
Abstract
Racing performance traits are the main indicators for evaluating the performance and value of sport horses. The aim of this study was to identify the key genes for racing performance traits in Yili horses by performing a genome-wide association study (GWAS). Breeding values for racing performance traits were calculated for Yili horses (n = 827) using an animal model. Genome-wide association analysis of racing performance traits in horses (n = 236) was carried out using the Blink, and FarmCPU models in GAPIT software, and genes within the significant regions were functionally annotated. The results of GWAS showed that a total of 24 significant SNP markers (P < 6.05 × 10− 9) and 22 suggestive SNP markers (P < 1.21 × 10− 7) were identified. Among them, the Blink associated 16 significant SNP loci and FarmCPU associated 12 significant SNP loci. A total of 127 candidate genes (50 significant) were annotated. Among these, CNTN6 (motor coordination), NIPA1 (neuronal development), and DCC (dopamine pathway maturation) may be the main candidate genes affecting speed traits. SHANK2 (neuronal synaptic regulation), ISCA1 (mitochondrial protein assembly), and KCNIP4 (neuronal excitability) may be the main candidate genes affecting ranking score traits. A common locus (ECA1: 22698579) was significantly associated with racing performance traits, and the function of the genes at this locus needs to be studied in depth. These findings will provide new insights into the detection and selection of genetic variants for racing performance and will help to accelerate the genetic improvement of Yili horses.
Similar content being viewed by others
Introduction
Regional preferences for certain traits have resulted in phenotypic variation, which may result from adaptations to the local racing ecosystem1. Although racing traits are complex, selecting racehorses with traits common to winners in a given environment for breeding can increase the probability of genetic variation in those traits in the offspring. Over time, systematic selection can optimize the population’s genome2. Therefore, knowledge of association between traits and influential genotypes will help breeders produce healthier more sustainable, and better-performing horses3.
Equine research and breeding have encountered major changes due to the rapid development of molecular genetics technology4,5. Genome-wide association studies (GWAS) have been successfully deployed to identify quantitative trait loci (QTLs) for complex traits using relatively modest sample sizes6,7,8. Today, there are 302 horse traits listed on the Online Mendelian Inheritance in Animals (OMIA) website, while the HorseQTLdb lists 2216 QTLs representing 61 traits. A total of 431 QTLs were identified as being related to racing ability, gait, and jumping ability of horses. These key genetic markers offer the possibility of applications for genetic testing and selection in horses9,10,11,12.
The thoroughbred, developed relatively quickly over the last three centuries through crossbreeding of local British mares with Middle Eastern stallions, has become the world’s most successful racehorse. Most thoroughbreds compete in races over much shorter distances (1000–3200 m) and are bred for both speed and stamina13. Fewer founders, large populations, stronger selection pressures, and lower genetic diversity make the racing traits and genomic structure of Thoroughbreds suitable for study13,14,15. A wealth of genetic information related to racing distance16, speed3, rankings17, and longevity of participation18 have been reported. Today, thoroughbreds are widely used to improve the racing performance of other horse breeds.
The Yili horse, originating from the Yili Kazakh Autonomous Prefecture in the Xinjiang Uygur Autonomous Region of China, was developed during the last century by crossing native Kazakh mares with stallions of Orlov, Budyonny, and Don River breeds19. To meet different production needs, there are several phenotypically and genetically distinct subgroups of Yili horses that are used for meat, milk, and racing. The Yili racehorse group includes several types of galloping, trotting and pacing. The gallop type was developed through the crossbreeding of Yili mares with thoroughbred stallions, to combine their best qualities20. This group has become one of the most influential horse racing groups in China due to the standardized holding of racing events and breeder preferences. Horse racing is held annually during the Xinjiang Tianma Cultural and Tourism Festival, where young (2–3-year-old) and adult Yili horses compete in races over short, medium, and long distances (1000–5000 m) and are bred for both speed and stamina attributes.
In recent years, the racing performance of the Yili horse has been improving from the crossbreeding with thoroughbred stallions and the strong selection for racing capability. The focus of our team’s research has gradually shifted from analyzing physiological and biochemical indicators in racehorses to genomics21,22,23. Previous studies found some key polymorphisms in the MSTN, GH, DMRT3, COMT genes and sought to determine the relationship between these genes and body size, gait, racing performance and cardiac function24,25,26,27. We obtained some inferential conclusions but lacked large data samples to demonstrate significant effects. Consequently, in the present study, we hypothesized that the enhancement of racing performance in the racing population of Yili horses is genetically influenced by thoroughbred stallions and that there are some genes or genomic regions associated with race performance traits. Therefore, we first analyzed the phenotypic data of Yili racehorses (gallop type), using the breeding values of race performance traits of Yili horses and thoroughbred stallions as the phenotypic data. Genotype data were obtained through 5x and 10x whole-genome resequencing. Lastly, GWAS technology was used to identify genetic markers that were closely related to racing performance, which provides a reference for the selective breeding of Yili horses.
