
Abstract
Chronic obstructive pulmonary disease (COPD) is a prevalent respiratory disorder with environmental factors being the primary risk determinants. However, genetic factors also substantially contribute to the susceptibility and progression of COPD. Although genome-wide association studies (GWAS) have identified several loci associated with COPD susceptibility, the specific pathogenic genes underlying these loci, along with their biological functions and roles within regulatory networks, remain unclear. This lack of clarity constrains our ability to achieve a deeper understanding of the genetic basis of COPD. This study leveraged the FinnGen R11 genetic dataset, comprising 21,617 cases and 372,627 controls, along with GTEx V8 eQTLs data to conduct a cross-tissue transcriptome-wide association study (TWAS). Initially, we performed a cross-tissue TWAS analysis using the Unified Test for Molecular Signatures (UTMOST), followed by validation of the UTMOST findings in single tissues using the Functional Summary-based Imputation (FUSION) method and conditional and joint (COJO) analyses of the identified genes. Subsequently, candidate susceptibility genes were screened using Multi-marker Analysis of Genomic Annotation (MAGMA). The causal relationship between these candidate genes and COPD was further evaluated through summary data-based Mendelian randomization (SMR), colocalization analysis, and Mendelian randomization (MR). Additionally, the identified results were validated against the COPD dataset in the GWAS Catalog (GCST90399694). GeneMANIA was employed to further explore the functional significance of these susceptibility genes. In the cross-tissue TWAS analysis (UTMOST), we identified 17 susceptibility genes associated with COPD. Among these, a novel susceptibility gene, G protein-coupled receptor kinase 4 (GRK4), was validated through single-tissue TWAS (FUSION) and MAGMA analyses, with further confirmation via SMR, MR, and colocalization analyses. Moreover, GRK4 was validated in an independent dataset. This study identifies GRK4 as a potential novel susceptibility gene for COPD, which may influence disease risk by exacerbating inflammatory responses. The findings address gaps in previous single-tissue GWAS studies, revealing consistent expression and potential function of GRK4 across different tissues. However, considering the study’s limitations, further investigation and validation of GRK4’s role in COPD are warranted.
Similar content being viewed by others


Introduction
Chronic obstructive pulmonary disease (COPD) is a progressive and heterogeneous disorder, primarily characterized by persistent airflow limitation, often accompanied by respiratory symptoms such as emphysema and chronic bronchitis, as well as pathological changes including small airway remodeling and abnormal alveolar structures1. The predominant symptoms of COPD include dyspnea, chronic cough, and sputum production, frequently associated with lung hyperinflation, which significantly impairs patients’ quality of life2. Globally, COPD affects approximately 300 million individuals and is responsible for an estimated 3.2 million deaths annually3,4. The disease is marked by a prolonged course, high rates of morbidity and mortality, and substantial treatment costs. This burden is particularly severe in low- and middle-income countries, where COPD imposes increasing economic and psychological stress on individuals and their families, leading to a serious public health challenge5. Although smoking, household environment, air pollution, and population aging are recognized as primary risk factors for COPD6,7, genetic factors may also heighten the risk by contributing to lung damage and inflammatory responses8. However, the pathogenesis of COPD remains incompletely understood, and research on therapeutic strategies is not yet comprehensive. In recent years, the potential of natural compounds and their derivatives in anti-inflammatory, anti-fibrotic, and anti-tumor resistance applications has garnered increasing attention9,10,11,12,13. However, the precise therapeutic targets of these compounds remain unknown. Therefore, identifying additional effective drug targets is of significant importance for the prevention and treatment of COPD.
Although diseases such as COPD and asthma are commonly attributed to environmental factors like smoking or exposure to pollutants, research has demonstrated that genetic factors also play a significant role in the risk of developing chronic respiratory diseases14,15. The NHGRI-EBI GWAS Catalog has listed 1,150 genetic variants associated with COPD16. Additionally, the COPDGene study identified several genetic loci associated with emphysema, gas trapping, and airway traits through computed tomography, including loci near HHIP, 15q25, and AGER, as well as novel genetic associations near SERPINA10 and DLC1. The Z variant in SERPINA1 (rs28929474T) exemplifies how GWAS can provide additional insights into COPD risk variants, even though traditional GWAS methods did not identify it as a risk gene17,18. However, many of the disease-associated loci identified by GWAS are located in non-coding regions, posing challenges in assessing their functional significance19. Moreover, the complexity of linkage disequilibrium (LD) may obscure the identification of causal variants driving these associations20.
Compared with traditional GWAS, transcriptome-wide association studies (TWAS) enhance the precision of identifying candidate genes associated with complex traits by integrating expression quantitative trait loci (eQTL) with GWAS summary statistics20. This approach not only enables the identification of trait-related genes with greater accuracy but also provides preliminary insights into their regulatory roles, thus addressing GWAS limitations in functional annotation. Although TWAS offers distinct advantages in exploring gene-phenotype associations, conventional single-tissue TWAS methods have notable limitations, particularly in their inability to comprehensively capture the extensive gene expression patterns and regulatory characteristics across multiple tissues in complex, multi-system diseases such as COPD. This constraint may result in the incomplete identification of cross-tissue key genes and their regulatory networks, thereby hindering a comprehensive understanding of the disease’s genetic basis. Consequently, employing cross-tissue TWAS is crucial for investigating the genetic background of multi-system diseases like COPD, as it captures coordinated expression features across tissues, revealing more intricate and systemic pathobiological mechanisms. A more advanced method, known as the Unified Test for Molecular Signatures (UTMOST), extends TWAS by conducting gene-level association analyses across multiple tissues21. Unlike single-tissue approaches, UTMOST integrates results from multiple tissues into a unified metric within a summary statistics-based framework, thereby enhancing the accuracy of expression imputation across all available tissues and improving the quantification of overall gene-trait associations. In recent years, cross-tissue association analyses have been widely utilized to identify candidate susceptibility genes for complex multisystem diseases, such as frailty22, lung cancer23, and ulcerative colitis24.
