
Abstract
Identifying the active natural compounds remains a challenge for drug discovery, and new algorithms need to be developed to predict active ingredients from complex natural products. Here, we proposed Meta-DEP, a Meta-paths-based Drug Efficacy Prediction based on drug-protein-disease heterogeneity network, where Meta-paths contain all the shortest paths between drug targets and disease-related proteins in the network and drug efficacy is measured by a predictive score according to drug disease network proximity. Experiments show that Meta-DEP performs better than traditional network topology analysis on drug-disease interaction prediction task. Further investigations demonstrate that the key targets identified by Meta-DEP for drug efficacy are consistent with clinical pharmacological evidence. To prove that Meta-DEP can be used to discover active natural compounds, we apply it to predict the relationship between the monomeric components of traditional Chinese medicine included in the TCMSP database and diseases. Results indicate that Meta-DEP can accurately predict most of the drug-disease pairs included in the TCMSP database. In addition, biological experiments are directly used to demonstrate that Meta-DEP can mined active compound from traditional Chinese medicine with integrating disease transcriptomic data. Overall, the model developed in this study provides new impetus for driving the natural compound into innovative lead molecule. Code and data are available at https://github.com/t9lex/Meta-DEP.
Similar content being viewed by others

Introduction
Throughout history, natural compounds have played a pivotal role in the prevention and treatment of diseases. Traditional Chinese medicine (TCM), a product of millennia of clinical practice, boasts remarkable therapeutic effects and high safety, thus emerging as a rich source of natural compounds for innovative drug research1. Typically, TCM comprises a meticulous blend of multiple herbs, harboring numerous potentially therapeutic active ingredients2. However, the complexity nature of TCM poses significant challenges in the identification and isolation of these active components through wet-lab experiments3. Therefore, the development of efficient approaches for identifying active compounds in TCM holds immense potential in in facilitating the transition of TCM into modern therapeutics.
Currently, many computational approaches have been proven to greatly facilitate the discovery of active compounds from TCM4. These methods are mainly divided into two categories, including single-target based and network-based. Single-target based methods, such as ligand-based virtual screening and protein structure-based virtual screening, directly predict the interactions between ligands and disease proteins, thereby identifying active molecules in TCM5. Nevertheless, this approach may overlook the intricate and multifaceted nature of TCM, which often involves numerous components and targets.
Conversely, network-based approaches offer a comprehensive understanding of the pharmacological mechanisms of action of drugs and enable researchers to systematically investigate the intricate interactions between drugs and biological systems6. Network pharmacology, as a network-based approach, is currently extensively utilized for the identification of active molecules in TCM. Within this framework, network construction and network analysis are two pivotal steps. Network construction primarily involves the seamless integration of three crucial types of databases: compound-target related databases, disease-gene related databases, and protein-protein interaction databases. By analyzing the topological properties of drug nodes within the constructed network, the active substances in TCM can be ultimately determined7,8. To further enhance the accuracy of predictions, a new network analysis method called Network-based Proximity has been developed to predict drug efficacy9, and some studies have demonstrated that this method can better identify active molecules in TCM10. Nevertheless, the complexity of the analysis steps in network pharmacology limits its application to a certain extent.
The recent decades have witnessed the remarkable success of artificial intelligence (AI) technology in the field of drug discovery. With the improvement of computing power and accumulation of big data, AI is revolutionizing the traditional approach to drug research and development, significantly enhancing efficiency and success rates11,12. Based on drug-protein-disease interaction heterogeneous network, many deep learning approaches proposed for predicting drug-disease interactions13,14,15,16. These studies primarily focus on enhancing the accuracy and robustness of model predictions by enriching the information of network nodes, or leveraging the mechanistic information of drugs within the network to improve the interpretability of the models. However, it remains unknown whether deep learning models can directly quantify the drug-disease relationships solely based on the heterogeneous network of drug-protein-disease interactions, as well as discover active compounds from TCM.
To address the aforementioned research challenges, this study proposes a model called Meta-DEP, which stands for Meta-paths-based Drug Efficacy Prediction leveraging the drug-protein-disease heterogeneity network. This model is built upon the previously reported deep learning frameworks for predicting drug-disease interactions17, with the aim of directly and quantitatively evaluating drug efficacy. Experimental results demonstrate that the model developed in this study can accurately predict the relationships between most active compounds in traditional Chinese medicine (TCM) and diseases listed in the TCMSP database18. Furthermore, by integrating disease transcriptomic data, it is capable of identifying the active monomeric components within the TCM compound prescription Xin-Ji-Er-Kang19,20,21 which exert a protective effect against myocardial ischemia through exerting anti-inflammatory or mitochondrial damage-reducing mechanisms. In summary, the model proposed in this study offers fresh momentum for facilitating the transformation of traditional Chinese medicine formulas into innovative drugs, thus paving the way for novel therapeutic approaches.
Materials and methods
Data
Protein-protein interaction network
The Protein-Protein Interaction (PPI) network is derived from the Human Interactome dataset established by Albert-László Barabási and colleagues, which constitutes a comprehensive collection of 332,749 pairwise interacting bindings among 18,508 distinct human protein species22. We use the Python Networkx software package23 to obtain the largest connected subgraph of the Protein-Protein Interaction (PPI) network as our protein network. Following preprocessing steps, the final PPI network encompasses 311,210 interaction pairs connected by 17,329 unique proteins.
Drug-protein associations
Drug-protein associations are derived from the Drug-Gene Interaction Database (DGIdb)24, a compendium integrating drug-gene interactions from a variety of publications, databases, and online resources. The DGIdb24 employs a hybrid methodology combining expert curation with text-mining techniques to standardize information across 41 distinct sources, ultimately assembling 54,591 drug-gene interactions involving 41,102 individual genes and 14,449 separate drugs. In our study, we retrieved the coding genes for target proteins associated with specific drugs from the DGIdb24, followed by mapping these genes to their corresponding protein entities. Consequently, our constructed drug-protein interaction network details 7,713 distinct drug-protein engagements among a set of 1,161 drugs and 2,019 proteins.
