
Abstract
DNA methylation modifications are an important mechanism affecting the process of atherosclerosis (AS). Previous studies have shown that Galectin-8 (GAL8) DNA methylation level is associated with sudden death of coronary heart disease or acute events of coronary heart disease. However, the mechanism of GAL8 DNA methylation and gene expression in AS has not been elucidated, prompting us to carry out further research on it. ApoE-/- mice were used to establish an atherosclerosis model, and DNA methylation inhibitor DO05 and MAPK/mTOR inhibitor UO126 were used for intervention. Pyrosequencing was used to detect changes in GAL8 DNA methylation levels of the mouse aorta between groups. ROC curve analysis was performed to assess the relationship between GAL8 DNA methylation and atherosclerosis. Aortic staining with hematoxylin and eosin (H&E) was used to observe the aortic intima, plaque area, and characteristics of secondary lesions within the plaque. Oil Red O staining was used to detect lipid deposition in mouse arterial plaques or macrophages. Movat staining was used to detect the number of foam cells in the plaque. Immunohistochemistry (IHC) and Western blot were used to quantify the localization and expression levels of DNA methyltransferase1 (DNMT1), GAL8, MAPK/mTOR pathway proteins, Light Chain3 (LC3), Beclin1, Sequestosome1 (p62), Tumor Necrosis Factor-α (TNF-α), and other proteins. Immunofluorescence (IF) was used to detect the fluorescence intensity of GAL8, LC3, Monocyte chemoattractant protein-1(MCP-1), and other proteins. Detection of autophagosomes in macrophages by transmission electron microscopy was also performed. The foam cell model was induced with human monocytes (THP-1) and co-cultured with foam cells using siRNAs targeting GAL8, DO05, and UO126. The level of DNMT1 was detected by Western blot; Oil red O staining was used to detect lipid deposition in foam cells in each group, and the localization and expression levels of GAL8, MAPK/mTOR pathway proteins, LC3, Beclin1, p62, and TNF-α were quantitatively determined by Western blot. Immunofluorescence (IF) was used to detect the fluorescence intensity of GAL8, MAPK/mTOR pathway protein, LC3, p62, TNF-α, and other proteins. The GAL-8 promoter region harbors six CpG sites susceptible to DNA methylation. Following DNMT1 inhibition, the DC05 group displayed a significant decrease in methylation across all six CpG sites compared to the C57 and AS groups. Conversely, the UO126 group exhibited increased methylation at the first three CpG loci relative to the AS group. ROC curve analysis revealed GAL8 DNA methylation as an independent risk factor for atherosclerosis: GAL8, along with inflammation-related proteins MCP-1, MMP9, and TNF-α, were upregulated in the mouse lesion group, while expression of autophagy-related proteins LC3 and Beclin1 was downregulated. Additionally, phosphorylated MAPK/mTOR pathway proteins were detected in the mouse model of atherosclerosis. After inhibiting the methylation level of GAL-8 DNA, the expression of GAL-8 was up-regulated, macrophage autophagy was inhibited, inflammation was increased, and atherosclerotic lesions in mice were aggravated. After direct inhibition of the activity of the MAPK/mTOR pathway, macrophage autophagy was further weakened, the inflammatory response was further aggravated, and the atherosclerotic lesions of mice were further aggravated. After the specific knockdown of GAL-8 using siRNA GAL-8 using foam cells, the above phenomenon was reversed, macrophage autophagy was promoted, the inflammatory response was reduced, and the degree of atherosclerosis was alleviated. The degree of GAL8 DNA methylation is related to the progression of atherosclerosis, and its hypomethylation can aggravate atherosclerotic lesions. The mechanism may be through the regulation of MAPK/mTOR pathway to slow down the autophagy of macrophages, and then aggravate the inflammation in plaques. Targeting GAL8 DNA methylation may be a new target for the diagnosis and treatment of atherosclerosis.