This study aims to identify novel susceptibility genes for COPD through cross-tissue TWAS and validate these findings using multiple independent approaches. Specifically, we integrated COPD GWAS data from FinnGen R11 (discovery set) and the GWAS Catalog (validation set), along with eQTL data from the Genotype-Tissue Expression (GTEx) project V8, to perform cross-tissue TWAS analyses. Subsequently, we evaluated associations within each tissue using Functional Summary-based Imputation (FUSION) and conditional and joint (COJO) analyses25, and further validated candidate genes using Multi-marker Analysis of Genomic Annotation (MAGMA)26. Additionally, we assessed the causal relationships between candidate genes and COPD through summary-data-based Mendelian Randomization (SMR) and colocalization analyses, as well as Mendelian Randomization (MR). Finally, we explored the biological functions of these genes via GeneMANIA bioinformatics analysis27. This multi-layered approach systematically identifies and validates novel pathogenic genes associated with COPD, thereby offering new insights into its underlying pathomechanisms.
Materials and methods
Statement on ethical approval
This study utilized publicly available databases, including the GWAS Catalog, FinnGen R11, and GTEx V8, and did not involve any direct experimentation on human subjects or human tissue samples. As such, ethical approval or consent was not required for this research. All analyses were conducted using secondary data derived from these publicly accessible resources.
Study design
The analysis process is shown in Fig. 1.

Flowchart of the study design.
COPD data sources
The GWAS data for the COPD discovery cohort were obtained from the FinnGen database, version R1128. This database integrates genotypic data from Finnish biobanks with digital health records from the Finnish health registry to explore the relationships between genetic variations and disease trajectories. The FinnGen project aims to expand its study population to 500,000 individuals by the end of 2023, thereby broadening the scope of research. In the FinnGen database, the total sample size for COPD is 394,224, including 21,617 cases and 372,627 controls. Among the 21,617 cases, 5,695 were female, and 15,922 were male. (Data access link: https://r11.risteys.finregistry.fi/endpoints/J10_COPD; Download link: https://storage.googleapis.com/finngen-public-data-r11/summary_stats/finngen_R11_J10_COPD.gz).
The GWAS data for the COPD validation cohort were sourced from the study conducted by Wei Zhou et al.29, and are available in the GWAS Catalog database (GCST ID: GCST90399694). This study, conducted under the Global Biobank Meta-analysis Initiative, integrates data across continental biobanks, thereby enhancing the ability to validate diverse GWAS results and improving disease research and risk prediction. The total sample size for COPD in this dataset is 995,917, comprising 58,559 cases and 937,358 controls. (Data access link: https://www.ebi.ac.uk/gwas/studies/GCST90399694; Download link: https://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics/GCST90399001-GCST90400000/GCST90399694/GCST90399694.tsv.gz).
eQTL data sources
The eQTL data were derived from GTEx database30,31, a large-scale genomics project aimed at investigating how gene expression and genomic variation operate across different human tissues. For this study, we utilized the GTEx V8 dataset, the eighth edition, which includes samples from 49 distinct human tissues. This dataset encompasses gene expression data, genetic variation data, gene co-expression networks, and eQTL analysis results. The GTEx data are widely employed in disease research, gene function studies, and drug development, as they facilitate the understanding of the relationships between genetic variation and disease, elucidate gene expression patterns across various tissues, and support the advancement of personalized medicine.
UTMOST analysis
In this study, we employed UTMOST to conduct cross-tissue TWAS, a method frequently used to estimate cross-tissue gene expression in TWAS analysis21. UTMOST integrates multiple single-tissue association results into a robust metric that quantifies the overall gene-trait association. This approach enhances the ability to identify genes associated with complex traits, thereby overcoming the limitations of single-tissue sample size and increasing the statistical power of the analysis. Subsequently, we applied the Generalized Berk-Jones (GBJ) test to integrate gene-trait associations across single-tissue statistics by accounting for covariance32. After adjusting for the false discovery rate (FDR), a significance threshold of FDR < 0.05 was considered statistically significant (UTMOST available at: https://github.com/Joker-Jerome/UTMOST?%20tab=readme-ov-file).
FUSION analysis
For single-tissue TWAS analysis, we utilized FUSION method, which combines GWAS data for COPD with eQTL data from 49 tissues provided by GTEx V8 to estimate gene-disease associations33. We employed genomic data from 1,000 European individuals to estimate LD between single nucleotide polymorphisms (SNPs) at each locus. FUSION integrates multiple statistical models, including BLUP, BSLMM, LASSO, Elastic Net, and Top 1, to evaluate the contribution of each SNP to gene expression. By leveraging different approaches to predict and weight each SNP’s contribution, FUSION provides a comprehensive and accurate assessment34. We then combined the genetic effects of COPD (Z-scores from COPD GWAS) with these gene weights to conduct TWAS for COPD (FUSION available at: http://gusevlab.org/projects/fusion/).
COJO analysis
Following FUSION analysis, it is possible to identify multiple associated traits within a single locus. To determine which of these traits are conditionally independent, we performed COJO analysis, a post-FUSION processing method designed to identify independent genetic signals33. COJO analysis accounts for LD between markers, thereby providing a comprehensive understanding of the genetic architecture underlying trait variation. After testing, genes exhibiting independent associations are labeled as “jointly significant,” while those that are no longer significant are considered “marginally significant”35.