Materials and methods
Experimental animals and phenotypic data
The studied populations consisted of Yili horse (gallop types, n = 827), and thoroughbreds (studs, n = 134) from Xinjiang Uygur Autonomous Region, Northwest China. A total of 2576 flat racing records and 12,546 g-pedigree data entries from 827 Yili horses for 9 years (2015 to 2023) were used to estimate breeding values for racing performance traits. Based on the tracing of the horse information, a total of 212 Yili horses (118 stallions and 94 mares) with qualified race records (n ≥ 6) and 41 Thoroughbreds (studs) with progeny numbers (n ≥ 50) were selected for DNA re-sequencing. The sequencing depth was 5X and 10X, respectively.
Racing performance data collection was carried out from February to November each year using a standardized 2000 m sand track and electronic timing system. The study traits were as follows: (1) Average speed (AS) was the average speed of the horse completing the race. Ranking score (RS) was the sum of the ranking score of the horses in the competition and the time gap between the horse and the leading horse using the following formula: RS = ( K - KX )*100 + ( RTX - RTF ), where K is the total number of horses in the race; KX is the ranking of the x horse; RTX is the race time of the x horse, and RTF is the race time of the winning horse.
Estimated breeding values
The significance (P < 0.05) of the fixed effects of racing performance traits in Yili horses was tested using the GLM process (SAS 8.1). We considered that the age of racing, racing distance, year of birth, gender of horse, month of racing, and level of racing to be fixed effects, and individual additive genetic effects as random effects. The results of descriptive statistics (Tables S1) and fixed effects significance test (Tables S3-9) are provided in the Supplementary Material.
The estimates of genetic parameters and breeding values (EBVs) for the speed and ranking score traits were determined using the single trait repeatability model from the DMU software. The genetic and phenotypic correlations were determined using multi-trait animal models (DMUs), and the standard errors (SEs) of genetic and phenotypic correlations were estimated using the method of Klei and Tsuruta28. The single trait breeding values + residuals as phenotypic values29 were used to carry out the GWAS. Thesingle trait repeatability model equation is as follows:
where Y is the vector of observations, β is the vector of fixed effects, a is the vector of additive genetic effects, pe is the vector of permanent environmental factors of individuals for speed and ranking score trait, and e is the vector of residuals. X and Z are the incidence matrices corresponding to fixed and additive effects, respectively, and W is the permanent environmental effect incidence matrix.
DNA resequencing data
Blood samples from the Yili horses (n = 212) and Thoroughbreds (n = 41) were collected with the owner’s consent between 2021 and 2023. DNA was extracted from the blood samples using a GenoPrep animal tissue DNA extraction kit with magnetic beads (Mix-V4.0, Boridi, Hebei, China). The DNA fragments were end-repaired, A-tailed, adaptor-ligated, and amplified using the Dongshengxing ETC821 bioanalyzer (Dongsheng, Jiangsu, China, USA). QC of DNA samples was performed by agarose gel electrophoresis to determine the extent of DNA degradation and the presence of heterobands, RNA, and protein contamination; Qubit 2.0 fluorometry was used to measure DNA concentration.
DNA resequencing libraries were constructed using the GenoBaits DNA library prep kit for ILM (BioVision, San Francisco, CA, USA) on quality-checked DNA. Sequencing was performed using an MGI-2000/MGI-T7 sequencing platform (Shenzhen UW Smart Technology, Shenzhen, China). In this study, each base sequence was quality-checked (-w 4 -q 20 -n 2 -u 30) using Fastp software (ver. 0.20.0)30. The paired-end sequences were localized to the equine reference genome (Equus caballus 3.0) using BWA (ver. 0.7.17)31. Variant detection was performed using the HaplotypeCaller module of GATK (ver. 4.0.4.0)32.
Variant site filtering
PLINK software33 was used for QC of the sequencing data with the following criteria: minor allele frequency (MAF) < 5%, individual detection rate < 95%, SNP missing rate < 90%, and Hardy-Weinberg equilibrium P value > 10− 4. Sequencing yielded 22,039,238 SNPs, and 10,741,200 SNPs were obtained for genotypic analysis after data quality control. We calculated marker intervals and linkage disequilibrium (LD) to estimate R2 for all markers and plotted the marker distribution (Fig. 1). The frequency, MAF and heterozygosity values are shown in the supplementary material (Figure S1-2).

Genotype analysis including R2 (a, b and c) and distance (d, e and f).
Population structure
Based on the SNP markers obtained by quality control, Phylogenetic evolutionary trees were constructed using the IQ-TREE 2 and iTOL34,35 (Fig. 2).

Phylogenetic evolutionary trees of the samples (TB, thoroughbreds; YH, Yili horses).