Disease-protein associations
Disease-protein linkages were obtained from the DisGeNET25 repository, a compendium that constitutes one of the most comprehensive assemblies of genes implicated in human pathologies. DisGeNET25 amalgamates data from expert-curated databases with knowledge harvested by means of text-mining techniques applied to the scientific literature, thereby harmonizing and standardizing information on disease-associated genes and genetic variations from various origins. This resource covers the entire breadth of human diseases along with both physiological and pathological phenotypes. The present iteration of DisGeNET25 enumerates over 24,000 unique diseases and traits, involves 17,000 genes, and catalogues 117,000 genomic variations. In our study, we identified protein-coding genes associated with specific diseases and proceeded to map these genes onto their corresponding protein entities. Consequently, the systematically constructed disease-protein interaction network encompasses 11,482 disease-protein associations across 634 diseases and 9,109 proteins.
Drug–disease associations
In this study, we compiled a dataset of 1948 known drug indications from the repoDB database26. Only small molecule drugs approved by the United States Food and Drug Administration (FDA) were considered, and each drug’s generic name was standardized using Medical Subject Headings (MeSH) and the Unified Medical Language System (UMLS) vocabulary. A majority (75%) of the drugs were indicated for fewer than three disease conditions; conversely, only 4% of the drugs had therapeutic indications spanning over ten distinct diseases. Regarding disease coverage, 70% of the diseases had fewer than five associated drugs; 16% of the diseases were treated by a range of 5 to 10 drugs; and finally, 14% of the diseases had more than ten drugs available for treatment.
Drug-disease pairs score
Network-based proximity between drugs and diseases
A drug-disease proximity metric algorithm9 has been developed that quantifies the relationship between a drug and disease proteins based on their interactions within a network context, thereby facilitating the estimation of a drug’s therapeutic potential. The specific details of this measure are as follows:
The closeness between a drug and a disease was systematically quantified by employing distance metrics that consider the shortest path distances among drug targets and disease-associated proteins within a biological network. Given \(\:S\), which represents the ensemble of proteins implicated in the disease state, \(\:T\) denoting the collection of proteins targeted by the drug, and \(\:d(s,t)\) signifying the shortest path length between nodes \(\:s\) and \(\:t\) in the network, this methodology establishes a proximity metric to assess the relational proximity between drugs and diseases.
To evaluate the statistical significance of the proximity between a drug and a disease \(\:\left(T,S\right)\), this methodology created a reference distance distribution corresponding to the expected distances between two randomly selected groups of proteins matching the size and the degrees of the original disease proteins and drug targets in the network. The reference distance distribution was generated by calculating the proximity between these two randomly selected groups, a procedure repeated 1,000 times. The mean \(\:{{\upmu\:}}_{d(S,T)}\) and \(\:{\sigma\:}_{d(S,T)}\) of the reference distribution were used to convert an observed distance to a normalized distance, defining the proximity measure:
This algorithm defines a drug as being proximal to a disease (suggesting a therapeutic effect) when the computed proximity metric \(\:z\le\:-0.15\)[9].
Construct the training data set
We map drug targets and disease proteins to the human interaction group network, and then use the drug-disease proximity9 measure to calculate the score based on the network relationship to quantify the therapeutic effect of drugs on diseases. If the drug-disease proximity score \(\:z\le\:-0.15\), the drug is defined as the proximal end of the disease, that is, the drug has a therapeutic effect on the disease. Otherwise regarded as no efficacy. In this study, 1785 unknown pairs were selected as negative samples through a random negative sampling strategy. And we utilized 1487 pairs of therapeutic drug-disease associations with scores less than − 0.15 out of 1948 pairs as true positive data. Simultaneously, we considered 461 pairs of drug-disease associations with scores greater than − 0.15 among 1785 pairs of negatively sampled drug-disease associations as true negative data. Then, the true positive data and the true negative data were merged as the training set (Fig. S1). Subsequently, in order to facilitate the training of the model in subsequent steps, this study will convert the obtained network proximity scores in accordance with the following formula:
After conversion, if \(\:{z}_{new}\ge\:0.065\), the drug is considered to be at the proximal end of the disease.
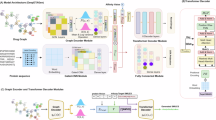
The overall framework of Meta-DEP
The Meta-DEP framework is presented in Fig. 1. Meta-DEP model discerns the pathways related to drug-disease interactions by traversing the shortest connections between drug targets and diseases within a complex, heterogeneous network composed of drugs, proteins, and diseases. To comprehend the overarching connectivity within this diverse graph structure, it initially utilizes Metapath2vec27 to generate feature vector for the constituent nodes. Then, to capture the detailed mechanism of drug action patterns, the embeddings of the nodes along the shortest paths between a drug and a disease are fed into an RNN28 module to model their sequential dependencies. Moreover, Meta-DEP incorporates a dual attention29 mechanism consisting of path attention and node attention. This design allows for the intelligent aggregation of node embeddings along the identified pathways, assigning relative significance to each node’s contribution to the overall pathway and variably weighting the relevance of distinct paths in influencing the final prediction outcome. Finally, to enhance the predictive capability of the model, the Meta-DEP architecture employs a multitask learning strategy involving both regression and classification tasks. The regression task predicts the proximity score between drugs and diseases, while the classification task performs binary prediction of drug efficacy.

Architecture of Meta-DEP.
Metapath related to drug disease effects
To accurately model the effects of a drugs, it is imperative to identify the paths conveying drug-disease interaction information that effectively represent the mechanism of action. We prioritize these shortest paths because the shorter the distance between a drug and its disease target, the higher the likelihood of a therapeutic effect30,31,32. Utilizing the shortest paths algorithm from the Python Networkx software package23, we can systematically discern such pathways. Given a drug and a disease, the set of shortest paths connecting them within the context of a protein-protein interaction network comprises a series of \(\:PATH=\{{path}_{1},{path}_{2},\:\dots\:\:,\:{path}_{L}\}\), where each \(\:{path}_{i}=\{{node}_{m1}\to\:{node}_{m2}\to\:\dots\:\:{node}_{disease}\}\). Here, \(\:{node}_{m1}\) and \(\:{node}_{m2}\) denote intermediate nodes along a specific path, with \(\:L\) denoting the number of distinct shortest paths. These paths consist of interconnected nodes that bridge the gap between the starting drug node and the end disease node, thereby elucidating potential routes through which the drug may exert its effect on the disease.