MAGMA analysis
We conducted gene analysis using MAGMA software (version 1.08). This tool is utilized for gene-based or gene-set-based association analyses, enabling the identification of functional genes or modules (e.g., gene regulatory pathways) associated with specific traits. MAGMA is also effective in detecting genes associated with multiple low-effect SNPs. In this study, we applied default parameters to aggregate SNP-level association statistics into gene scores, thereby quantifying each gene’s association with the phenotype36,37. For detailed information on parameter settings and methodological explanations, please refer to the original MAGMA documentation26 (MAGMA available at: https://cncr.nl/research/magma/).
SMR and bayesian colocalization analysis
We employed SMR to investigate the pleiotropic relationships between gene expression and traits using summary-level data from COPD and eQTL studies. This approach facilitates the identification of COPD causative genes and enhances our understanding of the gene-trait relationship38,39,40. We selected significant SMR probes based on an FDR-corrected SMR P < 0.05 and used the HEDI test with a P > 0.05 to indicate the absence of heterogeneity40 (SMR available at: https://yanglab.westlake.edu.cn/software/smr/#SMR&HEIDIanalysis).
Subsequently, we performed Bayesian colocalization analysis using the “coloc” R package (version 5.2.3) to determine whether GWAS and eQTL signals overlap at causal variant sites41,42. This analysis computes posterior probabilities for five hypotheses (PPH): (1) no association with either trait (H0), (2) association with only trait 1 (H1), (3) association with only trait 2 (H2), (4) association with both traits due to different causal variants (H3), and (5) association with both traits due to the same causal variant (H4). Following the literature, we defined colocalization when PPH4 > 0.8 and moderate colocalization when PPH4 > 0.543 (“coloc” R package available at: https://github.com/chr1swallace/coloc).
MR analysis
We conducted MR analysis using the “TwoSampleMR” R package (version 0.6.6). In this analysis, we used cis-eQTL SNPs as instrumental variables (IVs), gene expression as the exposure, and COPD GWAS data as the outcome. IVs were required to have an R2 < 0.001 and LD = 10,000 kb. Since only one independent IV was available, we employed the Wald ratio as the primary method for estimating MR effects, with a significance threshold set at P < 0.0544 (“TwoSampleMR” R package available at: https://github.com/MRCIEU/TwoSampleMR?tab=readme-ov-file).
GeneMANIA analysis
Finally, we used GeneMANIA to analyze the complex relationships and biological functions among genes. The GeneMANIA platform integrates various datasets on genetic interactions, pathways, and co-expression of target genes27 (GeneMANIA platform available at: https://genemania.org/).
Results
Cross-tissue and single-tissue TWAS analysis
In the discovery set for COPD, cross-tissue TWAS analysis identified a total of 352 genes with P < 0.05 (Supplementary Material 1, S1), of which 17 genes remained significant after stringent FDR correction (PFDR < 0.05) (Table 1). These significant genes suggest potential roles in the pathophysiological processes of COPD and provide candidate targets for further investigation. To enhance the rigor and reliability of our analysis, we conducted validation through single-tissue TWAS, identifying a total of 638 genes that remained significant post-FDR correction (PFDR < 0.05) in at least one tissue (Supplementary Material 1, S2), potentially reflecting tissue-specific regulatory mechanisms involved in COPD.
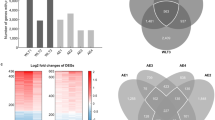
Further analysis revealed that the 17 genes identified through cross-tissue TWAS also demonstrated consistent significance in single-tissue TWAS, as detailed in Fig. 2. This concordance not only reinforces the findings from the cross-tissue analysis but also highlights the broad expression and potential functions of these genes across different tissues. Among the genes achieving strict significance thresholds in both cross-tissue and single-tissue analyses, seven candidates were identified, comprising six protein-coding genes (MFSD10, GRK4, PPA2, TET2, XPNPEP3, and HTT) and one long non-coding RNA (lncRNA) gene (NOP14-AS1) (Supplementary Material 1, S3). The identification of these genes provides valuable insights into the molecular mechanisms underlying COPD, particularly regarding the roles of protein-coding and non-coding RNAs in disease pathology.

Heatmap (Note: The heatmap of 17 genes identified by UTMOST is separated by tissue in FUSION; gray boxes indicate PFDR > 0.05 in FUSION.). Note: The heatmap was generated using R (version 4.4.0) with the R packages: pheatmap (version 1.0.12) and reshape2 (version 1.4.4).
COJO analysis
In the COPD discovery set, seven candidate genes primarily located on chromosomes 4 and 22 (MFSD10, GRK4, PPA2, TET2, XPNPEP3, HTT, and NOP14-AS1) were further analyzed using COJO analysis within each tissue to validate their associations with COPD and eliminate potential false positives due to LD. The results indicated that GRK4 remained significantly associated independently of other genes, suggesting its possible key role in COPD pathogenesis (Supplementary Material 1, S4 and Supplementary Material 2).
Notably, while PPA2, TET2, and HTT exhibited significance in single-tissue TWAS analyses, the presence of LD may have confounded these results. Consequently, these genes did not meet the stringent criteria for multi-layer validation and were excluded from subsequent in-depth analysis. This selection process underscores the rigorous control strategies employed in this study to ensure the reliability of candidate genes and the accuracy of our findings. By mitigating potential false positives, these analyses offer more precise genetic targets for COPD molecular studies and lay a robust foundation for future investigations.