Genome-wide association study
To assess the potential associations between genetic loci and traits at the genomic level, genome-wide association studies (GWASs) were performed using GAPIT (ver 3)36, which integrates multiple algorithms for association analysis and ensures that plausible associations of loci are screened by multiple methods that corroborate each other. The GWAS models used in this study include Bayesian information and linkage disequilibrium iterative nested keyway (Blink)37, and fixed stochastic cyclic probabilistic uniform (FarmCPU)38. The GCTA software (ver 1.92.4) was used to determine population stratification and relatedness in Yili horses, and the results were used as random effects in a GWAS.
Genome-wide association analysis significance thresholds (6.05 × 10− 9) and suggestive significance thresholds (1.21 × 10− 7) were determined using a value of 0.05/n, 1/n, where n (8235197) is the number of independent SNPs computed using the genetic type 1 error calculator (v.0.2; https://pmglab.top/gec/#/)39,40.
The proportion of variance explained (PVE): was calculated as follows41:
where \(\:\varvec{\beta\:}\) is the effect of SNP markers, MAF is the frequency of SNP marker minor alleles, \(\:\varvec{s}\varvec{e}\left(\varvec{\beta\:}\right)\:\)is the standard error of the effect of SNP markers, and N is the number of samples analyzed by GWAS.
Gene function annotation
The, reference genome (EquCab3.0) of the horse, Equus caballus, was downloaded from the National Center for Biotechnology Information (NCBI) site, and the 100 kb region before and after the significant locus was annotated by ANNOVAR42. GO and KEGG enrichment analyses were performed using DAVID (https://david.ncifcrf.gov/summary.jsp)43,44,45,46. The animal QTLdb NR database was used to find significant loci and gene functions, and the database was also used for functional gene mining of associated intervals47.
Data availability statement
Sequences are available from GSA with the BioProject accession number PRJCA023926 (https://www.cncb.ac.cn/).
Results
Descriptive statistics
In this study, a total of 2576 speed and ranking score records were used as data for genetic parameter estimation. The results of variance component estimation are given in Table 1, which shows that speed and ranking score had moderate heritability (0.347, 0.156). We also found a highly significant positive genetic correlation (0.920) and phenotypic correlation (0.735), which is shown in the Supplementary Material (Tables S2). The frequency distribution of data of EBVs for speed and ranking score traits were normally distributed (Fig. 3).

Frequency distribution of estimated breeding values (EBVs) for speed (A) and ranking score (B) traits in Yili horses.
Resequencing of Yili horses
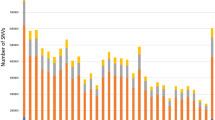
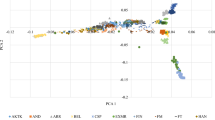
The sequence alignment to the reference genome was 99.34%, and the average depth of sequencing was 7.59X, with 71.58% at 5X coverage, and 21.30% at 10X coverage (see supplementary material Table S10-11). The results of the genome testing are shown in Fig. 4. The sequencing data were evenly distributed throughout the genome, with good sequencing randomness, and the SNPs had a high-density distribution on ECA 20, 29, and X. Kinship matrices as random effects and principal component analysis (PCA) as covariates were added to the GWAS analysis model. (Fig. 5). Additional pca results are included in supplementary Material (Figure S3-5). According to the Kinship, PCA, and evolutionary relationship between populations, we finally selected 212 Yili horses and 24 thoroughbreds for GWAS analysis.

Density distribution of re-sequenced SNP sites in Yili horses.

Kinship (A) and population stratification (B) of sample groups (TB, thoroughbreds; YH, Yili horses).
Association analysis
The association loci were screened by GWAS, P values were -log10 transformed, and Manhattan plots were drawn (Figs. 6 and 7), with a total of 24 significant loci (P < 6.05 × 10− 9) and 22 suggested SNP markers (P < 1.21 × 10− 7). In the Blink model (Figs. 6A and 7A), eight SNP loci were found to be associated with the speed trait (P < 1.21 × 10− 7), of which five were significantly associated (P < 6.05 × 10− 9), and 18 SNP loci were found to be associated with the ranking score trait (P < 1.21 × 10− 7), with 13 SNP loci being significantly correlated (P < 6.05 × 10− 9). In the FarmCPU model (Figs. 6B and 7B), four SNP loci were found to be associated with the speed trait (P < 1.21 × 10− 7), with three of them significantly associated (P < 6.05 × 10− 9); 22 SNP loci were found to be associated with the ranking score trait (P < 1.21 × 10− 7), of which 10 SNP loci were significantly correlated (P < 6.05 × 10− 9).

Manhattan plots of speed trait base on Blink (A) and FarmCPU (B). The values in the upper right corner of the image indicate the distribution of SNP density on the chromosomes.

Manhattan plots of ranking score trait base on Blink (A) and FarmCPU (B). The values in the upper right corner of the image indicate the distribution of SNP density on the chromosomes.
