
Abstract
Ferroptosis is linked to various pathological conditions; however, the specific targets and mechanisms through which traditional Chinese medicine influences ischemic stroke (IS)-induced ferroptosis remain poorly understood. In this study, data from the Gene Expression Omnibus and disease target databases (OMIM, GeneCards, DisGeNet, TTD, and DrugBank) were integrated with ferroptosis-related gene datasets. To identify key molecular targets of Chuanxiong Rhizoma (CX), drug ingredient databases, including PubChem and TCMBank, were employed to map CX-related targets (CX-DEGs-FRG and CX-IS-FRG). Gene targets and relevant signaling pathways were analyzed using weighted gene co-expression network analysis, protein-protein interaction networks, Gene Ontology, and Kyoto Encyclopedia of Genes and Genomes pathway enrichment. The least absolute shrinkage and selection operator regression and support vector machine methods were utilized to identify intersecting genes, and the predictive accuracy of core targets was evaluated through receiver operating characteristic curve analysis. Immune cell infiltration in the IS microenvironment was assessed using CIBERSORT, followed by molecular docking of CX’s active components with key targets. The JAK-STAT3 pathway was identified as a critical regulatory mechanism, and five key targets (ALOX5, PTGS2, STAT3, G6PD, and HIF1A) emerged as central to the IS-induced ferroptosis. Elevated infiltration of CD8 + T cells and neutrophils was significantly correlated with IS. Notably, the active components mandenol and myricanone demonstrated strong binding affinities with these five targets, which validated the results from network-based analysis. In conclusion, the JAK-STAT3 pathway, through its regulation of ALOX5, PTGS2, STAT3, G6PD, and HIF1A, could play a crucial role in modulating ferroptosis and immune responses in IS. These findings suggest that CX could serve as a potential therapeutic approach for IS, targeting the regulation of IS-induced ferroptosis and the immune microenvironment.
Similar content being viewed by others

Introduction
Stroke is the second leading cause of mortality and the third primary cause of impairment globally. Recent statistics indicated that the incidence of stroke has risen by 70.0% over the past two decades, with fatalities increasing by 43.0% and overall incidence rising by 102.0%1. Ischemic stroke (IS), accounting for 80–85% of all stroke cases worldwide, is a significant contributor to human mortality2. Consequently, there is a pressing need for rigorous research into stroke treatment. IS, characterized by an abrupt deficit in neurological function, results from a disruption in blood supply to a specific area of the brain3. The pathophysiology of IS is complex that comprises various cellular and molecular processes, including neuroinflammation, oxidative stress, autophagy, and apoptosis. These mechanisms may lead to dysfunction of the blood-brain barrier, alterations in cerebral blood flow, damage to brain tissue, and neurological impairments4. Traditional Chinese medicine (TCM) plays a crucial role in the treatment of IS5,6. Previous studies have demonstrated the pathophysiological effects of TCM on IS through machine learning techniques7,8.
The Stockwell Laboratory introduced the concept of ferroptosis in 2012, highlighting its significance in lipid and amino acid metabolism, reactive oxygen species (ROS) management, and iron regulation. Previous studies have established a link between ferroptosis and various neurological disorders, including neurodegenerative diseases, hemorrhagic stroke, and IS. Two hallmark phenomena associated with ferroptosis, lipid peroxidation and iron accumulation, have been identified, suggesting that ferroptosis may contribute to neuronal damage following stroke. This indicates that targeting ferroptosis could represent a viable therapeutic option for IS.
Further studies on the drugs and mechanisms associated with IS-induced ferroptosis may lead to significant advancements in its detection and treatment. Previous research on Chuanxiong Rhizoma (CX) for the treatment of stroke revealed the mechanisms of ferroptosis; however, the effective components, pathways, and targets of CX in the context of IS-induced ferroptosis remain unexplored.
The immunological microenvironment plays a critical role in modulating immunosuppression, peripheral immune system activation, and neuroinflammation following stroke9. Significant disparities exist in overall gene expression and biological function in IS, accompanying by marked impairments observed in inflammatory and immune functions10. Signal transducer and activator of transcription 3 (STAT3), a key component of the acute phase response factor complex activated by interleukin-6 (IL-6), was identified over 20 years ago11,12. Both excessive activation and insufficient activity of STAT3 can lead to disease, highlighting the importance of precise regulation of STAT3 function to maintain health. During stroke, STAT3 activation exerts both pro-inflammatory and anti-inflammatory effects across various cell types and stages. STAT3 regulates the production of key regulatory transcription proteins, including Eomes, BCL-6, and Blimp-1, as well as the cytokine signaling inhibitor SOCS3. This regulation is evidenced by the reduced levels of these molecules in memory CD8 + T cells deficient in STAT3.
This research examined the molecular mechanisms of CX in IS-induced ferroptosis and the associated immunological microenvironment through network pharmacology and molecular docking techniques. Data of patients with IS were extracted from public databases, revealing eight models of IS-induced ferroptosis-related genes (FRGs). Additionally, functional enrichment and immune infiltration analyses were performed to explore potential underlying mechanisms. Two machine-learning techniques were employed to forecast illness while identifying genes with notable characteristics. Following screening and verification, the effects of the TAK/STAT3 pathway on ferroptosis, CD8 + T cell activity, and neutrophil infiltration in IS were analyzed, thereby providing a theoretical foundation for future experimental research.