MAGMA analysis
In the COPD discovery set, MAGMA analysis identified a total of 426 genes significantly associated with COPD (PFDR < 0.05) (Supplementary Material 1, S5). These genes may play crucial roles in COPD pathogenesis, providing numerous candidate targets for further exploration of COPD molecular mechanisms. To enhance the robustness and consistency of our findings, we performed an integrative analysis combining the UTMOST cross-tissue analysis results with key genes detected by FUSION and MAGMA, aiming to identify core genes highly relevant to COPD. This integrative approach ultimately confirmed four critical candidate genes: MFSD10, GRK4, TET2, and HTT (Fig. 3). These genes demonstrated significance across multiple analytical methods, suggesting consistent genetic associations and potential biological functions under varied tissues and conditions.

Venn Diagram (Note: MAGMA identified 426 significant genes associated with COPD, FUSION identified 638, and UTMOST cross-tissue analysis identified 17. Among these, four genes—MFSD10, GRK4, TET2, and HTT—were commonly identified across all analyses.).
SMR and colocalization analysis
SMR analysis provides estimates of causal effects of genetic variation on phenotypes, while colocalization analysis further assesses whether these causal associations share the same genetic signals between gene expression and phenotype, thereby enhancing the credibility and rigor of causal inference. In the COPD discovery set, we selected four key genes—MFSD10, GRK4, TET2, and HTT—for SMR and colocalization analyses within their respective tissues to strengthen the robustness of the results. SMR analysis revealed that MFSD10, TET2, and HTT did not exhibit significant causal effects on COPD in their corresponding tissues (PSMR−FDR > 0.05), nor did they share the same genetic signals with COPD in colocalization analysis (PP.H4 < 50%), suggesting that changes in their expression may not directly contribute to COPD pathogenesis (Supplementary Material 1, S6 and S7).
In contrast, GRK4 demonstrated significant causal effects on COPD across 16 different tissues, supporting its potential biological role in COPD. Moreover, colocalization analysis indicated that GRK4 shares the same genetic signal with COPD, specifically SNP rs624833 (moderate colocalization, PP.H4 > 50%). This finding suggests that GRK4 may play a crucial role in the pathological mechanisms of COPD. Detailed data are presented in Table 2; Figs. 4 and 5, and 6. The integration of SMR and colocalization analyses thus further substantiates the significance of GRK4, providing strong support for its potential as a therapeutic target for COPD.

Effect plots from SMR analysis. (Note: A: in Adipose Visceral Omentum; B: in Adrenal Gland; C: in Artery Aorta; D: in Artery Coronary; E: in Artery Tibial; F: in Brain Cerebellar Hemisphere; G: in Brain Cerebellum; H: in Breast Mammary Tissue; I: in Colon Sigmoid; J: in Colon Transverse; K: in Esophagus Gastroesophageal Junction; L: in Esophagus Muscularis; M: in Ovary; N: in Pituitary; O: in Skin Sun Exposed Lower leg; P: in Thyroid).

Locus plots from the SMR analysis. (Note: A: in Adipose Visceral Omentum; B: in Adrenal Gland; C: in Artery Aorta; D: in Artery Coronary; E: in Artery Tibial; F: in Brain Cerebellar Hemisphere; G: in Brain Cerebellum; H: in Breast Mammary Tissue; I: in Colon Sigmoid; J: in Colon Transverse; K: in Esophagus Gastroesophageal Junction; L: in Esophagus Muscularis; M: in Ovary; N: in Pituitary; O: in Skin Sun Exposed Lower leg; P: in Thyroid).

Colocalization analysis plots. (Note: A: in Adipose Visceral Omentum; B: in Adrenal Gland; C: in Artery Aorta; D: in Artery Coronary; E: in Artery Tibial; F: in Brain Cerebellar Hemisphere; G: in Brain Cerebellum; H: in Breast Mammary Tissue; I: in Colon Sigmoid; J: in Colon Transverse; K: in Esophagus Gastroesophageal Junction; L: in Esophagus Muscularis; M: in Ovary; N: in Pituitary; O: in Skin Sun Exposed Lower leg; P: in Thyroid).
MR analysis
MR analysis demonstrated significant positive causal effects of GRK4 on COPD across multiple tissues, further supporting its potential pathogenic role in the disease. Specifically, significant positive causal associations between GRK4 and COPD were observed in the brain anterior cingulate cortex, minor salivary gland, pancreas, and stomach (P < 0.05, OR > 1), indicating that upregulation of GRK4 expression in these tissues may increase the risk of COPD. These findings suggest that GRK4 may influence COPD development through multiple pathways across different tissues (Fig. 7, Supplementary Material 1, S8).

Forest plot of the causal effect of GRK4 on COPD.
Notably, MR analysis did not show a significant causal effect of GRK4 on COPD in the brain caudate basal ganglia (P > 0.05), which may reflect tissue-specific effects of GRK4 or differential mechanisms in COPD pathogenesis across tissues. This observation underscores the need for further investigation to elucidate the functional differences of GRK4 in various tissues and physiological contexts.
Validation of GRK4 efficacy
To further validate the role of the GRK4 gene in COPD, we conducted a cross-tissue TWAS analysis in the COPD validation set, identifying 301 candidate genes with P < 0.05 (Supplementary Material 1, S9), among which 27 genes remained significant after FDR correction (PFDR < 0.05), as detailed in Table 3. Additionally, for single-tissue TWAS validation, 980 significant genes (PFDR < 0.05) were identified in at least one tissue (Supplementary Material 1, S10). Cross-validation between cross-tissue and single-tissue analyses revealed 11 candidate genes meeting stringent significance thresholds, including nine protein-coding genes (GPD2, GRK4, HHIP, IL18R1, MAP4K4, MFSD10, MSANTD1, SLC9A2, and TCF4) and two lncRNAs (NOP14-AS1 and TEX41) (Table 4, Supplementary Material 1, S11).