P value expansion detection
To test for population inflation in the results of this GWAS, we compared the observed P values with the randomized expected P values (Figs. 8 and 9). The results show that most SNPs are on the diagonal (red symbols), which indicates that the population structure of this GWAS calculation was well controlled. The upward movement of significant loci was observed in both Blink and FarmCPU models, and the combined Q-Q results of the three models proved that the upward movement of loci was not caused by inflated P values and confirmed the validity of the results.

QQ plots of speed trait base on Blink (A) and FarmCPU (B).

QQ plots of ranking score trait base on Blink (A) and FarmCPU (B).
Significant site information
Gene annotation was performed based on the screened SNP loci. Yili horses showed 125 functional genes for the racing performance traits of speed and ranking score, of which 48 were significant (p < 6.05 × 10− 9) (Tables 2 and 3; Fig. 10); detailed gene information is provided in the Supplementary Material( Tables S12-S13). There were two intersecting genes associated with the speed and 17 intersecting genes associated with the ranking score. The most significant loci were all related to speed and ranking score traits (ECA1: 22698579), with annotated genes LOC102148475 and LOC106782040, and the proportion of variance explained (PVE) equal to 5.789.

Venn Diagram of significant loci (A, B) and genes (C) for speed and ranking score traits.
Gene function enrichment analysis
The results of GO enrichment of genes related to speed trait showed (Fig. 11) that they were significantly enriched in magnesium ion transport, magnesium ion transmembrane transporter activity, and axon guidance. The KEGG results showed significant enrichment mainly in axon guidance and regulation of the actin cytoskeleton. The results of GO enrichment of genes related to ranking score trait showed (Fig. 12) significant enrichment mainly in immune regulation (natural killer cell activation in the immune response and T cell activation in the humoral immune response), cytokine receptor binding (type I interferon receptor binding, cytokine receptor binding). KEGG results showed significant enrichment mainly in the RIG-I-like receptor signaling pathway, the JAK-STAT signaling pathway, and cytokine-cytokine receptor interaction.

Enrichment determination of speed (A) and ranking score (B) traits associated genes by GO analysis.

Enrichment determination of speed (A) and ranking score (B) traits associated genes by KEGG analysis.
Discussion
As an integrated tool for genomic association and prediction, GAPIT is being widely used in genome research due to its varied analytical strategies and functions48,49,50. In GAPIT (ver 3.0), FarmCPU and BLINK were evaluated and found to have extraordinary computational speed and statistical power38. In a comparison of several GWAS models, Jiabo Wang et al. used a comparison function to evaluate the computational power, FDR, and type I error of GLM, MLM, and FarmCPU models, and the results showed that FarmCPU outperformed MLM and GLM36. The BLINK, FarmCPU model used in this study had high sensitivity and statistical functionality, locating 46 genomic regions associated with racing performance traits in the Yili horse. These helped to identify mutated loci for higher racing performance and provided new insights on methods for detecting and selecting desirable genetic variations.
The locus most significantly associated with both speed and ranking score traits in Yili horses was ECA1: 22,698,579 (BLINK, FarmCPU, 1.61 × 10− 11, 7.60 × 10− 15; 1.91 × 10− 13, 7.15 × 10− 9), which is linked to the annotated genes LOC102148475 and LOC106782040, whose functional roles are poorly understood or currently unknown. However, this SNP locus is closer to the only known functional locus for the speed trait (ECA1: 25885857) reported in the horse QTLdb database51. It is likely to be closely related to the racing performance of Yili horses and is thus valuable for further in-depth study.
The most significant locus for the speed trait was ECA16: 15,545,322 (BLINK, 2.65 × 10− 11), an SNP locus 100 kb away from the significant locus reported in the horse QTLdb database for the racing performance trait (ECA16: 15645555). This locus is located 190 kb downstream of the CNTN6 gene, which encodes a glycosylphosphatidylinositol (GPI)-anchored neuronal membrane protein that is a member of the immunoglobulin superfamily and may play a role in the formation of axonal connections in the developing nervous system52. Studies have shown that a deficiency of CNTN6 in mice leads to severe motor coordination abnormalities and learning difficulties53. Motor coordination is crucial for high-speed performance in Yili horses, especially during a race, where the ability to coordinate between limbs is essential for the fastest speed.
The second significant site is ECA1: 114,866,932 (FarmCPU, 2.76 × 10− 10). This locus is located 15 kb upstream of NIPA1. The NIPA1 gene encodes a magnesium transporter, which is associated with early nuclear endosomes and cell surfaces in various types of neurons and in epithelial cells. The protein may play a role in the development and maintenance of the nervous system. It has been shown that mutations in this gene are associated with degenerative motor neuron diseases54. Therefore, the NIPA1 gene may be closely related to motor neuron development and control during high-intensity activity in Yili horses.