Similar content being viewed by others


Introduction
Coronary heart disease (CHD) is the most prevalent cardiovascular disease (CVD) globally and the primary cause of death. Epidemiological data indicates that 8.88 million people succumbed to CHD in 2019, solidifying its position as the leading cause of sudden cardiac death (SCD)1. Studies have documented a 50% incidence of SCD, with coronary artery disease accounting for up to 80% of SCD cases in patients aged 50 years or older2,3,4. Atherosclerosis arises from a complex interplay of factors, with established risk factors for CHD including smoking, hypertension, type 2 diabetes, hyperlipidemia, elevated triglyceride (TG) and low-density lipoprotein cholesterol (LDL) levels, and low high-density lipoprotein cholesterol (HDL) levels5,6. Despite this knowledge, a complete understanding of CHD pathogenesis and its underlying atherosclerotic process remains elusive. The burgeoning field of omics has yielded a growing body of evidence implicating epigenetics, encompassing DNA methylation, histone modification, and non-coding RNA in atherogenesis7. DNA methylation, in particular, plays a critical role in stem cell maintenance and differentiation, individual aging, developmental anomalies, and the onset and progression of complex diseases such as tumors, diabetes, psychiatric disorders, and neurological diseases8,9,10,11,12,13,14. DNA methylation (DNAm) is an epigenetic mechanism essential for cell and tissue differentiation. This process involves the enzymatic addition of a methyl group to cytosine (C) residues, converting them to 5-methylcytosine (5-mC), by DNA methyltransferase (DNMT) using S-adenosylmethionine (SAM) as a methyl donor. A growing body of research suggests a link between DNA methylation and the risk of developing coronary heart disease across diverse populations, making it a valuable tool for understanding CHD development. For instance, Jiang et al.15,16. Employed peripheral blood mononuclear cell studies to demonstrate that both 5-mC and 5hm function as risk factors for CHD. Additionally, Agha et al.17. Conducted an 11-year follow-up study of over 11,000 individuals free of CHD at baseline. Their findings revealed a strong correlation between methylation levels at 52 CpG island loci and the subsequent development of CHD or myocardial infarction. Furthermore, several studies have investigated the relationship between blood DNA methylation and various CHD risk factors, including smoking, lipid metabolism, and inflammation18,19. Based on the current study, the analysis only considered the overall level of methylated cytosine in the genome at CpG loci and its correlation with coronary heart disease. It did not examine differential methylation of specific loci or its impact on the disease. To explore the mechanism of differential methylation genes acting on CHD. The research group collected human coronary artery tissue and used 850 k methylation chip experiments to construct a dataset of susceptibility differential methylation genes for sudden death of human CHD, and found that the significantly different hypomethylated genes included GAL-8, LTF, RFPL3, etc., while the hypermethylated genes included TMEM9B, ANK3, C6orF48, etc. This suggests that these genes DNA methylation was closely related to sudden death of CHD. In addition, we have confirmed that the level of GAL-8DNA methylation is closely related to the level of coronary plaque inflammation and risk factors for coronary heart disease. We found that MAPK was closely related to sudden coronary death through Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment of differential methylation sites20. In addition, we also confirmed that blockade of CXCR4 promotes macrophage autophagy through the PI3KAKT/mTOR pathway to alleviate coronary heart disease21. Building on these findings, Zhou H et al. demonstrated that mTOR inhibition can participate in regulating autophagy in atherosclerosis22. Additionally, Seeger et al. confirmed the MAPK signaling pathway as a key regulator of the inflammatory response and showed that its inhibition can slow atherosclerotic progression23,24,25. However, current studies on GAL8 mainly focus on tumor and bone diseases26,27, and no relevant studies on the etiology and mechanism of coronary heart disease involving GAL8 have been found. Therefore, to investigate whether the degree of GAL8 DNA methylation can be involved in altering atherosclerotic lesions through the MAPK/mTOR pathway, we employed ApoE-/- mice to establish models of atherosclerosis with GAL8 DNA methylation and MAPK/mTOR inhibition. We further used THP-1 cells to induce foam cells and established models with GAL8 inhibition, GAL8 DNA methylation inhibition, and MAPK/mTOR inhibition. These models were used to detect the localization and expression of proteins associated with the MAPK/mTOR pathway, inflammatory responses, and autophagy in macrophages. Additionally, we investigated changes in autophagosomes to explore how GAL8 DNA methylation affects macrophage autophagy and inflammation through the MAPK/mTOR pathway, ultimately influencing the degree of atherosclerosis.