Results
Gene targets
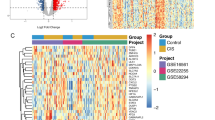
The GSE16561 dataset comprised 39 patients who were diagnosed with IS, as well as 24 healthy controls. The analysis of differentially expressed genes (DEGs) revealed a total of 96 upregulated genes and 324 downregulated genes (Fig. 1), including ALOX5 and PTGS2, which were specifically downregulated (Supplementary Table S1). The GeneCards database was queried for obtaining 8914 gene targets associated with ischemic cerebral apoplexy, yielding 1090 highly expressed genes with a deep score of greater than 2.36. Additionally, 20 disease targets were acquired from the OMIM database, 17 from the TTD, 1159 from DisGeNet, and 144 from DrugBank. After combining and eliminating duplicates, a total of 2245 unique targets were identified. A subsequent search in FerrDb V2, followed by the removal of duplicates, resulted in the identification of 1993 FRGs, which were utilized for further analyses.

Gene Targets in GSE16561 for Disease. (a) Volcano plot displaying genes with significant differential expression. Filtering criteria include LogFC ≥ 0.58 and P < 0.05. Genes significantly downregulated are represented by blue dots, significantly upregulated by red dots, and those with no significant change by black dots. (b) Heatmap of differentially expressed genes (DEGs) in the GSE16561 dataset. Red indicates increased gene expression, while blue indicates decreased gene expression.
Drug component targets of Chuanxiong
The active components of the drug were identified through searching for Canonical SMILES in PubChem, followed by target identification using Swiss TargetPrediction. A diagram summarizing the active ingredients and their corresponding targets is presented in Supplementary Table S2. Figure 2 illustrates the application of drug active component targets using Cytoscape software, highlighting the top five drug components with the highest screening degree values, which were marked in red, involving citronellyl acetate, nerol acetate, linoleic acid, mandenol, and myricanone (Table 1).

Components and targets of Chuanxiong Rhizoma. A schematic representation of components and targets of Chuanxiong Rhizoma. The purple section highlights the active chemical components, the green section represents the associated targets, and the main components are highlighted in red.
Weighted gene co-expression network analysis (WGCNA)
The GSE16561 dataset was utilized to perform WGCNA, revealing modules composed of highly interconnected genes. A scale-free co-expression network was constructed using a soft threshold of 0.18, resulting in favorable mean connectivity (Fig. 3a). Modules exhibiting strong correlations were merged using a clustering height threshold of 10. This process led to the identification of 14 distinct signature modules, each assigned a unique color (Fig. 3b). The association between each module and clinical characteristics was evaluated. The results revealed a negative correlation between the MEbrown module and controls (r = − 0.6, P = 5e-07), as well as a positive correlation between the MEbrown module and stroke (r = 0.6, P = 5e-07). In contrast, the MEyellow module exhibited a negative correlation with stroke (r = − 0.75, P = 1e-11) and a positive correlation with healthy controls (r = 0.75, P = 1e-11) (Fig. 3c). These findings suggest that the MEbrown and MEyellow modules have clinical significance.

Weighted gene co-expression network analysis. (a) Determination of the optimal soft threshold through network topology analysis. (b) Dendrogram showing the clustering of genes, with different colors representing distinct modules. (c) Heatmap illustrating the correlation between gene modules and ischemic stroke (IS) traits. The MEbrown module demonstrates the highest correlation with IS.
Intersection genes
Intersection genes from the DEGs, FRGs, and Chuanxiong drug target datasets (CX-DEGs-FRGs) were identified using a Venn diagram. As illustrated in Fig. 4a, two intersection genes, ALOX5 and PTGS2, were highlighted. In Fig. 4b, a Venn diagram was employed to identify the common genes between the IS disease targets, FRGs, and Chuanxiong drug targets (CX-IS-FRGs), resulting in the identification of 42 intersection genes.

Venn diagram of the intersectional targets. (a) The intersectional targets among CX, DEGs, and FRGs. (b) The intersectional targets among CX, IS, and FRGs.
CX-IS-FRG intersections in protein-protein interaction (PPI) network analysis
The PPI network of CX-IS-FRGs consisted of 42 nodes and 381 edges. Network analysis was performed to assess two topological properties of the 42 targets, aiming to identify the most significant nodes in the network. Using Cytoscape software, the 42 nodes were visualized based on their degree values (Fig. 5a). Further analysis in the subnetwork facilitated the identification of key target genes (Fig. 5b). Ultimately, five genes were identified as the primary intersection gene targets: IL-6, serum albumin (ALB), peroxisome proliferator-activated receptor gamma (PPARG), hypoxia-inducible factor 1 alpha (HIF1A), and STAT3 (Table 2).

Protein-protein interaction (PPI) network and module analysis. (a) PPI network of CX-IS-FRGs, consisting of 42 nodes and 381 edges. (b) PPI network analysis of core targets identified in the CX-IS-FRGs.
Gene Ontology (GO) and Kyoto Gene and Genome Database (KEGG) pathway enrichment analyses
To further elucidate the functions and regulatory pathways associated with central intersection gene targets, GO and KEGG pathway enrichment analyses were conducted on these gene targets. The biological processes (BP), cellular components (CC), and molecular functions (MF) of 42 potential genes were examined, ultimately leading to identify five critical genes. The most significant statistical results are presented in Fig. 6a, b, and c. The GO analysis revealed that CX-IS-FRGs were predominantly concentrated in the following categories: GO:0031667 (response to nutrient levels), GO:1,904,813 (ficolin-1-rich granule lumen), and GO:0008134 (transcription factor binding). Additionally, KEGG pathway analysis of shared elements indicated that CX-IS-FRGs were primarily involved in the following signaling pathways: hsa05200 (pathways in cancer), hsa05167 (Kaposi sarcoma-associated herpesvirus infection), and hsa05208 (chemical carcinogenesis-ROS), as illustrated in Fig. 7a. A chord diagram was generated using R language for the enrichment analysis (Fig. 7b), and the core genes were labeled in the signaling pathways. Notably, STAT3 and HIF1A exhibited significantly higher gene expression levels (Fig. 7c).