The third significant site is ECA8: 74314792 (BLINK, 1.58 × 10− 9), which is located within the DCC gene, near the 5’ end. The product of DCC gene expression is a transmembrane phosphoprotein, which is a member of the immunoglobulin superfamily of cell adhesion molecules. The amino acid sequence of DCC shares homology with neural cell adhesion factor (NCAM) and other related cell surface glycoproteins, which suggests that loss of DCC function may lead to decreased cell-to-cell contact and adhesion, thus enhancing the metastatic ability of cancer cells55. It has been shown that DCC can encode netrin 1 receptors and mediate axon guidance of neuronal growth cones towards the source of netrin 1 ligands56, a process that has been linked to the development of adolescent dopamine neurons57. Horse racing is a high-intensity sport with critical neuronal involvement, and DCC may be involved in neuronal development in racehorses by regulating the excitatory conduction mechanism, which in turn could affect racing performance.
The most significant locus in the ranking score trait is ECA12: 33662348 (BLINK, FarmCPU, 3.88 × 10− 15, 2.51 × 10− 18), which is 26 kb upstream of SHANK2, near the 5’ end. The SHANK2 gene enables ionotropic glutamate receptor binding activity, which is involved in the regulation of chemical synaptic transmission and synaptic organization of multiple processes, including learning and memory. Located in a variety of cellular components and expressed in the cerebral cortex, SHANK2 encodes a scaffolding protein in the postsynaptic membrane of excitatory neurons and is involved in the induction and maturation of dendritic spines58. Competitive racing performance is also an ability acquired with constant practice, and the SHANK2 gene may be associated with competitive neurotransmission and reinforcement processes, the lack of which could result in loss of competitive racing performance in racehorses.
The second significant site is ECA23: 4362189 (BLINK, FarmCPU, 1.07 × 10− 13, 1.13 × 10− 14), which is 33 kb upstream of the ISCA1 gene near the 5’ end. The ISCA1 gene codes for a mitochondrial protein involved in the biogenesis and assembly of iron-sulfur clusters, which play a role in electron transfer. Studies have shown that ISCA1 gene deletion leads to abnormal morphology and impaired enzyme activity of mitochondrial respiratory chain complexes I, II and IV, and reduced ATP synthesis, concurrent with signs of dilated cardiomyopathy59. In horse racing, the strength of cardiac function tends to determine the magnitude of the ability to exercise–a strong heart is a prerequisite for high-intensity exercise. Thus, the ISCA1 gene may influence heart function by affecting mitochondrial proteins, enzyme activities, and ATP synthesis, which in turn indirectly affects horse racing performance.
The third significant site is ECA3: 104971599 (FarmCPU, 6.03 × 10− 13), near the 5’ end within the KCNIP4 gene, which is a member of the family of potassium-ion (Kv) channel-interacting proteins (KCNIPs), which share similarities with the calcium-binding proteins. It regulates neuronal excitability in response to changes in intracellular calcium ions by modulating A-type currents and thus neuronal excitability60. Related studies have reported that the KCNIP4 gene is associated with growth traits in broiler chickens61, sheep62, and beef cattle63. Potassium ion channels are involved in the regulation of a variety of neuronal functions. Strenuous exercise is accompanied by a complex physiological regulatory process. During intense exercise of short duration, the body enhances the loading capacity of exercise vectors through neurotransmitter release, accelerated heart rate, insulin secretion, and modulation of neuronal excitability. KCNIP4 may be involved in this process by improving the efficiency and capacity of neural activity, thus allowing a rapid burst of physical exertion.
In addition, the ranking score trait is associated with genes such as GOT2 (glutamic acid transaminase) and SLC38A7 (amino acid transporter protein), which are involved in the metabolism of amino acids64, which may provide an energy source for intense exertion.
This study has some limitations, such as the small sample size and the lack of information about SNP loci. The functions of the significant genes, LOC102148475, LOC106782040, LOC111774796, and LOC111774797, have not yet been identified, and further studies are needed to investigate the significant SNP loci.
Conclusions
In this study, two GWAS models, BLINK, and FarmCPU, were used to analyze the racing performance traits of Yili horses, and a total of 46 SNP markers (24 significant markers) were associated with the BLINK and FarmCPU models, including 50 significant candidate genes. The discovery of some associated candidate genes (CNTN6, NIPA1, DCC, SHANK2, ISCA1, KCNIP4) will help us to understand the genetic mechanism. In addition, this study identified a locus (ECA1, 22698579) that is significantly associated with the traits of speed and ranking score in Yili horses. However, the specific function of this locus is not well understood and needs to be further explored. In conclusion, further research is needed to validate and expand upon the associations revealed in this study, as well as to explore the potential of using these genes to improve the genetics of racing performance in Yili horses.
Data availability
The data that support the findings of this study are available from the corresponding author, C.W., upon reasonable request.
References
Han, H. et al. Common protein-coding variants influence the racing phenotype in galloping racehorse breeds. Commun. Biology 5, 1320. https://doi.org/10.1038/s42003-022-04206-x (2022).
Mikko, S. Recent advances in genomics of equine health, welfare and performance. Proc. Proc. 12th World Congress Genet. Appl. Livest. Prod. (WCGALP). 3147–3150 https://doi.org/10.3920/978-90-8686-940-4 (2022).