Metapath2vec captures global connectivity information of drug-protein-disease heterogeneous networks
In the heterogeneous network of drugs, proteins, and diseases, which incorporates multiple node types and edges representing their interactions, Metapath2vec employs the strategic definition of metapaths to model the complex relationships among these entities and thereby encapsulate the global connectivity information (TSNE33 dimensionality reduction clustering visualization of feature representations obtained by different representation learning methods27,34,35,36,37,38, see Fig. S3). Within this drug-protein-disease heterogenous network, a specific metapath is defined as \(\:\left(\text{drug},\text{to},\text{protein}\right),\left(protein,toprotein\right),(protein,to,disease)\), which signifies a trajectory that conceptually connects a disease node through one or more intervening protein nodes back to another disease node; here, \(\:\text{drug}\) denotes a disease node, \(\:protein\) represents a protein node, and \(\:disease\) indicates a disease node. Based on this predefined metapath, Metapath2vec27 generates sequences of nodes by employing random walks throughout the network topology. Subsequently, it utilizes the Skip-gram model during the training process to learn low-dimensional vector embeddings \(\:E\in\:{R}^{N\times\:d}\) for every individual node, where \(\:N\) refers to the total number of nodes in the network and \(\:d\) is the dimensionality of the embedding space. This approach allows Metapath2vec27 to capture meaningful representations that encode the structural and relational properties inherent within the network structure.
Recurrent neural network
Given a drug–disease pair, the embeddings generated by the Metapath2vec27 and the shortest path set \(\:PATH\), we employ RNN to encode both long-term and short-term dependencies in a Metapath. Such sequential dependencies are crucial to the model intelligibility. Given a node \(\:{node}_{m}\) and a path \(\:{path}_{i}\), the input of the RNN recurrent neural network is the node embedding \(\:X\) generated by Metapath2vec27. After the network accepts the input \(\:{X}_{t}\) at time \(\:t\), the value of the hidden layer is \(\:{S}_{t}\), and the output value is \(\:{O}_{t}\). The value of \(\:{S}_{t}\) depends not only on \(\:{X}_{t}\), but also on \(\:{S}_{t-1}\). Specifically, it can be expressed by the following formula:
Where \(\:X\) is a vector, which represents the value of the input layer; \(\:S\) is a vector, which represents the value of the hidden layer. \(\:U\) is the weight matrix from the input layer to the hidden layer, \(\:O\) is also a vector, which represents the value of the output layer; \(\:V\) is the weight matrix from the hidden layer to the output layer. \(\:f\) is the nonlinear activation function of ReLU, and \(\:g\) denotes the Softmax function. The output of each node is aggregated to the attention module for each path to represent the entire path and the final prediction. Because the length of the shortest path is not equal, we use the filling method as follows. Assuming that the maximum length of a path is set to \(\:{l}_{max}\), for the path that shorter than \(\:{l}_{max}\), we use the padding value \(\:pad\) (such as 0) to fill the path, and the following processing ignores these padding positions to avoid affecting performance.
Node attention
In the context of a shortest Metapath \(\:{path}_{p}\) connecting a drug-disease pair, for each node produced by the RNN (Recurrent Neural Network)28, the output includes hidden state variables denoted as \(\:{O}_{p}\in\:{R}^{{l}_{max}\times\:d}\), \(\:{O}_{p}=\:\left\{{o}_{1},{o}_{2},\dots\:,{o}_{pad},{o}_{pad}\right\}\) and \(\:{o}_{pad}\) are filled hidden states. Initially, all the hidden values at these filled positions are transformed to negative infinity. Subsequently, a linear layer is applied, followed by a Softmax activation function, to combine and condense the embeddings into a single value that signifies the importance or weight of the node in the Metapath context.
Where \(\:{W}_{n}\in\:{R}^{d\times\:1}\) is a learnable parameter, \(\:{\varOmega\:}_{p}\in\:{R}^{{l}_{max}\times\:1}\) represents the weight of each node in the path \(\:{path}_{p}\), where \(\:{\varOmega\:}_{p}=\:\left\{{w}_{1},{w}_{2},\dots\:,{w}_{max}\right\}\). For a node \(\:j\) in \(\:{path}_{p}\), its weight is calculated as follows:
Then we aggregate the hidden states of these nodes weighted by \(\:{w}_{i}^{{\prime}}\) to get the embedding of \(\:{path}_{p}\):
Path attention
Following the aggregation step, for a given drug-disease pair, the embedding representing the shortest Metapath is obtained: \(\:{E}_{PATH}=\:\left\{{e}_{{path}_{1}},{e}_{{path}_{2}},\dots\:,{e}_{{path}_{L}}\right\}\). The path attention is similar to the node attention:
Where \(\:{\sigma\:}_{p}\) is the softmax function, \(\:f\) is the nonlinear activation layer. The final predicted outcome \(\:{y}^{{\prime}}\), denoted as, represents the probability that the drug will be effective in treating the disease. Additionally, the score \(\:{y}_{s}\), quantifies the closeness or proximity between drugs and diseases, serving as an indicator of the drug’s therapeutic impact on the disease.
Objective function
Within the scope of this research, the Meta-DEP model has been designed to be concurrently trained on two distinct learning objectives: regression and classification tasks. Regarding the regression component, the model employs the Mean Squared Error (MSE) as its loss function, an extensively adopted metric for gauging the discrepancy between estimated outcomes and actual targets. The MSE is mathematically defined as the mean of the sum of squared discrepancies between each predicted value and its true counterpart, with its functional form expressed thusly:
And the categorical prediction task is approached as a two-class classification problem, where the chosen loss function for optimization is the CrossEntropyLoss.