To eliminate potential false-positive results due to LD for the 11 candidate genes located on chromosomes 2, 4, 18, and 22, we performed COJO analysis in their respective tissues (Supplementary Material 1, S12). Additionally, MAGMA analysis confirmed 279 genes significantly associated with COPD (PFDR < 0.05) (Supplementary Material 1, S13). To enhance the reliability of our results, we integrated the UTMOST cross-tissue analysis findings with significant genes detected by FUSION and MAGMA, ultimately identifying four key candidate genes: MAP4K4, GRK4, MSANTD1, and TCF4 (Table 5; Fig. 8).

Venn diagram (Note: MAGMA identified 279 key genes associated with COPD, FUSION identified 980, and UTMOST cross-tissue analysis identified 27, of which 4 are common: MAP4K4, GRK4, MSANTD1, and TCF4).
In subsequent causal validation analyses, we investigated the potential causal roles of these four key candidate genes in COPD using SMR, colocalization, and MR analyses (Supplementary Material 1, S14, S15, and S16). SMR analysis indicated that GRK4 met the significance threshold in 22 tissues (Fig. 9). Colocalization analysis demonstrated that GRK4 shared the same genetic signal with COPD, specifically SNP rs12647713 (Fig. 10C and F), in the artery tibial and cultured fibroblasts, corroborating the SMR findings (Fig. 10A, B, D, and E).

Forest plot of SMR analysis.

SMR analysis effect and locus plots, and colocalization plots (Note: A, B, C: SMR analysis effect plot, locus plot, and colocalization plot in Artery Tibial; D, E, F: SMR analysis effect plot, locus plot, and colocalization plot in Cells Cultured Fibroblasts).
Moreover, MR analysis showed positive causal effects of GRK4 on COPD in the adipose subcutaneous tissue, brain caudate basal ganglia, and minor salivary gland (P < 0.05, OR > 1) (Fig. 11). These findings were also validated in an independent database, further supporting the hypothesis that GRK4 is a key susceptibility gene for COPD. Collectively, GRK4 demonstrated significant associations with COPD across multiple analyses, suggesting its potential involvement in the genetic susceptibility and pathophysiological mechanisms of COPD.

Forest plot of MR analysis.
GeneMANIA analysis
The potential interaction gene network centered around GRK4 is illustrated in Fig. 12 (Supplementary Materials 3). The most prominent functions within the GRK4-associated gene network include the G protein-coupled receptor signaling pathway, coupled to cyclic nucleotide second messengers, G protein-coupled receptor activity, and adenylate cyclase-modulating G protein-coupled receptor signaling pathways (Supplementary Materials 1 S17).

GeneMANIA gene network centered on GRK4.
Discussion
In this study, we systematically evaluated the relationship between genetic susceptibility of gene expression and COPD risk using GWAS data from Finnish R11 and eQTL data from GTEx V8. Through cross-tissue and single-tissue TWAS analyses, as well as MAGMA validation, we identified GRK4 as a COPD susceptibility gene and further validated its significance using SMR, MR, and colocalization analyses. Moreover, validation in an independent database reinforced the potential role of GRK4 as a COPD susceptibility gene. Finally, GeneMANIA analysis provided deeper insights into the potential functions of GRK4, offering critical directions for future research.
COPD is a complex, multifactorial disease influenced by both environmental factors and genetic susceptibility. For instance, significant variability in COPD incidence among individuals with similar smoking histories suggests a strong association with genetic variations45. To date, GWAS and gene expression analyses have identified numerous genes and molecular pathways implicated in COPD pathogenesis. Notably, SNPs at the α-nicotinic acetylcholine receptor (CHRNA3/5) locus have been shown to be significantly associated with COPD risk and lung function, with these findings validated in multiple independent cohorts46. Furthermore, research by Michael H. Cho et al.47 identified a novel COPD susceptibility locus, 19q13, through a GWAS involving 3,499 COPD cases and 1,922 controls across four distinct cohorts, with additional validation in familial datasets. These studies provide valuable genetic targets for early screening and personalized treatment strategies for COPD.
With the gradual elucidation of the molecular mechanisms underlying COPD, the G protein-coupled receptor kinase (GRK) family has emerged as a potential contributor to its pathogenesis. GRKs, as serine/threonine protein kinases, have gained significant attention due to their pivotal roles in signal transduction. The GRK family is divided into two main subfamilies: GRK2/3 and GRK4/5/648. GRKs interact with agonist-activated G protein-coupled receptors (GPCRs), influencing receptor phosphorylation and thereby modulating signal transmission and desensitization49. During the pathological process of inflammatory responses, the expression levels of GRKs change notably. Studies have shown that GRK membrane activity, as well as the expression of GRK2 and GRK5, is markedly increased in IL-1-treated rat lung tissue, with these changes being completely reversed following treatment with the anti-inflammatory steroid dexamethasone50. Furthermore, lipopolysaccharide (LPS) signaling via the TLR4 pathway has been found to downregulate GRK2 and GRK5 expression in polymorphonuclear neutrophils (PMNs)51. Similarly, significant downregulation of GRK2 and GRK6 expression has been observed in peripheral blood mononuclear cells (PBMCs) from patients with rheumatoid arthritis (RA) or multiple sclerosis (MS)52,53,54.