GO enrichment analysis of CX-IS-FRGs. (a) GO analysis of biological processes associated with CX-IS-FRGs. (b) GO molecular function analysis. (c) GO cellular component analysis. Purple indicates lower logP-values, while blue indicates higher logP-values.

Results of the KEGG pathway enrichment analysis. (a),(b). KEGG enrichment analysis. (c). The mechanism by which CX active components affect IS through the “hsa05200 Pathways in cancer” pathway. Solid lines indicate promotion of signaling, while dashed lines indicate inhibition. Red targets enhance signaling, and green targets inhibit signaling.
Immune microenvironment analysis
Analysis of the immune microenvironment revealed a significant increase in the infiltration of CD8 + T cells and neutrophils, as illustrated in Fig. 8a. Heat maps depicting CX-IS-FRGs, CX-GES-FRGs, and immune infiltration are presented in Fig. 8b. Notably, ALOX5, STAT3, and PTGS2 exhibited significant correlations with CD8 + T cell levels, while ALOX5, G6PD, HIF1A, PTGS2, and STAT3 were significantly associated with neutrophil levels.

Results of immune infiltration by CIBERSORT. (a). Boxplot displaying the distinct immune cell infiltration profiles across 22 immune cell types. (b). Heatmap illustrating the relationship between intersectional targets and 22 immune cell types. “*” indicates P < 0.05, “**” represents P < 0.01, “***” indicates P < 0.001, and “****” represents P < 0.0001.
Screening feature genes
To improve the selection of junction genes, the least absolute shrinkage and selection operator (LASSO) regression and support vector machine (SVM) analysis were performed to identify unique genes. Following LASSO regression analysis, 7 of the 8 intersecting genes, ALOX5, PTGS2, STAT3, G6PD, HIF1A, ALB, and PPARG, were identified (Fig. 9a). After SVM regression analysis, 5 out of the 8 intersecting genes were selected: ALOX5, PTGS2, STAT3, G6PD, and HIF1A (Fig. 9b). Thereafter, shared genes were identified. Finally, 5 particular genes, ALOX5, PTGS2, STAT3, G6PD, and HIF1A, were discerned (Fig. 9c).

Feature gene selection. (a) Feature genes selected using the LASSO logistic regression algorithm, with penalty parameter tuning performed by 10-fold cross-validation, identifying 7 CX-IS-FRGs. (b) Feature gene selection using the SVM-RFE algorithm, filtering 8 core targets and identifying the optimal combination of feature genes. (c) Marker genes identified by both LASSO and SVM-RFE models.
Core CX-IS-FRGs target IS and receiver operating characteristic (ROC) curve analysis
The GSE16561 dataset revealed significant differences in the expression levels of ALOX5, PTGS2, STAT3, G6PD, and HIF1A between the IS and control groups (Fig. 10a). The identified genes were subsequently validated, and a ROC curve was plotted to evaluate their clinical utility. The area under the curve (AUC) values for STAT3, ALOX5, HIF1A, PTGS2, and G6PD were 0.875, 0.8194, 0.7906, 0.7873, and 0.7533, respectively, and STAT3 demonstrated the highest diagnostic value. Through a comprehensive analysis of these characteristic genes, a logistic regression model was developed and a ROC curve was plotted (Fig. 10b).

Core target expression and ROC analysis. (a) Boxplots displaying the expression levels of five core targets. (b) ROC curve analysis for the five core targets.
Molecular docking
Five targets, including STAT3, ALOX5, HIF1A, PTGS2, and G6PD, along with the drug components citronellyl acetate, nerol acetate, linoleic acid, mandenol, and myricanone were evaluated for molecular docking. The binding energies, presented in Table 3, illustrate the spontaneous interactions between these small pharmacological molecules and the target proteins. Among the five strongest interactions, myricanone exhibited binding to the amino acids ASP-333, GLU-146, ARG-143, ASP-156, and GLU-334 in ALOX5 via hydrogen bonds, with a binding energy of -7.3 kJ/mol (Fig. 11a). Additionally, myricanone bound to the amino acids THR-334, ARG-257, and ARG-175 of G6PD through hydrogen bonds, yielding a binding energy of -8.9 kJ/mol (Fig. 11b). Furthermore, myricanone interacted with the amino acids ASP-270 and ARG-754 of HIF1A through hydrogen bonds, resulting in a binding energy of -5.8 kJ/mol (Fig. 11c). In PTGS2, myricanone formed hydrogen bonds with TYR-136, leading to a binding energy of -7.1 kJ/mol (Fig. 11d). Finally, myricanone established a bond with LYS-370 in STAT3 through hydrogen bonds, which exhibited a binding energy of -6.9 kJ/mol (Fig. 11e).

Molecular docking results. (a) Molecular docking of Myricanone and ALOX5. (b) Molecular docking of Myricanone and G6PD. (c) Molecular docking of Myricanone and HIF1A. (d) Molecular docking of Myricanone and PTGS2. (e) Molecular docking of Myricanone and STAT3.
Discussion
Ferroptosis represents an emerging area of research, with the aim of identifying new biomarkers and treatment targets. Previous studies have demonstrated that abnormalities in iron metabolism play a significant role in IS. Specifically, iron metabolism is dysregulated, resulting in iron accumulation in the brain following IS13. The acidic environment of brain tissue following cerebral ischemia may hinder the binding of iron to transferrin, promoting its release from transferrin14. Consequently, neurons readily absorb this free iron, leading to the elevated cellular iron level15. Furthermore, ferritin level is significantly elevated in astrocytes and macrophages located at the periphery of the injury in the ischemic cortex16. However, the therapeutic targets of TCM for IS-induced ferroptosis remain obscure.