In our multi-objective training process, we aim to minimize the combination of two losses with \(\:{l}_{2}\) regularization:
Where \(\:\alpha\:\) is a hyperparameter that weighs the weight of classification tasks, \(\:\theta\:\) is the set of parameters to be learned in Meta-DEP, \(\:\lambda\:\) is the \(\:{l}_{2}\) regularizer to prevent over-fitting, \(\:{||\theta\:||}_{2}^{2}\) is the square of the second norm of \(\:\theta\:\).
Performance evaluation and experiment setup
Meta-DEP training is a dual task training of regression and classification (Regression and classification dual task ablation experiment, see Fig. S2A), given a drug disease pair, we input all the shortest paths between the drug and the disease into the Meta-DEP, and Meta-DEP will predict the probability that the drug will have a therapeutic effect on the disease and score the magnitude of its pharmacodynamic effect. In this study, Pearson correlation coefficient was used as a performance indicator to evaluate and adjust the optimization feature representation, model framework, and hyperparameter settings through five-fold cross-validation. First, keep the model architecture unchanged, set the hyperparameters in a moderate range, and adjust the feature representation to make the performance index reach a temporary optimal state (The influence of feature representation on model performance, see Fig. S2B). Then, the feature representation is fixed, the hyperparameters are still in the moderate range, and the model architecture is adjusted to find the temporary optimal performance index (The influence of model architecture on model performance, see Fig. S2C). After determining the feature representation and model architecture, the combination of hyperparameters of the model is optimized to achieve the best state.
Cell culture
The cell lines used in this study were purchased from the American Type Culture Collection (ATCC). AC16 cells were cultured in DMEM/F12 (Gibco) supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 U/mL streptomycin.
The cells were cultured at 37 °C with 5% CO2 in an atmosphere containing.
CCK-8 cell viability assay
Cell viability was measured by Cell Counting Kit-8 (CCK-8) assay kit (IV08-500 T, Invigentech, USA). Briefly, AC16 cells were cultured in 96-well plates and treated with various concentrations of Bifendate. After treatment for 24 h, each well added 10µL of CCK-8 reagent. This plate was incubated at 37 °C for 1 h in a humidified atmosphere with 5% CO2. The absorbance of each well was measured at 450 nm using a microplate reader (SPECTR AMAX190, MD Company, USA).
The content of ROS in mitochondria was detected by flow cytometry
AC16 cells were cultured in 6-well plates. When the confluence reached 70%, the cells were treated with 100µmol/L hydrogen peroxide and 10µmol/L bifedate for 24 h. After incubation, the cells were collected in a 1.5 ml ep tube, 500 µl working solution was added, and they were incubated at 37 °C for 20 min (mixed every 3–5 min). After removal of the working solution, the cells were washed twice with PBS and resuspended in 500 µL PBS. The MFI (Mean Fluorescence Intensity) was detected by flow cytometry (FACSCelesta, BD, USA).
The intracellular ROS content was detected by active oxygen detection kit
AC16 cells were cultured in 12-well plates. When the confluence reached 70%, the cells were treated with 100 µmol/L hydrogen peroxide and 10 µmol/L bifendate for 24 h. After incubation, the cells were washed twice with PBS. DCFH-DA (S0033S, Beyotime Biotechnology, China) fluorescent dye stock solution was diluted with serum-free medium to make a working solution (final concentration 10µM), 500 µl of working solution was added to each well, and incubated at 37 °C for 20 min. The working solution was removed and the cells were washed twice with PBS. After adding the complete medium, the fluorescence intensity was measured by high-content microscope (ImageXpress MicroConfocal. MD, USA).
Mitochondrial membrane potential was detected by mitochondrial membrane potential detection kit
AC16 cells were cultured in 12-well plates. When the confluence reached 70%, the cells were treated with 100 µmol/L hydrogen peroxide and 1 µmol/L bifendate for 24 h. After incubation, the cells were washed with PBS twice. JC-1 (HY-K0601, Med Chem Express, China) fluorescent dye stock solution was diluted with serum-free medium to make a working solution (final concentration 2µM), 500 µl of working solution was added to each well, and incubated at 37 °C for 20 min. The working solution was removed and the cells were washed twice with PBS. After adding the complete medium, the fluorescence intensity was detected by high-content microscope (ImageXpress MicroConfocal, MD, USA).
Apoptosis was detected by TUNEL detection
AC16 cells were seeded into 12-well plates. When the confluence reached 70%, AC16 cells were treated with 100 µmol/L hydrogen peroxide and 10 µmol/L bifendate for 24 h. After incubation, cells were washed with PBS once, fixed with 4% paraformaldehyde for 30 min, and washed with PBS twice after fixation. Add immunostaining strong penetrant (P0260, Beyotime Biotechnology, China) and incubate at room temperature for 5 min; after incubation, the cells were washed twice with PBS, and 50 µl TUNEL staining solution (C1088, Beyotime Biotechnology, China) was added to the sample, and incubated at 37 °C for 60 min in the dark; after the incubation, the cells were washed three times with PBS. DAPI staining solution (C1005, Beyotime Biotechnology, China) was incubated for 20 min, and washed three times with PBS after incubation. 500ul PBS was added to each well, and the fluorescence intensity was detected by high-content microscope (ImageXpress MicroConfocal, MD, USA).
Result
Meta-DEP exhibits superior evaluation capabilities for drug efficacy
To investigate whether Meta-DEP model can whether the model can learn the network proximity score, we conducted a correlation analysis on the prediction scores obtained from the Meta-DEP model and the network proximity algorithm9. The results show that for the true positive drug-disease pairs, there is a strong correlation between the prediction scores of Meta-DEP and the network proximity algorithm9 (Fig. 2A). However, for the artificially constructed negative drug-disease pairs, the predicted values of the two methods exhibit weak correlation (Fig. 2B). These suggest that Meta-DEP effectively learned information similar to that of the network proximity algorithm9 during drug efficacy prediction, while potentially learning additional information that is distinct from what is captured by the network proximity algorithm9. Therefore, we further investigated whether the drug efficacy predictive performance of Meta-DEP is superior to that of the network proximity algorithm9. The results showed that the Meta-DEP algorithm was able to accurately predict almost 82.3% of the drug-disease pairs in the dataset, whereas the network proximity algorithm9 could only predict almost 76.3% of them (Fig. 2C-D). This indicates that Meta-DEP exhibits superior drug efficacy predictive performance compared to the network proximity algorithm9.