Han, H. et al. Selection in Australian Thoroughbred horses acts on a locus associated with early two-year old speed. PLoS One 15 https://doi.org/10.1371/journal.pone.0227212 (2020).
Kalbfleisch, T. S. et al. Improved reference genome for the domestic horse increases assembly contiguity and composition. Commun. Biology 1, 197. https://doi.org/10.1038/s42003-018-0199-z (2018).
Beeson, S. K., Schaefer, R. J., Mason, V. C. & McCue, M. E. Robust remapping of equine SNP array coordinates to EquCab3. Anim. Genet. 50, 114. https://doi.org/10.1111/age.12745 (2019).
Andersson, L. S. et al. Mutations in DMRT3 affect locomotion in horses and spinal circuit function in mice. Nature 488, 642–646. https://doi.org/10.1038/nature11399 (2012).
Petersen, J. L. et al. Genome-wide analysis reveals selection for important traits in domestic horse breeds. PLoS Genet. 9 https://doi.org/10.1371/journal.pgen.1003211 (2013).
Ricard, A. et al. Endurance exercise ability in the horse: A trait with complex polygenic determinism. Front. Genet. 8, 89. https://doi.org/10.3389/fgene.2017.00089 (2017).
Hill, E. W., McGivney, B. A., Gu, J., Whiston, R. & MacHugh, D. E. A genome-wide SNP-association study confirms a sequence variant (g. 66493737C > T) in the equine myostatin (MSTN) gene as the most powerful predictor of optimum racing distance for thoroughbred racehorses. BMC Genom. 11, 1–10. https://doi.org/10.1186/1471-2164-11-552 (2010).
Kristjansson, T. et al. The effect of the ‘Gait keeper’ mutation in the DMRT3 gene on gaiting ability in Icelandic horses. J. Anim. Breed. Genet. 131, 415–425. https://doi.org/10.1111/age.12120 (2014).
Liu, X. & Liu, Z. Y. A single-nucleotide mutation within the TBX3 enhancer increased body size in Chinese horses. Curr. Biol. 32, 480–487. https://doi.org/10.1016/j.cub.2021.11.052 (2022).
Ayad, A. & Aissanou, B. O. Profiling of genetic markers useful for breeding decision in Selle Francais Horse. J. Equine Veterinary Sci. 116 https://doi.org/10.1016/j.jevs.2022.104059 (2022).
Sharman, P. & Wilson, A. J. Genetic improvement of speed across distance categories in thoroughbred racehorses in Great Britain. Heredity 1–7. https://doi.org/10.1038/s41437-023-00623-8 (2023).
Cunningham, E., Dooley, J. J., Splan, R. & Bradley, D. Microsatellite diversity, pedigree relatedness and the contributions of founder lineages to thoroughbred horses. Anim. Genet. 32, 360–364. https://doi.org/10.1046/j.1365-2052.2001.00785.x (2001).
McGivney, B. A. et al. Genomic inbreeding trends, influential sire lines and selection in the global Thoroughbred horse population. Sci. Rep. 10, 466. https://doi.org/10.1038/s41598-019-57389-5 (2020).
Hill, E. W. et al. MacHugh, D. A sequence polymorphism in MSTN predicts sprinting ability and racing stamina in thoroughbred horses. PLoS One 5 https://doi.org/10.1371/journal.pone.0008645 (2010).
Tozaki, T. et al. A cohort study of racing performance in Japanese Thoroughbred racehorses using genome information on ECA18. Anim. Genet. 43, 42–52. https://doi.org/10.1111/j.1365-2052.2011.02201.x (2012).
Bailey, E., Petersen, J. L. & Kalbfleisch, T. S. Genetics of Thoroughbred racehorse performance. Annu. Rev. Anim. Biosci. 10, 131–150. https://doi.org/10.1146/annurev-animal-020420-035235 (2022).
Bahmurati Tereguhaz A century of changes in the Yili Horses. XINJIANG XUMUYE. 9, 38–41. https://doi.org/10.3969/j.issn.1003-4889.2016.09.005 (2016).
Li, M. Study on the genetic structure of Yili horse population. Urumqi: Xinjiang Agricultural University. https://doi.org/10.27431/d.cnki.gxnyu.2022.000949 (2022).
Wang, D. A preliminary study on the effects of specialized training on speed performance and biochemical indexes of young Yili horses. Urumqi: Xinjiang Agricultural Univ. https://doi.org/10.27431/d.cnki.gxnyu.2017.000109 (2017).
Zhang, Y. Effects of different warm-up levels on blood gas, biochemistry and heart rate of Yili horses after 1000 m race. Urumqi: Xinjiang Agricultural Univ. https://doi.org/10.27431/d.cnki.gxnyu.2021.000355 (2021).