CX is a pharmaceutical agent that stimulates blood flow and prevents clotting. It enhances blood circulation, facilitates the movement of vital energy, alleviates pain, and helps manage symptoms associated with conditions involving neurological or vascular dysfunction. Consequently, CX is frequently employed in the treatment of IS17,18,19. However, the precise mechanism by which CX influences IS-induced ferroptosis remains unclear. CX significantly reduces cell survival, disrupts mitochondrial structure and function, and exacerbates ferroptosis induced by ROS through activation of the Nrf2/HO-1 signaling pathway in BC cells20. Furthermore, CX regulates ferritin autophagy, promotes autophagy activation, facilitates ferritin degradation, and contributes to the accumulation of excess iron and lipid oxidation in the cerebellum of mice21. In rat models, CX decreased concentrations of ferrous iron, ROS, and malondialdehyde while increasing levels of superoxide dismutase and glutathione, thereby deaccelerating the progression of ferroptosis22. Analysis of the chemical composition and target components of CX revealed that citronellyl acetate, nerol acetate, linoleic acid, mandenol, and myricanone exhibited the strongest correlation with IS-induced ferroptosis. Notably, citronellyl acetate can cross the blood-brain barrier and influence the central nervous system23. Additionally, genetically predicted levels of circulating linoleic acid are associated with a reduced risk of IS, suggesting that moderate consumption of linoleic acid may help prevent IS24. Network pharmacology analyses indicate that mandenol has therapeutic effects on chronic cerebral circulation25 and also provides therapeutic benefits for neuropathic pain26.
The immunological environment and inflammatory response are critical factors, influencing both incidence and progression of IS, as well as being closely associated with its prognosis and severity9. The inflammatory immune response plays a crucial role at all stages of IS pathology, which is characterized by the production of inflammatory cytokines and the migration of immune cells26. Numerous studies have demonstrated that stroke can adversely affect the activity of the peripheral immune system27. In the present study, the analysis of the immune environment revealed a distinct differential expression of CD8 + T cells and neutrophils. Following antigen activation, CD8 + T cells can release perforin and granzymes through cell-cell interactions or induce neuronal death via the Fas-FasL pathway. This mechanism directly contributes to the death of damaged cells during cerebral ischemia28,29. Notably, CD8 + T cells are mobilized within 24 h following an ischemic event and reach their peak concentration during the early inflammatory response, approximately 3–4 days post-injury30. Immune cells in the bloodstream adhere to the inner lining of blood vessels, stimulating the production of molecules, thereby promoting cell adhesion and recruiting additional immune cells. Neutrophils, a component of the innate immune system, become activated and migrate toward the area of reduced blood supply in response to chemical signals, penetrating the compromised blood-brain barrier to access the surrounding tissue31. Neutrophils, mast cells, and other immune cells secrete intracellular MMPs, which degrade the vascular basement membrane and tight junction proteins at the site of ischemic damage. This process accelerates the disruption of the blood-brain barrier and contributes to the expansion of the cerebral infarction area32. Coordinated M2-type microglia eliminate invading neutrophils, thereby preventing cytotoxic substances-induced neuronal damage33,34.
In the present study, five core targets were combined with eight CX-DEGs-FRGs targets identified through PPI network analysis, involving ALOX5, PTGS2, STAT3, G6PD, HIF1A, IL6, ALB, and PPARG. These targets were further validated by comparing the IS group with the healthy group. The results indicated that ALOX5, PTGS2, STAT3, G6PD, and HIF1A were significantly expressed. Notably, ALOX5 could play a substantial role in linking the inflammatory cascade with oxidative stress pathways, which is critical for the progression of IS35. HIF1-α is known to activate angiogenesis and neovascularization, thereby improving stroke prognosis36. Furthermore, a deficiency in HIF1-α reduces enhancements in neuronal viability and sensory functions in cases of hypoxic-ischemic damage and IS37,38. Meanwhile, the regulation of the HIF1-α/VEGF pathway following IS can protect the brain from the destruction of the blood-brain barrier, inflammation, neuronal apoptosis, brain edema, and neurological dysfunction39. Inhibition of PTGS2 has exhibited to promote angiogenesis in patients with IS40. In a rat model of middle cerebral artery occlusion, such treatment could reduce the size of the damaged area, alleviate neurological deficits, and improve the severity scores associated with neurological impairment following IS. Furthermore, this intervention could significantly enhance overall neurological outcomes in rats41. Adeficiency in G6PD is associated with an increased risk of adverse outcomes three months post-IS and may also elevate the likelihood of mortality during hospitalization42. However, age-related changes may contribute to the development of atherosclerosis43. Subsequently, the GO and KEGG pathway enrichment analyses were conducted, which revealed significant expression of the JAK-STAT3 pathway. Previous studies have demonstrated that the administration of JAK-STAT3 inhibitors could lead to a marked reduction in neurological deficits, infarct size, and astrocytic differentiation in the ipsilateral brain44. Additionally, these inhibitors have been reported to be associated with the suppression of the pro-inflammatory response, thereby mitigating IS injury45. Furthermore, the promotion of STAT3 phosphorylation has been demonstrated to modulate inflammation in peripheral neutrophils following ischemia46.