Model performance. (A) Correlation analysis between the score of Meta-DEP and the score of Network-based proximity on true positive drug-disease pair data. (B) Correlation analysis between the score of Meta-DEP and the score of Network-based proximity on true negative drug-disease pair data. (C) The proportion of true and false numbers predicted by Meta-DEP or Network-based proximity in real drug-disease pair data. (D) Comparing the prediction scores of Meta-DEP and Network-based proximity in real drug-disease pair data.
Exploring drug combination predictions with Meta-DEP
To further evaluate the performance of Meta-DEP in quantitatively assessing drug efficacy, we randomly extracted 100 pairs of drug combinations from the CDCDB database39, which have been reported to exhibit synergistic therapeutic effects when used in combination. Subsequently, we utilized the scoring mechanism of Meta-DEP to forecast the pharmacological effects of these drug combinations. Specifically, for a given disease, if Meta-DEP predicts a higher score for the combined drug efficacy than for any individual drug within the combination, it would deduce that the concurrent administration of these drugs could potentiate the therapeutic outcome for that particular disease. The findings revealed that Meta-DEP accurately discerned 65 pairs of drug combinations (Fig. 3A, Table S1), including the combined usage of Acetaminophen and Acetylsalicylic acid for treating Migraine Disorders40,41, the combined usage of Vildagliptin and Nateglinide for type 2 diabetes mellitus42, Entacapone and Carbidopa, in combination with Droxidopa, are used for the treatment of Parkinson’s Disease43,44 (Fig. 3B-D). In contrast, traditional Network-based proximity algorithms were only able to predict 34 of these drug combinations (Fig. 3A).

Using Meta-DEP score to explain drug combinations. (A) Evaluation of Drug Combination Efficacy for 100 Pairs of Drug Combination Data from CDCDB. (B) The prediction of drug combinations for Migraine Disorders. (C) The prediction of drug combinations for diabetes mellitus type 2. (D) The prediction of drug combinations for Parkinson’s Disease.
Meta-DEP identified the critical target related to drug efficacy
Within the human body, drugs typically interact with multiple protein targets. Therefore, the systematic evaluation of the key protein targets that underlie the therapeutic effects of drugs on diseases is crucial for pharmacodynamic research. In this case study, we found that the node attention weights of Meta-DEP can accurately identify the critical protein targets responsible for the pharmacological effects of drugs. As illustrated in (Fig. 4A-B), Selegiline and Levodopa are two commonly used drugs in the clinical treatment of Parkinson disease. Meta-DEP is capable of precisely identifying and assigning greater node attention weights to the key targets—monoamine oxidase and dopamine receptors—responsible for the therapeutic effects of these two drugs among the direct targets of drugs45,46,47,48. Furthermore, we analyzed 1828 drug-disease pairs in our training dataset, where the pharmacological targets were clearly documented in the Drugbank database49. Meta-DEP was able to successfully identify the crucial targets for 768 of these pairs (Fig. S4, Table S2), highlighting its effectiveness in target identification for drug-disease relationships.

Explain the pathways linking levodopa and selegidol to Parkinson disease, respectively. (A) Sankey diagram of the critical targets connecting Levodopa and Parkinson disease identified by Meta-DEP. (B) Sankey diagram of the critical targets connecting Selegiline and Parkinson disease identified by Meta-DEP.
Meta-DEP demonstrates remarkable abilities in accurately predicting active natural compounds
The TCMSP database18, a comprehensive system pharmacology platform specializing in archiving natural products, holds extensive information regarding natural compounds and their intricate relationships with drug targets and diseases. To assess Meta-DEP’s proficiency in identifying bioactive natural products, we curated a subset of 15 diseases from the TCMSP database18. Notably, these diseases were represented in our training set and exhibited interactions with over 50 distinct natural product molecules each. Compared to Network-based proximity algorithms, our findings revealed that scoring mechanism of Meta-DEP exhibited remarkable accuracy in predicting the majority of natural products associated with these diseases (Fig. 5). To further validate its generalization capabilities, we challenged Meta-DEP with seven diseases that were not part of our original training set. Remarkably, Meta-DEP was able to successfully predict the natural products linked to these diseases within the TCMSP database18, further validating its robust abilities for active natural compound discovery.

Meta-DEP score demonstrate impressive power for predicting the efficacy material basis of traditional Chinese medicine. The transverse axis represents the number of disease-related Chinese medicine monomer components collected in the TCMSP Chinese medicine database, and the longitudinal axis represents the diseases existing in the TCMSP Chinese medicine database. Meta-DEP assigns a score of 0.065 or higher, it signifies an association between the natural products and the disease. On the other hand, a network analysis score of -0.15 or lower indicates an association between the natural products and the disease.