Materials and methods
Experimental animals and groups
Thirty 7-week-old male ApoE-/- mice (SPF, 20–24 g) were purchased from Beijing Huafukang Biotechnology Co., Ltd. (Certificate No.: SCXK (Beijing) 2019–0010) along with 10 C57 male mice. All procedures involving animals were preapproved by the Institutional Animal Care and Use Committee of Guizhou Medical University (No.202304465). And all mice treated in this study were performed in accordance with the relevant guidelines and regulations, and all methods used were reported in accordance with the ARRIVE guidelines, In accordance with the Animal Ethics Committee of Guizhou Medical University. After one week of feeding with a standard diet, the 30 ApoE-/- mice were randomly divided into three groups: the atherosclerosis group, the GAL8DNA methylation inhibition group (DC05), the MAPK/mTOR pathway inhibition group (UO126), and the negative control group (C57, consisting of the 10 C57 mice). Subsequently, all mice were fed a high-fat diet (3% cholesterol, 0.2% propylthiouracil, 0.5% sodium cholate, 10% lard, 5% sugar, and 81.3% basal diet. The mice were fed in a standardized environment with light and dark cycles where they had free access to food and water)21until they reached 13 weeks of age. For the following three weeks, the tail vein of each mouse (including those in the C57 group) received continuous injections of normal saline, DNA methylation inhibitor DC05 (10 μM), or MAPK/mTOR pathway activity inhibitor UO126 (5 μg/10 μl)28twice a weekand all mice were humanely killed with an overdose of sodium pentobarbital (80 mg/kg, i.p.)29. At 16 weeks of age, all mice were anesthetized and euthanized. To minimize blood interference in subsequent experiments, the following procedures were performed: whole body perfusion, followed by fixation of the aortic root with 4% paraformaldehyde fixative solution for freezing and paraffin sectioning. To remove residual blood, a PBS buffer wash was performed before quick fixation with liquid nitrogen for 3 min. Finally, the tissues were cryopreserved at −80 °C for protein extraction.
HE, Oil Red O, and Movat stains for assessment of aortic plaque burden, intima thickness, lipid content, and foam cell presence in mice
Mouse aortic tissue was paraffin-embedded, and 2–3 μm thick sections were prepared using standard methods. These sections were then stained with HE to observe changes in aortic structure under a microscope. We measured coronary intima-media thickness and plaque area to assess atherosclerotic burden. For Oil Red O staining, the mouse aorta was placed flat in a special box, covered with an appropriate amount of OCT embedding medium, and then rapidly frozen in liquid nitrogen. Frozen tissue Sects. (3 μm) were obtained using a constant cooling chamber microtome. Lipid deposition in the aorta and foam cells were then observed under light microscopy following Oil Red O staining. Movat staining was used to visualize changes in coronary artery structural components. Image J software was employed to quantify the number of foam cells and the extent of lipid deposition within plaques.
Pyrosequencing to detect the DNA methylation level of GAL8 in the mouse aorta
We retrieved the GAL8 gene sequence from the NCBI GenBank database and used MethPrimer software to predict methylation within its promoter region. Primers were designed based on the predicted methylation sites. Following DNA extraction and bisulfite conversion, the methylated region was amplified by PCR using primers with 5’-biotin labels. Pyrosequencing was then performed to identify the methylation status of each CpG site. The methylation percentage of each CpG site was calculated using QCpG software, which treats methylated cytosines as artificial C/T single nucleotide polymorphisms (SNPs). Pyrophosphate primers are shown in Table 1..