Currently, studies directly investigating GRK4 in COPD are limited; however, there is substantial evidence linking GRK4 genetic variation to hypertension, where it plays a critical role in regulating receptor expression and function related to blood pressure. By modulating sodium handling in the kidneys, arterial function, and blood pressure control, GRK4 inhibition can restore normal blood pressure regulation55. This evidence suggests that GRK4 may similarly influence COPD pathophysiology through analogous signaling mechanisms. It is well-established56 that cardiovascular disease significantly contributes to the onset and progression of COPD, with both conditions sharing risk factors such as smoking, low socioeconomic status, and sedentary lifestyle. Notably, COPD patients have a higher prevalence of diabetes and hypertension compared to healthy individuals, a trend particularly pronounced in the GOLD (Global Initiative for Chronic Obstructive Lung Disease) stage III and IV subgroups of the Atherosclerosis Risk in Communities (ARIC) study57. Given the interplay between COPD and hypertension, it is plausible that GRK4 may play a cross-functional role in the pathogenesis of both conditions via shared pathophysiological pathways. One of the hallmarks of COPD is elevated oxidative stress58, which is crucial in lung tissue damage and sustained inflammation. Recent studies have shown that GRK4 expression is inherently upregulated in the kidneys and arteries of patients with essential hypertension and is significantly influenced by environmental factors such as cold stress, particulate matter (PM) exposure, and infection. These factors induce an increase in reactive oxygen species (ROS) levels, thereby upregulating GRK4 expression and affecting oxidative stress-related signaling pathways. Long-term exposure to PM2.5 has been demonstrated to increase ROS production, blood pressure, and GRK4 expression in Sprague-Dawley (SD) rats. This phenomenon is closely associated with oxidative stress, and the administration of the antioxidant Tempol, which inhibits ROS production, significantly reduces GRK4 expression in the kidneys, alleviating excessive phosphorylation of the D1 receptor (D1R). This process not only improves sodium excretion but also effectively lowers blood pressure in PM2.5-exposed SD rats, further underscoring the crucial role of oxidative stress in GRK4 regulation59,60. This mechanism may also be relevant to COPD pathogenesis, where GRK4 could influence the disease through the regulation of oxidative stress and chronic inflammatory responses. Therefore, GRK4 likely exerts significant roles beyond the cardiovascular system, potentially impacting COPD through shared physiological pathways. Future research should focus on the cross-system regulatory role of GRK4 in the cardiopulmonary system, particularly its specific functions in oxidative stress and inflammatory responses. Unraveling GRK4’s role in COPD could provide valuable insights for developing novel therapeutic targets.
Given the known role of GRK5 in regulating inflammatory responses and the general involvement of the GRK family in inflammation, we hypothesize that GRK4 may modulate the inflammatory response in COPD by influencing oxidative stress, thereby contributing to disease onset and progression. However, significant limitations persist in current research. Firstly, the specificity of GRK4’s function remains unclear. Although it may be involved in the inflammatory response, the precise regulatory mechanisms are yet to be elucidated and may be influenced by multiple factors. Secondly, many studies rely on model systems; results from animal and cell experiments may not fully represent the true pathological state in humans. Lastly, there may be redundancy and functional complementarity among GRK family members. Even if GRK4 plays a pivotal role in COPD, designing specific therapeutic strategies targeting it poses challenges.
In light of these limitations, we propose several directions for future research. First, validating the specific role of GRK4 in COPD through gene knockout or overexpression studies in cell and animal models, focusing on its impact on the expression of inflammatory markers, immune cell migration, and lung function changes. Second, employing molecular docking and molecular dynamics simulations61,62 to analyze interactions between GRK4 and associated pathways, exploring how these pathways might influence inflammatory responses in COPD. Finally, assessing GRK4 expression levels in samples from COPD patients to investigate its correlation with disease severity or prognosis, thereby validating its potential as a therapeutic target. These efforts would provide a foundation for evaluating the feasibility of GRK4 as a target for COPD treatment and offer support for future targeted therapeutic strategies.
Conclusions
This study systematically evaluated the potential role of GRK4 in COPD susceptibility, incorporating cross-tissue analysis and multi-database validation to reveal that GRK4 may play a critical role in the pathophysiological processes of COPD, particularly in oxidative stress and inflammation regulation pathways. This finding not only broadens our understanding of the systemic mechanisms underlying COPD but also offers new perspectives and entry points for multi-level, cross-tissue research on the disease. The results of this study provide essential evidence for future genetic marker screening in COPD, suggesting that GRK4 could be a valuable candidate for early detection, risk assessment, and targeted intervention. Moreover, the functional characteristics of GRK4 and its potential synergistic interactions with other GRK family members present new avenues for exploring the complex pathogenesis of COPD. Future research should focus on clarifying the specific regulatory pathways of GRK4 in oxidative stress and inflammatory responses through gene editing and molecular mechanism analysis and examining its cross-system impact in COPD and coexisting cardiovascular diseases. Overall, this study supports GRK4 as a potential target for personalized treatment strategies for COPD and lays the groundwork for the development of novel targeted therapies. Future investigations should aim to validate GRK4 expression changes at different stages of COPD and assess its clinical relevance, thereby solidifying its application as a therapeutic target and providing a scientific basis for precise treatment and intervention strategies.
Data availability
Data are publicly available.
References
Janssen, D. J. A. et al. European Respiratory Society clinical practice guideline: palliative care for people with COPD or interstitial lung disease. Eur. Respir. J. 62, 2202014 (2023).
Christenson, S. A., Smith, B. M., Bafadhel, M. & Putcha, N. Chronic obstructive pulmonary disease. Lancet 399, 2227–2242 (2022).
GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. (2018). Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392, 1789–1858
Lozano, R. et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2095–2128 (2012).
