, Y. DNA methylation in human diseases. Genes Dis. 5, 1–8 (2018).
Campagna, M. P. et al. Epigenome-wide association studies: current knowledge, strategies and recommendations. Clin. Epigenetics. 13, 214 (2021).
Pidsley, R. et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 17, 208 (2016).
Kaur, D. et al. Comprehensive evaluation of the Infinium human MethylationEPIC v2 BeadChip. Epigenetics Commun. 3, 6 (2023).
Peters, T. J. et al. Characterisation and reproducibility of the HumanMethylationEPIC v2.0 BeadChip for DNA methylation profiling. BMC Genom. 25, 251 (2024).
Noguera-Castells, A., García-Prieto, C. A., Álvarez-Errico, D. & Esteller, M. Validation of the new EPIC DNA methylation microarray (900K EPIC v2) for high-throughput profiling of the human DNA methylome. Epigenetics. 18, 2185742 (2023).
Salas, L. A. et al. An optimized library for reference-based deconvolution of whole-blood biospecimens assayed using the Illumina HumanMethylationEPIC BeadArray. Genome Biol. 19, 64 (2018).
Houseman, E. A. et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 13, 86 (2012).
Aryee, M. J. et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 30, 1363–1369 (2014).
Tian, Y. et al. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics. 33, 3982–3984 (2017).
Xu, Z., Niu, L., Li, L. & Taylor, J. A. ENmix: a novel background correction method for Illumina HumanMethylation450 BeadChip. Nucleic Acids Res. 44, e20 (2016).
Müller, F. et al. RnBeads 2.0: comprehensive analysis of DNA methylation data. Genome Biol. 20, 55 (2019).
Min, J. L., Hemani, G., Davey Smith, G., Relton, C. & Suderman, M. Meffil: efficient normalization and analysis of very large DNA methylation datasets. Bioinforma Oxf. Engl. 34, 3983–3989 (2018).
Peters, T. J. et al. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 8, 6 (2015).
Daenekas, B. et al. Conumee 2.0: enhanced copy-number variation analysis from DNA methylation arrays for humans and mice. Bioinformatics. 40, btae029 (2024).
Konigsberg, I. R. et al. Host methylation predicts SARS-CoV-2 infection and clinical outcome. Commun. Med. 1, 1–10 (2021).
Teschendorff, A. E. et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 29, 189–196 (2013).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 8, 118–127 (2007).
Graw, S., Henn, R., Thompson, J. A. & Koestler, D. C. pwrEWAS: a user-friendly tool for comprehensive power estimation for epigenome wide association studies (EWAS). BMC Bioinform. 20, 218 (2019).
Zhang, Q. et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 370, eabd4570 (2020).
Obando-Pereda, G. Can molecular mimicry explain the cytokine storm of SARS-CoV-2? An in silico approach. J. Med. Virol. 93, 5350–5357 (2021).
Bizzotto, J. et al. SARS-CoV-2 Infection Boosts MX1 Antiviral Effector in COVID-19 Patients. iScience. 23, 101585 (2020).
Maksimovic, J., Oshlack, A. & Phipson, B. Gene set enrichment analysis for genome-wide DNA methylation data. Genome Biol. 22, 173 (2021).
Kim, D. D. Y. et al. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 14, 1719–1725 (2004).
Samuel, C. E. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology. 411, 180–193 (2011).
Di Giorgio, S., Martignano, F., Torcia, M. G., Mattiuz, G. & Conticello, S. G. Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci. Adv. 6, eabb5813 (2020).
Stossel, T. P. et al. Filamins as integrators of cell mechanics and signalling. Nat. Rev. Mol. Cell. Biol. 2, 138–145 (2001).
Srivastava, R., Daulatabad, S. V., Srivastava, M. & Janga, S. C. Role of SARS-CoV-2 in Altering the RNA-Binding Protein and miRNA-Directed Post-Transcriptional Regulatory Networks in Humans. Int. J. Mol. Sci. 21, 7090 (2020).
Rigotti, A., Miettinen, H. E. & Krieger, M. The role of the high-density lipoprotein receptor SR-BI in the lipid metabolism of endocrine and other tissues. Endocr. Rev. 24, 357–387 (2003).
Wei, C. et al. HDL-scavenger receptor B type 1 facilitates SARS-CoV-2 entry. Nat. Metab. 2, 1391–1400 (2020).
Mahalanobis, P. C. On the generalized distance in Statistics. (1936).
Wickham, H. Ggplot2: Elegant Graphics for Data Analysis (Springer, 2009). https://doi.org/10.1007/978-0-387-98141-3
Moss, J. et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 9, 5068 (2018).
Zindler, T., Frieling, H., Neyazi, A., Bleich, S. & Friedel, E. Simulating ComBat: how batch correction can lead to the systematic introduction of false positive results in DNA methylation microarray studies. BMC Bioinform. 21, 271 (2020).
Smith, M. L., Baggerly, K. A., Bengtsson, H., Ritchie, M. E. & Hansen, K. illuminaio: An open source IDAT parsing tool for Illumina microarrays. F1000Research 2, 264 (2013).
Zhou, W., Laird, P. W. & Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 45, e22 (2017).
Nordlund, J. et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 14, r105 (2013).
Maksimovic, J., Gordon, L. & Oshlack, A. S. W. A. N. Subset-quantile Within Array Normalization for Illumina Infinium HumanMethylation450 BeadChips. Genome Biol. 13, R44 (2012).
Fortin, J. P. et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 15, 503 (2014).
Pidsley, R. et al. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genom. 14, 293 (2013).
Triche, T. J. Jr, Weisenberger, D. J., Van Den Berg, D., Laird, P. W. & Siegmund, K. D. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 41, e90 (2013).
Leek, J. T. & Storey, J. D. Capturing Heterogeneity in Gene Expression Studies by Surrogate Variable Analysis. PLOS Genet. 3, e161 (2007).
Teschendorff, A. E., Zhu, T., Breeze, C. E. & Beck, S. EPISCORE: cell type deconvolution of bulk tissue DNA methylomes from single-cell RNA-Seq data. Genome Biol. 21, 1–33 (2020).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Jaffe, A. E. et al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int. J. Epidemiol. 41, 200–209 (2012).
Kolde, R., Märtens, K., Lokk, K., Laur, S. & Vilo, J. seqlm: an MDL based method for identifying differentially methylated regions in high density methylation array data. Bioinformatics. 32, 2604–2610 (2016).
Suderman, M. et al. dmrff: identifying differentially methylated regions efficiently with power and control. 508556 Preprint at (2018). https://doi.org/10.1101/508556
Pedersen, B. S., Schwartz, D. A., Yang, I. V. & Kechris, K. J. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics. 28, 2986–2988 (2012).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Kanehisa, M. & Goto, S. K. E. G. G. Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Olshen, A. B., Venkatraman, E. S., Lucito, R. & Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 5, 557–572 (2004).
Mermel, C. H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41 (2011).
Acknowledgements
This research was supported by an Incheon National University Research Grant in 2023.
Author information
Authors and Affiliations
Contributions
HHY contribute to methodology, software, investigation, writing of original draft, and visualization. MRH contribute to conceptualization, funding acquisition, data curation, resources, review and editing of original draft, and supervision. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, HH., Han, MR. MethylCallR : a comprehensive analysis framework for Illumina Methylation Beadchip. Sci Rep 14, 27026 (2024). https://doi.org/10.1038/s41598-024-77914-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-77914-5