Protein localization and mean absorbance of GAL8, p62, LC3, MMP9, TNF-ɑ and MCP-1 in the aorta of IHC mice
IHC staining was performed using an IHC kit (PV-9000, Zhongshan Jinqiao Biotechnology Co., Ltd., China). Following deparaffinization and hydration, tissue sections were incubated dropwise with 3% H2O2 for 10 min at 37 °C. Antigen retrieval was then performed by heating for 4 min, followed by blocking with 10% goat serum for 40 min at room temperature. Arterial paraffin sections were incubated overnight at 4 °C with primary antibodies against GAL-8 (1:100 dilution, 100 μL, Abcam, UAS, cat# EPR48), LC3 (Proteintech, China, Cat: No. 18725–1-AP), p62 (Proteintech, China, Cat: No. 18420–1-AP), TNF-α (1:100 dilution, 100 μL, Immunoway, China, Cat# YM3478), MMP-9 (1:100 dilution, 100 μL, Proteintech, China, Cat# 10,375–2-AP), and MCP-1 (1:100 dilution, 100 μL, Santa Cruz Biotechnology, USA, Cat # Sc-365870). Sections containing the primary antibody were then incubated with a biotinylated secondary antibody, and finally chromogenic using diaminobenzidine (DAB). The results were analyzed using Image-ProPlus 6.0 software.
Western blot to detect the changes in key proteins in GAL8 and MAPK/mTOR pathways
Frozen arterial samples were ground into a powder with liquid nitrogen and lysed with RIPA buffer containing a protease inhibitor cocktail to extract total coronary tissue proteins. Protein concentration was then determined. Equal amounts of protein samples were loaded onto an SDS-PAGE gel for electrophoresis, followed by transfer to a PVDF membrane. After blocking, the membrane was incubated with primary antibodies overnight at 4 °C. These primary antibodies included GAL-8, TNF-α, MCP-1, MMP-9, Beclin1 (Proteintech, China, CatNo11-1-AP), IL-1β (1:1000 dilution, 100 μL, Immunoway, China, Cat#YT2322), P62, LC3, P-MAPK (1:1000 dilution, 100 μL, Affinity, USA, Cat#AF4001), MAPK (1:100 dilution, 100 μL, Proteintech, China, Cat#14,064–1-AP), P-mTOR (1:1000 dilution, 100 μL, Proteintech, China, Cat#67,778–1-lg), mTOR (1:1000 dilution, 100 μL, Proteintech, China, Cat#: 67,778–1-lg), and DNMT1 (Biotechne, USA, Cat#NB100-56,519) at a standard dilution of 1:1000. Following washes, the PVDF membrane was incubated with horseradish peroxidase-conjugated secondary antibodies, either goat anti-mouse (1:5000 dilution, 100 μL, Absin, China, Cat#66,009–1-Ig) or anti-rabbit (1:6000 dilution, 100 μL, China, Cat#2594–1-AP) for 1 h at room temperature. Finally, the membrane was developed using ECL exposure solution (Liquid A:Liquid B = 1:1) in a chemiluminescence instrument. The resulting protein bands were analyzed using Image J software.
Immunofluorescence for the localization of key proteins in GAL8 and MAPK/mTOR pathways in mouse aorta and cell models
Tissue sections were routinely deparaffinized and processed as previously described. Cells were then removed, rinsed three times with cold PBS, fixed with 4% paraformaldehyde for 15 min, rinsed again with PBS, and permeabilized with 0.5% Triton-X 100 for 15 min. After another rinse, the sections were blocked with goat serum for 30 min. Subsequently, the sections were incubated with primary antibodies for GAL-8, DNMT1, LC3, P62, and TNF-α (all at a dilution of 1:200) overnight at 4 °C. Following the overnight incubation, the sections were incubated with an anti-rabbit fluorescent secondary antibody for 1 h at room temperature. Nuclei were then counterstained with DAPI for 10 min and sealed with an antifluorescence quencher. Finally, the sections were observed and photographed under a microscope. Interpretation of Results: In successful immunofluorescence staining, where autofluorescence did not significantly interfere with the signal, DAPI-stained nuclei appeared blue, while GAL-8, TNF-α, and LC3 displayed red fluorescence. P62, on the other hand, exhibited green fluorescence.