Beran, D. et al. Burden of asthma and chronic obstructive pulmonary disease and access to essential medicines in low-income and middle-income countries. Lancet Respir. Med. 3, 159–170 (2015).
Yang, I. A., Jenkins, C. R. & Salvi, S. S. Chronic obstructive pulmonary disease in never-smokers: risk factors, pathogenesis, and implications for prevention and treatment. Lancet Respir. Med. 10, 497–511 (2022).
Labaki, W. W. & Rosenberg, S. R. Chronic obstructive pulmonary disease. Ann. Intern. Med. 173, ICT17–ICT32 (2020).
Silverman, E. K. Genetics of COPD. Annu. Rev. Physiol. 82, 413–431 (2020).
Lu, Q.-Q. et al. Nitrogen-containing flavonoid and their analogs with diverse B-ring in acetylcholinesterase and butyrylcholinesterase inhibition. Drug Dev. Res. 81, 1037–1047 (2020).
Lin, X. et al. Regulation of oncoprotein 18/Stathmin signaling by ERK concerns the resistance to taxol in nonsmall cell lung cancer cells. Cancer Biother. Radiopharm. 31, 37–43 (2016).
Tang, J. et al. Spermidine-mediated poly(lactic-co-glycolic acid) nanoparticles containing fluorofenidone for the treatment of idiopathic pulmonary fibrosis. Int. J. Nanomed. 12, 6687–6704 (2017).
Lou, Y. et al. The mechanism of action of Botrychium (Thunb.) Sw for prevention of idiopathic pulmonary fibrosis based on 1H-NMR-based metabolomics. J. Pharm. Pharmacol. 76, 1018–1027 (2024).
He, J. et al. Graveoline attenuates D-GalN/LPS-induced acute liver injury via inhibition of JAK1/STAT3 signaling pathway. Biomed. Pharmacother.=Biomed. Pharmacother. 177, 117163 (2024).
Wilk, J. B. et al. Evidence for major genes influencing pulmonary function in the NHLBI family heart study. Genet. Epidemiol. 19, 81–94 (2000).
Palmer, L. J. et al. Familial aggregation and heritability of adult lung function: results from the Busselton Health Study. Eur. Respir. J. 17, 696–702 (2001).
Sollis, E. et al. The NHGRI-EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res. 51, D977–D985 (2023).
Cho, M. H. et al. A genome-wide association study of emphysema and airway quantitative imaging phenotypes. Am. J. Respir. Crit. Care Med. 192, 559–569 (2015).
Busch, R. et al. Genetic association and risk scores in a chronic obstructive pulmonary disease meta-analysis of 16,707 subjects. Am. J. Respir. Cell Mol. Biol. 57, 35–46 (2017).
Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012).
Tam, V. et al. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20, 467–484 (2019).
Hu, Y. et al. A statistical framework for cross-tissue transcriptome-wide association analysis. Nat. Genet. 51, 568–576 (2019).
Lin, D. et al. A cross-tissue transcriptome-wide association study identifies new susceptibility genes for frailty. Front. Genet. 15, (2024).
Zhu, M. et al. A cross-tissue transcriptome-wide association study identifies novel susceptibility genes for lung cancer in Chinese populations. Hum. Mol. Genet. 30, 1666–1676 (2021).
Ren, S., Sun, C., Zhai, W., Wei, W. & Liu, J. Gaining new insights into the etiology of ulcerative colitis through a cross-tissue transcriptome-wide association study. Front. Genet. 15, (2024).
Gui, J. et al. Identification of novel proteins for sleep apnea by integrating genome-wide association data and human brain proteomes. Sleep Med. 114, 92–99 (2024).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: Generalized Gene-Set Analysis of GWAS Data. Plos Comput. Biol. 11, e1004219 (2015).
Warde-Farley, D. et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 38, W214-220 (2010).
Kurki, M. I. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613, 508–518 (2023).
Zhou, W. et al. Global biobank meta-analysis initiative: Powering genetic discovery across human disease. Cell Genom. 2, 100192 (2022).
Aguet, F. et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
Lonsdale, J. et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Gaynor, S. M., Sun, R., Lin, X. & Quackenbush, J. Identification of differentially expressed gene sets using the Generalized Berk-Jones statistic. Bioinformatics (Oxford, England) 35, 4568–4576 (2019).
Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252 (2016).
Li, S. et al. Identifying causal genes for migraine by integrating the proteome and transcriptome. J. Headache Pain 24, 111 (2023).
Liao, C. et al. Transcriptome-wide association study of attention deficit hyperactivity disorder identifies associated genes and phenotypes. Nat. Commun. 10, 4450 (2019).
de Leeuw, C. A., Stringer, S., Dekkers, I. A., Heskes, T. & Posthuma, D. Conditional and interaction gene-set analysis reveals novel functional pathways for blood pressure. Nat. Commun. 9, 3768 (2018).
de Leeuw, C. A., Neale, B. M., Heskes, T. & Posthuma, D. The statistical properties of gene-set analysis. Nat. Rev. Genet. 17, 353–364 (2016).
Qi, T. et al. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat Commun. 9, 2282 (2018).
Wu, Y. et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat. Commun. 9, 918 (2018).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. Plos Genet. 10, e1004383 (2014).
Wallace, C. A more accurate method for colocalisation analysis allowing for multiple causal variants. Plos Genet. 17, e1009440 (2021).
Si, S., Liu, H., Xu, L. & Zhan, S. Identification of novel therapeutic targets for chronic kidney disease and kidney function by integrating multi-omics proteome with transcriptome. Genome Med. 16, 84 (2024).