Xixi, Y. Analysis of blood physiology, biochemistry, immunity indexes and transcriptome differences in different breeds of horses. Urumqi: Xinjiang Agricultural Univ. https://doi.org/10.27431/d.cnki.gxnyu.2023.000255 (2023).
Yuru H. Analysis of MSTN and GH gene polymorphisms and their association with body size traits in three breeds of horses. Urumqi: Xinjiang Agricultural Univ. https://doi.org/10.7666/d.Y2887129 (2015).
He, M. Correlation analysis of DMRT3, MSTN, ACTN3 gene polymorphisms and racing performance in Yili horses. Urumqi: Xinjiang Agricultural Univ. https://doi.org/10.7666/d.Y3101546 (2016).
Yali, X. Study on the association analysis of COMT exon 1 polymorphism with heart rate and racing performance in horses. Urumqi: Xinjiang Agricultural University. https://doi.org/10.27431/d.cnki.gxnyu.2018.000276 (2018).
Jinqiu, W. Analysis of the association between polymorphisms of MCT1, PPARα, NOS3, and HIF-1α genes and 2000m racing performance in Yili horses.Urumqi: Xinjiang Agricultural University. https://doi.org/10.27431/d.cnki.gxnyu.2022.000049 (2022).
Klei, B. T. S. Approximate variance for heritability estimates. Univ. Pittsburgh Med. Cent. 1–5 (2008).
Wang, X. et al. Using machine learning to improve the accuracy of genomic prediction of reproduction traits in pigs. J. Anim. Sci. Biotechnol. 13, 60. https://doi.org/10.1186/s40104-022-00708-0 (2022).
Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2, 107. https://doi.org/10.1002/imt2.107 (2023).
Kim, C. et al. Accelerating genome sequence mapping on commodity servers. Paper presented at the Proceedings of the 51st International Conference on Parallel Processing, Bordeaux, France. 8, 1–12. https://doi.org/10.1145/3545008.3545033 (2022).
Franke, K. R. & Crowgey, E. L. Accelerating next generation sequencing data analysis: an evaluation of optimized best practices for genome analysis Toolkit algorithms. Genomics Inf. 18, 10. https://doi.org/10.5808/GI.2020.18.1.e10 (2020).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4 https://doi.org/10.1186/s13742-015-0047-8 (2015).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37 (5), 1530–1534. https://doi.org/10.1093/molbev/msaa015 (2020).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. gkae268 https://doi.org/10.1093/nar/gkae268 (2024).
Wang, J., Tang, Y. & Zhang, Z. Performing genome-wide Association studies with multiple models using GAPIT. Springer eBooks 199–217 https://doi.org/10.1007/978-1-0716-2237-7_13 (2022).
Huang, M. & Zhou, L. X. BLINK: A package for the next level of genome-wide association studies with both individuals and markers in the millions. Gigascience 8, 154. https://doi.org/10.1093/gigascience/giy154 (2019).
Liu, X., Huang, M., Fan, B., Buckler, E. S. & Zhang, Z. Iterative usage of fixed and Random Effect Models for Powerful and efficient genome-wide Association studies. PLoS Genet. 12 https://doi.org/10.1371/journal.pgen.1005767 (2016).
Li, H. et al. Graph-based pan-genome reveals structural and sequence variations related to agronomic traits and domestication in cucumber. Nat. Commun. 13, 682. https://doi.org/10.1038/s41467-022-28362-0 (2022).
Li, N. et al. Super-pangenome analyses highlight genomic diversity and structural variation across wild and cultivated tomato species. Nat. Genet. 55, 852–860. https://doi.org/10.1038/s41588-023-01340-y (2023).
Shim, H. et al. A multivariate genome-wide association analysis of 10 LDL subfractions, and their response to statin treatment, in 1868 caucasians. PLoS One 10 https://doi.org/10.1371/journal.pone.0120758 (2015).
Yang, H. & Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 10, 1556–1566. https://doi.org/10.1038/nprot.2015.105 (2015).
Sherman, B. T. et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists. Nucleic Acids Res. 50, 216–221. https://doi.org/10.1093/nar/gkac194 (2022).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592. https://doi.org/10.1093/nar/gkac963 (2023).
Hu, Z. L. & Reecy, P. C. A. Developmental progress and current status of the animal QTLdb. Nucleic Acids Res. 44, 827–833. https://doi.org/10.1093/nar/gkv1233 (2016).
Tang, Y. et al. GAPIT Version 2: an Enhanced Integrated Tool for Genomic Association and Prediction. Plant. Genome 9(2). https://doi.org/10.3835/plantgenome2015.11.0120 (2016).
Wang, J. & Zhang, Z. GAPIT version 3: boosting power and accuracy for genomic association and prediction. Genomics Proteom. Bioinf. 19, 629–640. https://doi.org/10.1016/j.gpb.2021.08.005 (2021).
Enyew, M. et al. Genome-wide analyses using multi-locus models revealed marker-trait associations for major agronomic traits in Sorghum bicolor. Front. Plant Sci. 13 https://doi.org/10.3389/fpls.2022.999692 (2022).