Electron microscopy detection of autophagosomes
Tissues were pre-fixed with 3% glutaraldehyde, followed by post-fixation with 1% osmium tetroxide. Dehydration through an acetone series, infiltration with Epox 812 for an extended period, and embedding were performed. Semi-thin sections were then stained with methylene blue for light microscopy. Ultrathin sections were subsequently cut using a diamond knife and stained with uranyl acetate and lead citrate for observation under a JEM-1400-FLASH transmission electron microscope.
Cell model objects and grouping
The human monocyte line (THP-1) was purchased from Shanghai Fuhuasheng Biotechnology Co., Ltd. (Shanghai, China). The cells were cultured in RPMI-1640 medium supplemented with 12% fetal bovine serum under 5% CO2 at 37 °C. After 7 passages, cells were used for subsequent experiments. Cells were then treated with either PMA (160 nmol/mL) or ox-LDL (80 ng/L) and designated as the PMA and ox-LDL groups, respectively. The siRNA (1 μL/mL) negative control group was labeled as the si-NC group, the siRNA (1 μL/mL) silencing group was labeled as the si-GAL8 group, the GAL8 DNA methylation inhibition group was labeled as the DC05 group, and the MAPK/mTOR pathway inhibition group was labeled as the UO126 group. Finally, the cells were collected and used for oil red O staining, immunofluorescence staining, and western blotting.
Statistical analysis
Data were analyzed using SPSS 25.0 statistical software. Measurement data are expressed as mean ± standard error of the mean (X ± SEM). For comparisons of mean differences between two groups, a t-test was employed. One-way ANOVA was used for comparisons among multiple groups. We applied the LSD method for post hoc comparisons to control the error from multiple comparisons. Pearson correlation analysis assessed correlations between variables. ROC curve was used to analyze the predictive ability of GAL8 methylation for coronary heart disease. p < 0.05 was considered statistically significant. For each experiment, the average of at least 3 replicates was used.
Results
Changes in GAL8 gene methylation, GAL8 expression level after MAPK/mTOR inhibition, and atherosclerotic structure in mice
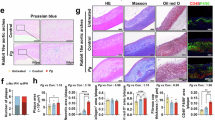
Histological analysis confirmed successful establishment of the mouse atherosclerosis model. The C57 group displayed normal aortic vascular morphology, with a continuous endothelial cell layer, no subendothelial lipid deposition, and tightly arranged smooth muscle cells (Fig. 1A). In contrast, the other three groups (AS, DC05, and UO126) exhibited significant atherosclerotic changes, including atheromatous plaque deposition, intimal hyperplasia, increased numbers of foam cells within plaques, and enhanced lipid deposition (P < 0.05) (Fig. 1A-G). Interestingly, GAL8 expression was up-regulated in the DC05 group compared to the AS group (P < 0.05) (Fig. 1H-K), despite DC05 treatment inhibiting GAL8 DNA methylation. Furthermore, this upregulation coincided with a worsening of the arterial lesions in the DC05 group compared to the AS group (P < 0.05) (Fig. 1A-G). Similarly, the UO126 group showed even higher GAL8 expression (P < 0.05) (Fig. 1H-K) and more severe arterial lesions (P < 0.05) (Fig. 1A-G) compared to the DC05 group.