Jiang, Y. et al. Endocrine and metabolic factors and the risk of idiopathic pulmonary fibrosis: A Mendelian randomization study. Front. Endocrinol. 14, 1321576 (2023).
Hobbs, B. D. & Hersh, C. P. Integrative genomics of chronic obstructive pulmonary disease. Biochem. Biophys. Res. Commun. 452, 276–286 (2014).
Pillai, S. G. et al. A Genome-Wide Association Study in Chronic Obstructive Pulmonary Disease (COPD): Identification of Two Major Susceptibility Loci. Plos Genet. 5, e1000421 (2009).
Cho, M. H. et al. A genome-wide association study of COPD identifies a susceptibility locus on chromosome 19q13. Hum. Mol. Genet. 21, 947–957 (2012).
Li, L. et al. G protein-coupled receptor kinases of the GRK4 protein subfamily phosphorylate inactive G protein-coupled receptors (GPCRs). J. Biol. Chem. 290, 10775–10790 (2015).
Métayé, T., Gibelin, H., Perdrisot, R. & Kraimps, J.-L. Pathophysiological roles of G-protein-coupled receptor kinases. Cell. Signal. 17, 917–928 (2005).
Mak, J. C. W., Hisada, T., Salmon, M., Barnes, P. J. & Chung, K. F. Glucocorticoids reverse IL-1beta-induced impairment of beta-adrenoceptor-mediated relaxation and up-regulation of G-protein-coupled receptor kinases. Br. J. Pharmacol. 135, 987–996 (2002).
Fan, J. & Malik, A. B. Toll-like receptor-4 (TLR4) signaling augments chemokine-induced neutrophil migration by modulating cell surface expression of chemokine receptors. Nat. Med. 9, 315–321 (2003).
Lombardi, M. S. et al. Decreased expression and activity of G-protein-coupled receptor kinases in peripheral blood mononuclear cells of patients with rheumatoid arthritis. FASEB: J. Off. Publ. Fed. Am. Soc. Exper. Biol. 13, 715–725 (1999).
Giorelli, M., Livrea, P. & Trojano, M. Post-receptorial mechanisms underlie functional disregulation of beta2-adrenergic receptors in lymphocytes from Multiple Sclerosis patients. J. Neuroimmunol. 155, 143–149 (2004).
Vroon, A. et al. G protein-coupled receptor kinase 2 in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Immunol. (Baltimore, Md: 1950) 174, 4400–4406 (2005).
Yang, J., Hall, J. E., Jose, P. A., Chen, K. & Zeng, C. Comprehensive insights in GRK4 and hypertension: From mechanisms to potential therapeutics. Pharmacol. Ther. 239, 108194 (2022).
Maclay, J. D. & MacNee, W. Cardiovascular disease in COPD. Chest 143, 798–807 (2013).
Mannino, D. M., Thorn, D., Swensen, A. & Holguin, F. Prevalence and outcomes of diabetes, hypertension and cardiovascular disease in COPD. Eur. Respir. J. 32, 962–969 (2008).
Barnes, P. J. Oxidative stress-based therapeutics in COPD. Redox Biol. 33, 101544 (2020).
Sun, D., Chen, K., Wang, J., Zhou, L. & Zeng, C. In-utero cold stress causes elevation of blood pressure via impaired vascular dopamine D1 receptor in offspring. Clin. Exper. Hypertens. (New York N.Y 1993). 42, 99–104 (2020).
Lu, X. et al. Long-term exposure of fine particulate matter causes hypertension by impaired renal D1 receptor-mediated sodium excretion via upregulation of G-protein-coupled receptor kinase type 4 expression in Sprague-Dawley rats. J. Am. Heart Assoc. 7, e007185 (2018).
Hasan, M. et al. Contriving a chimeric polyvalent vaccine to prevent infections caused by herpes simplex virus (type-1 and type-2): An exploratory immunoinformatic approach. J. Biomol. Struct. Dyn. 38, 2898–2915 (2020).
Roney, M. et al. Pharmacophore-based virtual screening and in-silico study of natural products as potential DENV-2 RdRp inhibitors. J. Biomol. Struct. Dyn. 41, 12186–12203 (2023).
Acknowledgements
Guanglei Chen: conceptualization, methodology, writing—original draft. Cancan Chu: methodology, software, investigation. Yaxian Jin: methodology, software, investigation. Yuhao Zheng: methodology, software, investigation. Changfu Yang: supervision, funding acquisition. Yunzhi Chen: writing—review and editing. Xing Zhu: conceptualization, writing—review and editing, supervision, funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This work received support from the National Natural Science Foundation of China (Grant No. 81960830), Guizhou University of Traditional Chinese Medicine Talent Innovation Team [Gui Traditional Chinese Medicine TD He Zi [2023] 002] and the Research Platform Team Project for Provincial Universities (Grant No. Qianjiaoji [2022] 023) with continuous backing from the Department of Education.
Author information
Authors and Affiliations
Contributions
G. C.: conceptualization, methodology, writing—original draft. C. C.: methodology, software, investigation. Y. J.: methodology, software, investigation. Y. Z.: methodology, software, investigation. C. Y.: supervision, funding acquisition. Y. C.: writing—review and editing. X. Z.: conceptualization, writing—review and editing, supervision, funding acquisition. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Ethical approval was obtained for all original studies.
Informed consent
Informed consent was obtained for all original studies. The requirement for patient consent was waived owing to the retrospective nature of this study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, G., Jin, Y., Chu, C. et al. A cross-tissue transcriptome-wide association study reveals GRK4 as a novel susceptibility gene for COPD. Sci Rep 14, 28438 (2024). https://doi.org/10.1038/s41598-024-80122-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-80122-w