Hu, Z. L. & Reecy, P. C. A. Bringing the animal QTLdb and CorrDB into the future: Meeting new challenges and providing updated services. Nucleic Acids Res. 50, 956–961. https://doi.org/10.1093/nar/gkab1116 (2022).
Mu, D. et al. Cntn6 deficiency impairs allocentric navigation in mice. Brain Behav. 8 https://doi.org/10.1002/brb3.969 (2018).
Hadj Amor, M. et al. Neuronal migration genes and a familial translocation t (3;17): Candidate genes implicated in the phenotype. BMC Med. Genet. 21, 26. https://doi.org/10.1186/s12881-020-0966-9 (2020).
Tanti, M., Cairns, D., Mirza, N., McCann, E. & Young, C. Is NIPA1-associated hereditary spastic paraplegia always ‘pure’? Further evidence of motor neurone disease and epilepsy as rare manifestations. Neurogenetics 21, 305–308. https://doi.org/10.1007/s10048-020-00619-0 (2020).
Marsh, A. P. L. et al. DCC mutation update: congenital mirror movements, isolated agenesis of the corpus callosum, and developmental split brain syndrome. Hum. Mutat. 39, 23–39. https://doi.org/10.1002/humu.23361 (2018).
Finci, L., Zhang, Y., Meijers, R. & Wang, J. H. Signaling mechanism of the netrin-1 receptor DCC in axon guidance. Prog Biophys. Mol. Biol. 118, 153–160. https://doi.org/10.1016/j.pbiomolbio.2015.04.001 (2015).
Vosberg, D. E., Leyton, M. & Flores, C. The Netrin-1/DCC guidance system: dopamine pathway maturation and psychiatric disorders emerging in adolescence. Mol. Psychiatry 25, 297–307. https://doi.org/10.1038/s41380-019-0561-7 (2020).
Caumes, R. & Thuillier, S. T. Phenotypic spectrum of SHANK2-related neurodevelopmental disorder. Eur. J. Med. Genet. 63 https://doi.org/10.1016/j.ejmg.2020.104072 (2020).
Ling, Y. et al. Novel rat model of multiple mitochondrial dysfunction syndromes (MMDS) complicated with cardiomyopathy. Anim. Models Experimental Med. 4, 381–390. https://doi.org/10.1002/ame2.12193 (2021).
Su, Z. J., Wang, X. Y., Zhou, C. & Chai, Z. Down-regulation of mir-3068-3p enhances kcnip4-regulated A-type potassium current to protect against glutamate-induced excitotoxicity. J. Neurochem. 153, 617–630. https://doi.org/10.1111/jnc.14932 (2020).
Cha, J. et al. Genome-Wide Association Study Identifies 12 Loci Associated with Body Weight at Age 8 weeks in Korean native chickens. Genes 12, 1170. https://doi.org/10.3390/genes12081170 (2021).
Mohammadi, H. & Moradi Shahrbabak, R. S. A. Genome-wide association study and gene ontology for growth and wool characteristics in Zandi sheep. J. Livest. Sci. Technol. 8, 45–55. https://doi.org/10.22103/jlst.2020.15795.1317 (2020).
Smith, J. L. et al. Genome-wide association and genotype by environment interactions for growth traits in U.S. Red Angus cattle. BMC Genom. 23, 517. https://doi.org/10.1186/s12864-022-08667-6 (2022).
Chan, K. et al. Loss of function mutation of the Slc38a3 glutamine transporter reveals its critical role for amino acid metabolism in the liver, brain, and kidney. Pflügers Archiv - Eur. J. Physiol. 468, 213–227. https://doi.org/10.1007/s00424-015-1742-0 (2016).
Acknowledgements
We greatly thank the staffs of Xinjiang Yili Kazak Autonomous Prefecture Zhaosu Horse Farm, Yili stud horse farm for providing us with working conditions.
Funding
This study was funded by the research programs Development of Key Technologies for the Horse Industry in Xinjiang, grant number [2022A02013], and The Innovation Environment (Talent, Base) Construction Project of Xinjiang, grant number [PT2311]. The APC was funded by Jun Meng and Xinkui Yao.
Author information
Authors and Affiliations
Contributions
Conceptualization, J.M. and X.Y.; methodology, J.W. and Y.Z.; statistical analysis, C.W. and Z.S.; investigation, C.W., T.W., X.L., and Z.S.; data curation, C.W.; writing—original draft preparation, C.W., and Z.S.; writing—review and editing, C.W., Y.Z., J.M. and X.Y.; supervision, X.Y. All authors have read and agreed to the submitted version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, C., Zeng, Y., Wang, J. et al. A genome-wide association study of the racing performance traits in Yili horses based on Blink and FarmCPU models. Sci Rep 14, 27648 (2024). https://doi.org/10.1038/s41598-024-79014-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-79014-w