100 × /400 × magnification of human coronary arteries with different degrees of lesions (A-C) HE, Movat staining, Oil red O staining at 100 × magnification, Bar = 200 μm; (E–G) Quantification of atherosclerotic area of mice, atherosclerotic lipid deposit area in mice and number of foam cells within the plaque. (D, H–K) Immunohistochemical staining at 400 × magnification, Bar = 50 μm and Western blot detection of GAL8 localization and expression. *P < 0.05, **P < 0.01, ***P < 0.001. Each group n = 10.
Changes in DNA methylation levels of GAL-8 promoter in each group in mouse arterial tissues
Pyrophosphate sequencing analysis revealed methylation of 6 CpG sites within the GAL-8 promoter region. Inhibiting DNMT1 resulted in significantly increased methylation levels (P < 0.05) at all 6 CpG sites in the DC05 group compared to both the C57 and AS groups. In the UO126 group, only the methylation levels of the first 3 CpG sites were significantly increased (P < 0.05) compared to the C57 and AS groups. Additionally, the DC05 group displayed significantly higher methylation levels (P < 0.05) at all 6 CpG sites compared to both the AS and UO126 groups. These findings are detailed in Table 2.
ROC curve analysis for assessment of the methylation degree of GAL 8 gene on atherosclerosis
The ROC curve analysis of GAL8 promoter region methylation demonstrated that GAL8 DNA methylation is an independent risk factor for atherosclerosis (AUC = 0.735, P < 0.01), as shown in Fig. 2.

ROC of GAL-8 DNA methylation levels.
Effect of GAL8 DNA methylation degree on GAL8 expression level and MAPK/mTOR pathway in atherosclerotic plaques of silent mice
To investigate the impact of GAL8 DNA methylation on the MAPK/mTOR pathway, we first silenced GAL8 DNA methylation and analyzed changes in key pathway proteins. Subsequently, the MAPK/mTOR pathway was inhibited for further verification. Immunohistochemistry and western blotting revealed that compared to the C57 group, the protein levels of MAPK, mTOR, p-MAPK, and p-mTOR were elevated in the AS, DC05, and UO126 groups (P < 0.05) (Fig. 3N, Q, R). Notably, DC05 displayed higher expression compared to AS, and UO126 showed the strongest upregulation after MAPK/mTOR pathway inhibition (P < 0.05) (Fig. 3N, Q, R). We further examined the expression of inflammatory factors (MCP-1, MMP9, TNF-α) and autophagy-related proteins (Beclin1, P62, LC3). Inhibiting GAL8 DNA methylation led to increased expression of inflammatory factors (P < 0.05) (Fig. 3A, G, D-M), decreased Beclin1 and LC3 (autophagy markers), and increased P62 expression (P < 0.05) (Fig. 3A-C, K–O), suggesting impaired autophagy. Additionally, this change was exacerbated by MAPK/mTOR pathway inhibition (Fig. 3). In conclusion, reduced GAL8 DNA methylation resulted in upregulated GAL8 expression, weakened autophagy, and enhanced inflammation. However, the effect was less pronounced compared to direct MAPK/mTOR pathway inhibition.

Expression and localization of Inflammatory cytokines and autophagy proteins among different groups (A). IHC detection of Inflammatory cytokines and autophagy proteins localization in foam at 400 × magnification, Bar = 50 μm; (G, L). Average protein level of Inflammatory cytokines and autophagy proteins; (B–F, H–K, M–O). Quantification of Inflammatory cytokines and autophagy proteins.*P < 0.05, **P < 0.01, ***P < 0.001. Each group n = 10.
GAL8 DNA effect of methylation degree on autophagy and inflammation
Immunofluorescence and electron microscopy corroborated the findings. Compared to the AS group, the DC05 and UO126 groups displayed increased fluorescence intensity of GAL8, MMP9, and MCP-1 (Fig. 4B), suggesting enhanced inflammation. Conversely, LC3 fluorescence in macrophages was reduced (Fig. 4B), indicating impaired autophagy. Electron microscopy confirmed a decrease in autophagosome numbers in the DC05 group compared to AS, with a further reduction observed in the UO126 group (Fig. 4A). These findings collectively support the hypothesis that reduced GAL8 DNA methylation leads to upregulated GAL8 expression, weakened autophagy, and a stronger inflammatory response. However, the overall effect was less pronounced compared to direct MAPK/mTOR pathway inhibition.