Excessive STAT3 activation in immune cells infiltrating tumors leads to immunosuppression by inhibiting both innate and adaptive immune responses. Increased STAT3 activity in specific subsets of the adaptive immune system can hinder the development of effector T cells47,48,49. Concurrently, the increase in STAT3 signaling in tumor cells diminishes the immune response of CD8 + T cells, impairing their ability to combat tumor growth50. Inhibition of the AKT-STAT3 pathway regulates microglial and astrocyte activation, the production of pro-inflammatory cytokines, and the continuous migration of neutrophils in the brain51. STAT3 also triggers the production of lipoprotein-2n, which is a glycoprotein released by N2-neutrophils. Furthermore, STAT3 stimulates the conversion of tumor cells from a mesenchymal to an epithelial state, promoting cancer cell metastasis and proliferation at distant sites52. HIF transcription factors are essential for embryonic development and play a role in wound healing following ischemia-reperfusion injuries, as well as in the resolution of inflammation in adults53. In low-oxygen conditions, HIF1-α is activated in CD4 + and CD8 + T lymphocytes upon activation, leading to metabolic changes that promote glycolysis and the initiation of pathways derived from glycolytic substrates54. The activation and migration of neutrophils rapidly deplete the available oxygen in the surrounding area, triggering localized activation of HIF signaling in epithelial cells. This activation enhances barrier function and prevents the infiltration of additional pro-inflammatory immune cells55. In zebrafish, HIF1A activation reduces neutrophil death, leading to delayed inflammation resolution and the persistence of pro-inflammatory characteristics56.
Following molecular docking analysis, the binding energies of mandenol with STAT3, ALOX5, HIF1A, PTGS2, and G6PD were found to be the highest. This suggests a strong affinity of CX for these therapeutic targets, indicating its potential as an effective drug for modulating IS-induced ferroptosis and influencing the immune microenvironment.
In summary, this research demonstrated that the active components of CX could interact with key proteins, including STAT3, ALOX5, HIF1A, PTGS2, and G6PD, which are critical in modulating IS-induced ferroptosis and the JAK-STAT3 signaling pathway. The stable interactions between CX’s active compounds and these targets may elucidate the underlying mechanisms that contribute to the efficacy of CX in this context.
Materials and methods
Data collection and acquisition
The Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo/) database was utilized to identify genes. Microarray data were obtained from IS samples57. Two databases, GSE195442 and GSE16561, were utilized, in which the GSE16561 dataset was ultimately selected, and probes were assigned to gene symbols using the GPL6883 platform. The keywords of “ischemic stroke,” “cerebral ischemic,” and “cerebral infarction” were used to extract the corresponding genes from the OMIM (https://www.omim.org), GeneCards (https://www.genecards.org), DisGeNet (https://www.disgenet.org/home/), TTD (https://db.idrblab.net/ttd/), and DrugBank (https://www.drugbank.com) databases. FRGs involved in driving, suppressing, marking, regulating, inducing, and inhibiting ferroptosis were obtained from the publicly available FerrDb V2 database (http://www.zhounan.org/ferrdb)58.
Drug components and targets of Chuanxiong
The active ingredients of the drug were identified using the TCMSP (https://old.tcmsp-e.com/tcmsp.php), PubChem (https://pubchem.ncbi.nlm.nih.gov) and TCM Bank (https://tcmbank.cn) databases. The potential active ingredients of CX were initially screened based on the following specific criteria: oral bioavailability (OB) ≥ 30% and drug likeness (DL) ≥ 0.18. Subsequently, canonical SMILES were searched to identify targets with probability scores greater than 0, utilizing Swiss TargetPrediction database (http://www.swisstargetprediction.ch). Duplicate drug component targets were removed, and the resulting network was visualized using Cytoscape 3.10.1 software (http://www.cytoscape.org/)59. The core chemical composition was selected based on the degree value.
DEGs screening
The “limma” package in R (version 3.48.3) was utilized to identify DEGs in the GSE16561 dataset between the control and IS groups, applying a threshold of |log2FC| > 0.58 and P < 0.05. The “pheatmap” package in R was employed to illustrate variations in gene expression through a heatmap (https://CRAN.R-project.org/package=pheatmap). The DEGs were integrated with the targets of CX components and FGRs, and the intersection genes were determined using Venn diagram analysis. Additionally, the intersection genes among all disease targets of IS, as well as the component targets of CX and FGRs, were also identified through Venn diagram analysis, leading to the derivation of CX-DEGs-FRGs and CX-IS-FRGs, respectively.
WGCNA
WGCNA is suggested for identifying gene clusters that exhibit strong associations with other clusters. Additionally, it can evaluate the correlation between these gene clusters and external variables. The correlation between modules and attributes of the genes or samples in the GSE16561 dataset was assessed using the “cor” function in R Studio. Samples and genes were filtered through rigorous assessments, while outlier samples were identified via hierarchical clustering and were subsequently removed, where a cutHeight of 70 was applied. A scale-free network was constructed by evaluating various power values (ranging from 1 to 30), and the appropriate power value was selected when the scale-free R² yielded 0.9. Following this, cluster analysis was carried out to identify significantly comparable modules, establishing a minimum module size of 30. To validate the relationship between the modules and characteristics, the gene module affiliation and the relevance of gene phenotypes in the selected modules were assessed.
PPI network construction
The Retrieval of Interacting Genes tool (STRING; http://string.embl.de/) was employed to construct a PPI network for the identified CX-IS-FRGs60. The protein type was specified as “Homo sapiens,” and the minimal interaction threshold was adjusted to “Medium confidence,” while all other parameters remained at their default settings. The PPI network was visualized using Cytoscape software (http://www.cytoscape.org/), where the nodes in the structure represented genes and the edges illustrated the connections between them.