Utilizing Meta-DEP to identify protective compounds of Xin-Ji-Er-Kang for myocardial ischemia
Xin-Ji-Er-Kang (XJEK)19,20,21, a traditional Chinese formula, has exhibited remarkable protective effects against myocardial ischemia. However, the specific natural compounds that are pivotal for its pharmacological activity remain elusive. In this real-world case study, we discovered that by integrating transcriptional data from myocardial infarction (MI)50 with the TCMSP database18, Meta-DEP was able to effectively predict the anti-inflammatory or mitochondrial-protective active compounds in XJEK19,20,21 that contribute to its myocardial ischemia-protective effects (Table S3). Inflammation and mitochondrial damage serve as significant contributors to myocardial injury caused by MI. To explore potential anti-inflammatory compounds in XJEK19,20,21, we initially employed reported MI transcriptomic data50 and conducted gene enrichment analysis to identify proteins associated with inflammatory signaling pathways. Subsequently, we utilized the TCSMP database18 to retrieve 262 compounds from XJEK19,20,21 and their corresponding direct targets. Surprisingly, based on these data, Meta-DEP predictions revealed that multiple compounds with steroidal ring structures were among the top-ranked compounds (Fig. S5, Table S3). Using Meta-DEP, we employed the same strategy to identify bifendate (Fig. 6A, Table S3), a monomeric component from XJEK19,20,21 that exhibits potential protective effects against mitochondrial damage. To validate this finding, we conducted relevant experiments at the cellular level. CCK-8 assays demonstrated that bifendate is capable of enhancing the vitality of AC16 cells treated with hydrogen peroxide, which simulates myocardial ischemia (Fig. 6B). Notably, compared to AC16 cells in the model group, bifendate significantly reduced the levels of mitochondrial reactive oxygen species (ROS), mitochondrial membrane potential, intracellular ROS, and apoptosis index in hydrogen peroxide-treated AC16 cells (Fig. 6C-J).

The wet-lab experiment verified the mitochondrial protective effect of bifendate, a monomer component of XJEK screened by mitochondrial damage-related pathway genes as disease proteins. (A) Structure of bifendate. (B) The effect of bifendate on the viability of H2O2-treated AC16 cells. (C,D) Flow cytometry was used to detect the effect of bifendate on ROS content in mitochondria of H2O2-treated AC16 cells. (E,H) The effect of bifendate on mitochondrial membrane potential of AC16 cells treated with H2O2 was detected by mitochondrial membrane potential detection kit (JC-1). (F,I) The effect of bifendate on intracellular ROS content of AC16 cells treated with H2O2 was detected by reactive oxygen species detection kit. (G,J) The effect of bifendate on the apoptosis of H2O2-treated AC16 cells was detected by TUNEL detection.
Discussion
Deeply rooted in Asian traditions, traditional Chinese medicine (TCM) has garnered global attention in pharmacology. TCM boasts a vast arsenal of natural medicinal compounds, widely recognized for their effectiveness and safety in drug discovery efforts. However, the intricate complexity of these compounds and their numerous associated targets poses a challenge in pinpointing the active ingredients within these herbal remedies. Network analysis, specifically focusing on drug-protein-disease interactions, emerges as a pivotal tool in overcoming this challenge. Nevertheless, traditional network pharmacology analysis involves numerous intricate steps and relies on a myriad of databases, rendering the process unduly cumbersome. To address this, our study proposed Meta-DEP, an end-to-end deep learning model, can directly predict drug efficacy on the drug-protein-disease heterogeneous network. Furthermore, validation results from the TCMSP database18 and the traditional Chinese formula Xin-Ji-Er-kang19,20,21 confirm that Meta-DEP can accurately identify bioactive natural compounds from TCM.
Guney et al. proposed network proximity9 has been widely recognized as an effective method for quantitatively predicting drug efficacy. The Meta-DEP model developed in this study focuses on predicting this network proximity values between drugs and diseases. Notably, our research found that the network proximity values learned by the Meta-DEP model exhibited higher accuracy in predicting known drug-disease pairs. This suggests that the Meta-DEP model possesses stronger predictive capabilities in drug efficacy research, providing more precise and reliable references for drug development. In addition, the attention module of Meta-DEP demonstrated good performance in identifying key proteins that play a crucial role in drug efficacy. This further validates the value of the Meta-DEP model in exploring drug efficacy mechanisms, helping us to gain a deeper understanding of the interactions between drugs and diseases, as well as the molecular mechanisms underlying drug efficacy.
We also used Meta-DEP to analysis synergistic effects of drug combinations, aiming to further evaluate the performance of Meta-DEP in quantitatively assessing drug efficacy. The research results indicate that Meta-DEP, to a certain extent, enables us to comprehensively evaluate the interactions between drugs and understand how they might work together to enhance treatment efficiency. Therefore, it would also be interesting to extend the application of Meta-DEP to predict potential combinations of active ingredients in TCM formulas for addressing complex diseases. This would provide valuable insights into the synergistic effects of various components within TCM formulas, potentially leading to the development of more targeted and effective treatment strategies.
Phenotype-based drug screening is one of the important avenues for new drug discovery, which screens candidate drugs with potential therapeutic effects by directly observing their impacts on the phenotype of organisms. Transcriptome data can comprehensively reflect the changes in gene activities within organisms under disease conditions. Therefore, screening active drugs through transcriptome data is an essential strategy for phenotype-based drug screening. Here, we found that that by incorporating disease-related specific signaling pathway proteins from the transcriptome data of diseases, along with natural compound targets from the TCSMP database18, into the Meta-DEP model, it can directly predict which natural products may be associated with specific signaling pathways related to diseases. However, due to limitations in the data used to train the Meta-DEP model, it is necessary to conduct further experimental validation to determine whether the compounds predicted by Meta-DEP are therapeutic or pathogenic.
Conclusion
In conclusion, based on the heterogeneous network of drugs, proteins, and diseases, this study innovatively proposes a meta-path-based deep learning model for drug efficacy prediction—Meta-DEP. Through multi-dimensional validation, this model not only demonstrates its powerful effectiveness but also highlights its significant advantages compared with traditional network topology analysis methods. Subsequently, we applied the Meta-DEP model to the identification of active ingredients in complex natural products and fully validated its excellent performance in discovering active natural compounds through experiments. Overall, the Meta-DEP model developed in this study provides new powerful momentum for promoting the transformation of natural compounds into innovative lead molecules and provides valuable insights for the field of drug discovery and development.
Data availability
Availability of data and materialsAll data generated or analyzed during this study are included in this article.Code availabilityThe source code is available from GitHub at https://github.com/t9lex/Meta-DEP.