(A) Detection of macrophage autophagosomes in aortic by projection electron microscopy. (B) Immunofluorescence localization of GAL8, LC3, MMP9, MCP-1 in mouse artery at 400 × magnification, Bar = 50 μm. Each group n = 6.
GAL8 silencing reduces foam cell lipid deposition
Silencing GAL8 with siRNA significantly reduced its expression in foam cells compared to the ox-LDL and si-NC groups, as confirmed by Western blotting (P < 0.05) (Fig. 5B). This reduction in GAL8 expression was accompanied by a significant decrease in intracellular lipid deposition (P < 0.05) (Fig. 5A). Conversely, the DC05 and UO126 groups, where GAL8 methylation was inhibited, displayed significantly increased GAL8 expression and lipid deposition compared to both the ox-LDL and si-NC groups (P < 0.05) (Fig. 5A, C-E). Notably, the UO126 group exhibited even higher levels of GAL8 expression and lipid deposition compared to the DC05 group (P < 0.05) (Fig. 5A, C, D). These findings suggest that reducing GAL8 expression is associated with reduced intracellular lipid accumulation in foam cells.

(A, C) Lipid deposition in foam cells was detected by Oil red at 400 × magnification, Bar = 50 μm. (B) Quantification of lipid deposition in foam cells. (B) Western blotting for DNMT1and GAL8 protein at different lesion sites. (D-E) Quantification of DNMT1and GAL8 protein. *P < 0.05, **P < 0.01, ***P < 0.001. Each group n = 6.
GAL8 silencing inhibits autophagy and enhances inflammatory response
Following GAL8 silencing, we employed immunofluorescence with single and double labeling to detect the fluorescence intensities of DNMT1 (red), GAL8 (red), MMP9 (red), TNF-α (red), p62 (green), and LC3 (red) (Fig. 6K-O). Additionally, Western blotting was used to assess the expression levels of GAL8 and its downstream key proteins in the MAPK/mTOR pathway, Beclin1 and p62-related autophagic proteins, and inflammatory factors like TNF-α (Fig. 6A,J). The results revealed that compared to the ox-LDL and si-NC groups, silencing GAL8 significantly reduced the protein expression levels of GAL8, p62, Mapk, mTOR, TNF-α, IL-β, and MMP9 in foam cells (P < 0.05) (Fig. 6B-I), with a corresponding decrease in the fluorescence intensity of GAL8, p62, TNF-α, and MMP9. Conversely, the DC05 and UO126 groups, where GAL8 methylation was inhibited, displayed increased expression of these proteins and enhanced fluorescence intensities of GAL8, p62, TNF-α, and MMP9 (P < 0.05) (Fig. 6B, D-I, L-O). Interestingly, Beclin1 protein expression increased in the si-GAL8 group, while it decreased in the DC05 and UO126 groups. Additionally, LC3 fluorescence decreased in these latter groups (Fig. 6C, M). These findings suggest that inhibiting GAL8 expression can suppress the activity of the MAPK/mTOR signaling pathway, thereby dampening autophagy and exacerbating the inflammatory response. However, the overall effect was less pronounced compared to directly inhibiting the MAPK/mTOR pathway.

Expression and localization of Inflammatory cytokines and autophagy proteins among different groups in foam cells (A, J). Western blotting for Inflammatory cytokines and autophagy proteins at different groups (B-I). Quantification of Inflammatory cytokines and autophagy proteins (K–O). Immunofluorescence localization of Inflammatory cytokines and autophagy proteins in foam cells at 400 × magnification, Bar = 50 μm. *P < 0.05, **P < 0.01, ***P < 0.001. Each group n = 6.