The GO and KEGG pathway enrichment analyses
For CX-IS-FRGs in BP, CC, and MF, the GO database (http://geneontology.org/) was utilized to elucidate their potential biological mechanisms. The CX-IS-FRGs underwent GO enrichment analysis to explore the underlying biological processes, cellular elements, and molecular activities. Additionally, the KEGG pathway analysis was employed to predict the involvement of protein interaction networks in various biological functions61,62,63. The CX-IS-FRGs were analyzed using the Metascape online tool (https://metascape.org/) for the GO enrichment analysis, which included MF, BP, and CC, as well as the KEGG pathway analysis. The resulting data were visualized using bubble mapping with GraphPad Prism 9.5.1 software (GraphPad Software Inc., San Diego, CA, USA). Additionally, the ‘clusterProfiler’ package in R was utilized for the KEGG pathway analysis on the significant DEGs, applying a selection criterion of P < 0.05.
Immune cell correlation analysis
The CIBERSORT utilizes a deconvolution technique to transform standardized gene expression matrices into the proportions of 22 immune cell types64. The CIBERSORT was employed to predict the relative abundances of these 22 immune cell categories infiltrating each tissue in the GSE16561 dataset. The relationships between specific genes and various invading immune cells were assessed using Spearman’s correlation analysis. To investigate the association between immune cell composition and the expression levels of specific genes, the ‘ggplot2’ package in R was utilized to conduct correlation analysis and generate figures. Additionally, the Pearson’s correlation coefficients among the immune cells were calculated via the ‘corrplot’ package in R, and the results were displayed in the corresponding heatmap.
Screening of feature genes
The LASSO algorithm, a regression-based method, improves predictive accuracy by applying normalization techniques to select relevant features. SVM is a supervised machine learning technology that is commonly utilized for both classification and regression tasks65. To prevent overfitting, the most appropriate genes were selected from the metadata dataset using the recursive feature elimination (RFE) approach. The distinctive genes were identified through both LASSO regression and SVM-RFE techniques. The genes identified by both methods were intersected, resulting in the identification of intersecting genes, which were considered as highlight genes.
ROC curve analysis
To investigate the impact of CX on the expression of IS-induced ferroptosis, the control and IS groups were compared using GraphPad Prism 9.5.1 software, concentrating on the expression levels of core targets identified in the GSE16561 database. The two groups were analyzed using independent sample t-test, with a significance threshold set at P < 0.05 to determine statistical significance. Additionally, ROC curve analysis66 was employed to evaluate the diagnostic efficacy of these targets in IS. Potential biomarkers were identified by screening targets with an AUC ≥ 7.0 and a P-value < 0.05.
Molecular docking
The targets exhibiting noteworthy features, as determined by ROC curve analysis, were selected for molecular docking, including the top five active elements of CX. Protein structures were obtained from the Uniprot and RCSB PDB databases, while the PubChem database was employed to acquire the structures of the active components. AutoDock 1.5.7 software was utilized for the dehydration, hydrogenation, and energy minimization of small molecule ligands, as well as for identifying active pockets and performing molecular docking between the compounds and their targets. Following the official AutoDock guidelines, 50 molecular docking procedures were conducted for each combination of effective constituents and targets67. The interaction avidity between each molecule and its target was considered satisfactory if the molecular docking binding energy was ≥ 5 kJ/mol68. The results were visualized using PyMOL 2.2.0 software.
Data availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
References
Zhang, H., Sun, X., Xie, Y., Zan, J. & Tan, W. Isosteviol Sodium Protects Against Permanent Cerebral Ischemia Injury in Mice via Inhibition of NF-κB-Mediated Inflammatory and Apoptotic Responses. J. Stroke Cerebrovasc. Dis. 26 (11), 2603–2614 (2017).
Campbell, B. C. V. et al. Ischaemic stroke. Nat. Rev. Dis. Primers. 5 (1), 70 (2019).
Haupt, M., Gerner, S. T., Bähr, M. & Doeppner, T. R. Neuroprotective Strategies for Ischemic Stroke-Future Perspectives. Int. J. Mol. Sci. 24 (5), 4334 (2023).
Dixon, S. J. et al. Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149 (5), 1060–1072 (2012).
Xu, A. et al. Elucidating the Synergistic Effect of Multiple Chinese Herbal Prescriptions in the Treatment of Post-stroke Neurological Damage. Front. Pharmacol. 13, 784242 (2022).
Xu, A. et al. Screening of key functional components of Taohong Siwu Decoction on ischemic stroke treatment based on multiobjective optimization approach and experimental validation. BMC Complement. Med. Ther. 23 (1), 178 (2023).
Liu, J. Q. et al. The identification and molecular mechanism of anti-stroke traditional Chinese medicinal compounds. Sci. Rep. 7, 41406 (2017).
Li, W. X. et al. Integrated analysis of ischemic stroke datasets revealed sex and age difference in anti-stroke targets. PeerJ 4, e2470 (2016).
Zera, K. A. & Buckwalter, M. S. The Local and Peripheral Immune Responses to Stroke: Implications for Therapeutic Development. Neurotherapeutics 17 (2), 414–435 (2020).
Li, W. X. et al. Different impairment of immune and inflammation functions in short and long-term after ischemic stroke. Am. J. Transl Res. 9 (2), 736–745 (2017).
Lütticken, C. et al. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science 263 (5143), 89–92 (1994).
Zhong, Z., Wen, Z. & Darnell, J. E. Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264 (5155), 95–98 (1994).
van Etten, E. S. et al. MRI Susceptibility Changes Suggestive of Iron Deposition in the Thalamus after Ischemic Stroke. Cerebrovasc. Dis. 40 (1–2), 67–72 (2015).
Lipscomb, D. C., Gorman, L. G., Traystman, R. J. & Hurn, P. D. Low molecular weight iron in cerebral ischemic acidosis in vivo. Stroke 29 (2), 487–492 (1998). discussion 493.