Abbreviations
- Metapath:
-
A path between two nodes where the edges can have different semantics
- Meta-DEP:
-
Meta-paths-based drug efficacy prediction based on drug-protein-disease heterogeneity network
- TCMSP database:
-
Traditional chinese medicine systems pharmacology database and analysis platform
- TCM:
-
Traditional chinese medicine
- AI:
-
Artificial intelligence
- XJEK:
-
Traditional chinese medicine compound prescription Xin-Ji-Er-Kang
- PPI:
-
Protein-protein interaction
- DGIdb:
-
Drug-gene interaction database
- DisGeNET repository:
-
A database of gene-disease associations
- repoDB database:
-
Drug repositioning database
- Metapath2vec:
-
Scalable representation learning for heterogeneous networks
- TSNE:
-
t-distributed stochastic neighbor embedding
- RNN:
-
Recurrent neural network
- ReLU:
-
Rectified linear unit
- Softmax:
-
Normalized exponential function
- MSE:
-
Mean squared error
- CrossEntropyLoss:
-
Cross-entropy loss function
- AC16:
-
Human cardiomyocyte cell line
- CCK-8:
-
Cell Counting Kit-8 Reagent
- ROS:
-
Reactive oxygen species
- TUNEL:
-
Terminal dexynucleotidyl transferase(TdT)-mediated dUTP nick end labeling
- CDCDB:
-
A large and continuously updated drug combination database
- MI:
-
Myocardial ischemia
References
Wang, X. et al. Traditional Chinese medicine: Current state, challenges, and applications. Serum Pharmacochem. Tradit. Chin. Med. 1–6 (2017).
Marshall, A. C. Traditional Chinese Medicine and Clinical Pharmacology (Springer, 2020).
Chen, Y-H. et al. Classification-based strategies to simplify complex traditional Chinese medicine (TCM) researches through liquid chromatography-mass spectrometry in the last decade (2011–2020): Theory, technical route and difficulty. J. Chromatogr. A. 1651, 462307 (2021).
Sliwoski, G., Kothiwale, S., Meiler, J. & Lowe, E. W. Computational methods in drug discovery. Pharmacol. Rev. 66(1), 334–395 (2014).
Agu, P. et al. Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management. Sci. Rep. 13(1), 13398 (2023).
Zhao, L. et al. Network pharmacology, a promising approach to reveal the pharmacology mechanism of Chinese medicine formula. J. Ethnopharmacol. 309, 116306 (2023).
Wu, J. et al. Network pharmacological analysis of active components of Xiaoliu decoction in the treatment of glioblastoma multiforme. Front. Genet. 13, 940462 (2022).
Miao, R., Meng, Q., Wang, C. & Yuan, W. Bibliometric analysis of network pharmacology in traditional chinese medicine. Evid.-Based Complem. Alternat. Med. (2022).
Guney, E., Menche, J., Vidal, M. & Barábasi, A-L. Network-based in silico drug efficacy screening. Nat. Commun. 7(1), 10331 (2016).
Gan, X. et al. Network medicine framework reveals generic herb-symptom effectiveness of traditional Chinese medicine. Sci. Adv. 9(43), eadh0215 (2023).
Mak, K-K., Wong, Y-H. & Pichika, M. R. Artificial intelligence in drug discovery and development. Drug Discov. Eval.: Saf. Pharmacokinetic Assays 1–38 (2023).
Mullowney, M. W. et al. Artificial intelligence for natural product drug discovery. Nat. Rev. Drug Discovery. 22(11), 895–916 (2023).
Zhao, B-W. et al. A multi-graph deep learning model for predicting drug-disease associations. Intelligent Computing Theories and Application: 17th International Conference, ICIC 2021, Shenzhen, China, August 12–15, 2021, Proceedings, Part III 17 580–590 (Springer, 2021).
Wang, Z., Zhou, M. & Arnold, C. Toward heterogeneous information fusion: Bipartite graph convolutional networks for in silico drug repurposing. Bioinformatics 36(Supplement_1), i525–i33 (2020).
Zhang, M-L. et al. RLFDDA: A meta-path based graph representation learning model for drug–disease association prediction. BMC Bioinform. 23(1), 516 (2022).
Zhao, B-W., Hu, L., You, Z-H., Wang, L. & Su, X-R. HINGRL: Predicting drug–disease associations with graph representation learning on heterogeneous information networks. Brief. Bioinform. 23(1), bbab515 (2022).
Yang, J. et al. Deep learning identifies explainable reasoning paths of mechanism of action for drug repurposing from multilayer biological network. Brief. Bioinform. 23(6), bbac469 (2022).
Ru, J. et al. TCMSP: A database of systems pharmacology for drug discovery from herbal medicines. J. Cheminform. 6, 1–6 (2014).
Ling, X. X., Chen, H., Fu, B. B., Ruan, C. S. & Gao, S. Xin-Ji-Er-Kang protects myocardial and renal Injury in Hypertensive Heart failure in mice. Phytomedicine 91(10184), 153675 (2021).
Hu, J. et al. Effects of Xin-Ji-Er-Kang on heart failure induced by myocardial infarction: role of inflammation, oxidative stress and endothelial dysfunction. Phytomedicine S0944711318300710 (2018).
Lian, F. PanRuan, Cheng-shaoLing, Xin-xinWang, Xiao-yunPan, MingChen, Mei-lingShen, Ai-zongGao, Shan. Xin-Ji-Er-Kang ameliorates kidney injury following myocardial infarction by inhibiting oxidative stress via Nrf2/HO-1 pathway in rats. Biomed. Pharmacother. 117(117) (2019).
Morselli Gysi, D. et al. Network medicine framework for identifying drug-repurposing opportunities for COVID-19. Proc. Natl. Acad. Sci. 118(19), e2025581118 (2021).
Hagberg, A., Swart, P. & Chult, S. D. Exploring Network Structure, Dynamics, and Function Using NetworkX (Los Alamos National Lab. (LANL), 2008).
Cotto, K. C. et al. DGIdb 3.0: A redesign and expansion of the drug-gene interaction database. Nucleic Acids Res. 46 (D1), D1068–D73. https://doi.org/10.1093/nar/gkx1143 (2018).