Palmer, C., Menzies, S. L., Roberts, R. L., Pavlick, G. & Connor, J. R. Changes in iron histochemistry after hypoxic-ischemic brain injury in the neonatal rat. J. Neurosci. Res. 56 (1), 60–71 (1999).
Ryan, F., Zarruk, J. G., Lößlein, L. & David, S. Ceruloplasmin Plays a Neuroprotective Role in Cerebral Ischemia. Front. Neurosci. 12, 988 (2019).
Pan, L. et al. Network pharmacology and experimental validation-based approach to understand the effect and mechanism of Taohong Siwu Decoction against ischemic stroke. J. Ethnopharmacol. 294, 115339 (2022).
Pang, H. Q. et al. Elucidating the chemical interaction effects of herb pair Danshen-Chuanxiong and its anti-ischemic stroke activities evaluation. J. Ethnopharmacol. ;318 (2024). (Pt B):117058.
Zhao, L. et al. A Network-Based Approach to Investigate the Neuroprotective Effects and Mechanisms of Action of Huangqi-Chuanxiong and Sanleng-Ezhu Herb Pairs in the Treatment of Cerebral Ischemic Stroke. Front. Pharmacol. 13, 844186 (2022).
Jing, S. et al. Levistilide a Induces Ferroptosis by Activating the Nrf2/HO-1 Signaling Pathway in Breast Cancer Cells. Drug Des. Devel Ther. 16, 2981–2993 (2022).
Zhou, L. et al. Ligustilide inhibits Purkinje cell ferritinophagy via the ULK1/NCOA4 pathway to attenuate valproic acid-induced autistic features. Phytomedicine 126, 155443 (2024).
Lou, T. et al. Integration of metabolomics and transcriptomics reveals that Da Chuanxiong Formula improves vascular cognitive impairment via ACSL4/GPX4 mediated ferroptosis. J. Ethnopharmacol. 325, 117868 (2024).
Nallasamy, P., Rajamohamed, B. S., Jeyaraman, J., Kathirvel, B. & Natarajan, S. Regenerative marine waste towards CaCO3 nanoformulation for Alzheimer’s therapy. Environ. Res. 225, 115631 (2023).
Ye, D. et al. Association Between Circulating Linoleic Acid and Risk of Ischemic Stroke. Front. Genet. 11, 582623 (2021).
Yu, Y., Zhang, G., Han, T. & Huang, H. L. Analysis of Herbal Mechanisms and Prescriptions for Chronic Cerebral Circulatory Insufficiency Based on Data Mining and Network Pharmacology. Comb. Chem. High. Throughput Screen. 25 (8), 1239–1253 (2022).
Cui, S., Feng, X. & Xia, Z. Ligusticum chuanxiong Hort. Ameliorates Neuropathic Pain by Regulating Microglial M1 Polarization: A Study Based on Network Pharmacology. J. Pain Res. 17, 1881–1901 (2024).
Ross, A. M., Lee, C. S. & Brewer, M. Peripheral immune response and infection in first-time and recurrent ischemic stroke or transient ischemic attack. J. Neurosci. Nurs. 46 (4), 199–206 (2014).
Jian, Z. et al. The Involvement and Therapy Target of Immune Cells After Ischemic Stroke. Front. Immunol. 10, 2167 (2019).
Mracsko, E. et al. Antigen dependently activated cluster of differentiation 8-positive T cells cause perforin-mediated neurotoxicity in experimental stroke. J. Neurosci. 34 (50), 16784–16795 (2014).
Gill, D. & Veltkamp, R. Dynamics of T cell responses after stroke. Curr. Opin. Pharmacol. 26, 26–32 (2016).
Maida, C. D., Norrito, R. L., Daidone, M., Tuttolomondo, A. & Pinto, A. Neuroinflammatory Mechanisms in Ischemic Stroke: Focus on Cardioembolic Stroke, Background, and Therapeutic Approaches. Int. J. Mol. Sci. 21 (18), 6454 (2020).
Mashaqi, S. et al. Matrix metalloproteinase-9 as a messenger in the cross talk between obstructive sleep apnea and comorbid systemic hypertension, cardiac remodeling, and ischemic stroke: a literature review. J. Clin. Sleep. Med. 17 (3), 567–591 (2021).
Saxena, S., Kruys, V., Vamecq, J. & Maze, M. The Role of Microglia in Perioperative Neuroinflammation and Neurocognitive Disorders. Front. Aging Neurosci. 13, 671499 (2021).
Kanazawa, M., Ninomiya, I., Hatakeyama, M., Takahashi, T. & Shimohata, T. Microglia and Monocytes/Macrophages Polarization Reveal Novel Therapeutic Mechanism against Stroke. Int. J. Mol. Sci. 18 (10), 2135 (2017).
Liu, D. et al. Genetic Variations of Oxidative Stress Related Genes ALOX5, ALOX5AP and MPO Modulate Ischemic Stroke Susceptibility Through Main Effects and Epistatic Interactions in a Chinese Population. Cell. Physiol. Biochem. 43 (4), 1588–1602 (2017).
Ewida, H. A., Zayed, R. K., Darwish, H. A. & Shaheen, A. A. Circulating lncRNAs HIF1A-AS2 and LINLK-A: Role and Relation to Hypoxia-Inducible Factor-1α in Cerebral Stroke Patients. Mol. Neurobiol. 58 (9), 4564–4574 (2021).
Barteczek, P. et al. Neuronal HIF-1α and HIF-2α deficiency improves neuronal survival and sensorimotor function in the early acute phase after ischemic stroke. J. Cereb. Blood Flow. Metabolism. 37 (1), 291–306 (2017).
McDonnell, E., Peterson, B. S., Bomze, H. M. & Hirschey, M. D. SIRT3 regulates progression and development of diseases of aging. Trends Endocrinol. Metab. 26 (9), 486–492 (2015).
Yang, X. et al. Sirt3 Protects Against Ischemic Stroke Injury by Regulating HIF-1α/VEGF Signaling and Blood-Brain Barrier Integrity. Cell. Mol. Neurobiol. 41 (6), 1203–1215 (2021).
Xu, S., Yang, J., Wan, H., Yu, L. & He, Y. Combination of Radix Astragali and Safflower Promotes Angiogenesis in Rats with Ischemic Stroke via Silencing PTGS2. Int. J. Mol. Sci. 24 (3), 2126 (2023).
Yuan, K. et al. Novel diagnostic biomarkers of oxidative stress, ferroptosis, immune infiltration characteristics and experimental validation in ischemic stroke. Aging (Albany NY). 16 (1), 746–761 (2024).
Ou, Z. et al. Glucose-6-phosphate dehydrogenase deficiency and stroke outcomes. Neurology 95 (11), e1471–e1478 (2020).
Li, J. et al. Glucose-6-phosphate dehydrogenase deficiency and intracranial atherosclerotic stenosis in stroke patients. Eur. J. Neurol. 29 (9), 2683–2689 (2022).
Cheng, X. et al. Astrocytic endothelin-1 overexpression promotes neural progenitor cells proliferation and differentiation into astrocytes via the Jak2/Stat3 pathway after stroke. J. Neuroinflammation. 16 (1), 227 (2019).
Zhu, H. et al. Janus Kinase Inhibition Ameliorates Ischemic Stroke Injury and Neuroinflammation Through Reducing NLRP3 Inflammasome Activation via JAK2/STAT3 Pathway Inhibition. Front. Immunol. 12, 714943 (2021).
Dhanesha, N. et al. PKM2 promotes neutrophil activation and cerebral thromboinflammation: therapeutic implications for ischemic stroke. Blood 139 (8), 1234–1245 (2022).
Rébé, C. & Ghiringhelli, F. STAT3, a Master Regulator of Anti-Tumor Immune Response. Cancers (Basel). 11 (9), 1280 (2019).
Huynh, J., Chand, A., Gough, D. & Ernst, M. Therapeutically exploiting STAT3 activity in cancer - using tissue repair as a road map. Nat. Rev. Cancer. 19 (2), 82–96 (2019).
Yu, H., Kortylewski, M. & Pardoll, D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 7 (1), 41–51 (2007).
Zou, S. et al. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer. 19 (1), 145 (2020).
Ryu, K. Y. et al. Dasatinib regulates LPS-induced microglial and astrocytic neuroinflammatory responses by inhibiting AKT/STAT3 signaling. J. Neuroinflammation. 16 (1), 190 (2019).
Tyagi, A. et al. Nicotine promotes breast cancer metastasis by stimulating N2 neutrophils and generating pre-metastatic niche in lung. Nat. Commun. 12 (1), 474 (2021).
Koh, M. Y. & Powis, G. Passing the baton: the HIF switch. Trends Biochem. Sci. 37 (9), 364–372 (2012).
Leone, R. D. & Powell, J. D. Metabolism of immune cells in cancer. Nat. Rev. Cancer. 20 (9), 516–531 (2020).
Corcoran, S. E. & O’Neill, L. A. HIF1α and metabolic reprogramming in inflammation. J. Clin. Invest. 126 (10), 3699–3707 (2016).
Elks, P. M. et al. Activation of hypoxia-inducible factor-1α (Hif-1α) delays inflammation resolution by reducing neutrophil apoptosis and reverse migration in a zebrafish inflammation model. Blood 118 (3), 712–722 (2011).
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30 (1), 207–210 (2002).
Zhou, N. & Bao, J. FerrDb: a manually curated resource for regulators and markers of ferroptosis and ferroptosis-disease associations. Database (Oxford). 2020, baaa021 (2020).
Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P. L. & Ideker, T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27 (3), 431–432 (2011).
Franceschini, A. et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41 (Database issue), D808–D815 (2013).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51 (D1), D587–D592 (2023).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 (1), 27–30 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28 (11), 1947–1951 (2019).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 12 (5), 453–457 (2015).
Li, W. X. et al. Screening of antibacterial compounds with novel structure from the FDA approved drugs using machine learning methods. Aging (Albany NY). 14 (3), 1448–1472 (2022).
Li, W. X. et al. Transcriptome integration analysis and specific diagnosis model construction for Hodgkin’s lymphoma, diffuse large B-cell lymphoma, and mantle cell lymphoma. Aging (Albany NY). 13 (8), 11833–11859 (2021).
Hetényi, C. & van der Spoel, D. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci. 11 (7), 1729–1737 (2002).
Gaillard, T. Evaluation of AutoDock and AutoDock Vina on the CASF-2013 Benchmark. J. Chem. Inf. Model. 58 (8), 1697–1706 (2018).
Funding
This work was support by Basic Research Project of Liaoning Provincial Department of Education in 2021 (Youth Project - Support Project)(No.LJKQZ2021066), National Natural Science Foundation of China funding project (No.82305391) and 2023 Joint Fund Project(No.2023-MSLH-197).
Author information
Authors and Affiliations
Contributions
Q. G., L. W., Y. C. participated in the design of the study, Q. G. carried out the study, X. F., J. L., X. Z., S. W. performed the statistical analysis, Q. G. , Z. W., J. Y. drafted the manuscript. Y. C. reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ge, Q., Wang, Z., Yu, J. et al. Chuanxiong Rhizoma regulates ferroptosis and the immune microenvironment in ischemic stroke through the JAK-STAT3 pathway. Sci Rep 14, 31224 (2024). https://doi.org/10.1038/s41598-024-82486-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-82486-5