Pinero, J. et al. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 45 (D1), D833–D9. https://doi.org/10.1093/nar/gkw943 (2017).
Brown, A. S. & Patel, C. J. A standard database for drug repositioning. Sci. Data. 4, 170029. https://doi.org/10.1038/sdata.2017.29 (2017).
Dong, Y., Chawla, N. V. & Swami, A. metapath2vec. In Proc. of the 23rd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining 135–144 (2017).
Grossberg, S. Recurrent neural networks. Scholarpedia 8(2), 1888 (2013).
Vaswani, A. et al. Attention is all you need. Adv. Neural. Inf. Process. Syst. 30 (2017).
Ren, Y., Ay, A. & Kahveci, T. Shortest path counting in probabilistic biological networks. BMC Bioinform. 19, 1–19 (2018).
Cheng, F. et al. Network-based approach to prediction and population-based validation of in silico drug repurposing. Nat. Commun. 9 (1), 2691 (2018).
Cheng, F., Kovács, I. A. & Barabási, A-L. Network-based prediction of drug combinations. Nat. Commun. 10(1), 1197 (2019).
Van der Maaten, L. & Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 9(11) (2008).
Perozzi, B., Al-Rfou, R., Skiena, S. & Deepwalk. Online learning of social representations. In Proc. of the 20th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining 701–710 (2014).
Grover, A. & Leskovec, J. node2vec: Scalable feature learning for networks. In Proc. of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining 855–864 (2016).
Kipf, T. N. & Welling, M. Variational graph auto-encoders. arXiv Preprint arXiv:161107308 (2016).
Tang, J. et al. Large-scale information network embedding. In Proc. 24th International Conference on World Wide Web 1067–1077 (2015).
Wang, D., Cui, P. & Zhu, W. Structural deep network embedding. In Proc. 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining 1225–1234 (2016).
Shtar, G., Azulay, L., Nizri, O., Rokach, L. & Shapira, B. CDCDB: A large and continuously updated drug combination database. Sci. Data. 9(1), 263. https://doi.org/10.1038/s41597-022-01360-z (2022).
Goldstein, J., Hagen, M. & Gold, M. Results of a multicenter, double-blind, randomized, parallel-group, placebo-controlled, single-dose study comparing the fixed combination of acetaminophen, acetylsalicylic acid, and caffeine with ibuprofen for acute treatment of patients with severe migraine. Cephalalgia 34(13), 1070–1078 (2014).
Diener, H., Pfaffenrath, V., Pageler, L., Peil, H. & Aicher, B. The fixed combination of acetylsalicylic acid, paracetamol and caffeine is more effective than single substances and dual combination for the treatment of headache: a multicentre, randomized, double-blind, single-dose, placebo-controlled parallel group study. Cephalalgia 25(10), 776–787 (2005).
Kudo-Fujimaki, K. et al. Efficacy and safety of nateglinide plus vildagliptin combination therapy compared with switching to vildagliptin in type 2 diabetes patients inadequately controlled with nateglinide. J. Diabetes Invest. 5 (4), 400–409 (2014).
Mizuno, Y. Definition and Classification of Parkinsonian Drugs 2823–2852 (NeuroPsychopharmacotherapy. Springer, 2022).
Beudel, M., de Bie, R. M. & Leenders, K. Treatment of Parkinson’s Disease: Early, Late, and Combined 2891–2915 (NeuroPsychopharmacotherapy. Springer, 2022).
Youdim, M. B. & Finberg, P. M. Pharmacological actions of l-deprenyl (selegiline) and other selective monoamine oxidase B inhibitors. Clin. Pharmacol. Ther. 56, 725–733 (1994).
Naoi, M., Maruyama, W. & Shamoto-Nagai, M. Neuroprotective function of rasagiline and selegiline, inhibitors of type B monoamine oxidase, and role of monoamine oxidases in synucleinopathies. Int. J. Mol. Sci. 23 (19), 11059 (2022).
Aubert, I. et al. Increased D1 dopamine receptor signaling in levodopa-induced dyskinesia. Annals Neurology: Official J. Am. Neurol. Association Child. Neurol. Soc. 57 (1), 17–26 (2005).
Guigoni, C. et al. Pathogenesis of levodopa-induced dyskinesia: Focus on D1 and D3 dopamine receptors. Parkinsonism Relat. Disord. 11, S25–S9 (2005).
Wishart, D. S. et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 46(D1), D1074–D82 (2018).
Dang, H., Ye, Y., Zhao, X. & Zeng, Y. Identification of candidate genes in ischemic cardiomyopathy by gene expression omnibus database. BMC Cardiovasc. Disord. 20 (1), 320. https://doi.org/10.1186/s12872-020-01596-w (2020).
Acknowledgements
Not applicable.
Funding
This work was supported by the Youth Science Fund of the National Natural Science Foundation of China (grant numbers 82304382, 82304855); the Research Startup Fund of Anhui Medical University (grant numbers 900206); Youth Science Fund of Anhui Province Natural Science (grant numbers 2208085QH276); College Students’innovation and Entrepreneurship Competition (grant numbers S202310366077); Research Enhancement Program of Anhui Medical University (grant numbers 2022xkjT001); Anhui Provincial Institute of Translational Medicine Funding Project (grant numbers 2021zhyx-C20).
Author information
Authors and Affiliations
Contributions
Author contributionsYL and QL conceived and designed the experiments.YL, and WS collected the data. YL, YS and YC contributed reagents/materials/analysis tools/codes. YL constructed the dataset. YL, YS, YC and QL wrote this manuscript. QL, SG, KZ, YL, YS, YC, YZ, JG, LH, and WS revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All the authors have agreed that the manuscript published in Chinese Medicine.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Y., Shen, Y., Cai, Y. et al. Integrating transcriptomic data with a novel drug efficacy prediction model for TCM active compound discovery. Sci Rep 15, 7688 (2025). https://doi.org/10.1038/s41598-024-82498-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-82498-1