Discussion
Epigenomics, transcriptomics, proteomics, metabolomics, genomics, and other methods have confirmed that gene changes and protein changes are related to atherosclerotic lesions. However, these methods cannot fully understand the complexity of their effects on coronary heart disease. Among these approaches, the role of epigenetics in atherosclerosis is becoming increasingly significant19,30,31. This study investigated the methylation level of GAL-8 DNA during atherosclerotic development. ROC curve analysis was used to analyze the ability of GAL8 DNA methylation to predict atherosclerosis in mice. The results showed that GAL8DNA methylation was an independent risk factor for atherosclerosis, which aligns with the research group’s previous findings20. However, Studies have shown its critical role in regulating capillary formation, endothelial cell migration, and angiogenesis in vivo32,33. Its specific mechanism in atherosclerosis remains largely unreported. Our previous results performed functional enrichment analysis on the differentially methylated genes. This analysis identified the MAPK signaling pathway and the mTOR signaling pathway as important enrichment pathways for these genes. This suggests a potential close correlation between autophagy, inflammation, and differentially methylated genes (DMGs)20. Additionally, previous research has shown that the MAPK/mTOR signaling pathway can alter the severity of atherosclerotic lesions by modulating changes in inflammation and autophagy34,35,36. At the same time, research has shown that DNA methylation can alter atherosclerotic lesions by modulating the expression of genes related to oxidative stress and inflammation37. However, the current understanding of the relationship between DNA methylation and coronary heart disease is primarily based on evidence linking the overall level of methylated cytosine in CpG loci across the genome to coronary heart disease. It does not consider the differential methylation and mechanisms of specific loci17,18,38.
Therefore, to verify whether GAL8 DNA methylation regulates coronary atherosclerotic heart disease lesions through the MAPK/mTOR pathway, we used ApoE-/- mice to establish an atherosclerosis model. We investigated the relationship between methyltransferase 1 (DNMT1) and the MAPK/mTOR pathway by inhibiting DNMT1 and the MAPK/mTOR pathway with DC05 and UO126, respectively, to further explore the role of the MAPK/mTOR pathway in GAL8 DNA methylation activity. The results of arterial pyrophosphate analysis in mice showed that after DNMT1 inhibition, the methylation levels of the six CpG sites in GAL8 DNA decreased. This was accompanied by upregulation of protein expressions for GAL8, MAPK, mTOR, phosphorylated-MAPK (p-MAPK) and phosphorylated-mTOR (p-mTOR). Notably, further upregulation of these proteins was observed after inhibition of the MAPK/mTOR pathway. Furthermore, we found that inhibiting the degree of GAL8 DNA methylation led to upregulation of inflammatory factors MCP-1, MMP9, and TNF-α, while downregulating autophagy-related proteins Beclin1 and LC3. Additionally, the expression of P62, a protein associated with autophagy, was upregulated after inhibiting GAL8 DNA methylation. This effect was further exacerbated after mTOR inhibition. Based on these findings, we hypothesized that a reduction in the degree of GAL8DNA methylation leads to upregulated GAL8 expression. This, in turn, weakens autophagy and strengthens inflammatory effects, ultimately resulting in an aggravation of atherosclerotic lesions. However, the magnitude of this effect appears to be less pronounced compared to directly inhibiting the MAPK/mTOR pathway. To further solidify these conclusions, we performed electron microscopy analysis. Compared to the control C57 group, the AS group exhibited an increase in autophagosomes. Conversely, the DC05 group displayed a decrease in autophagosomes compared to the AS group. Notably, the UO126 group showed an even greater decrease in autophagosomes compared to the DC05 group. In addition, Qiao L, et al. has shown that increasing the number of autophagosomes in the early stage of atherosclerosis may be a protective mechanism for vascular endothelial cells and smooth muscle cells39. For example, autophagosomes can clear excessive reactive oxygen species (ROS) and misfolded proteins in vascular endothelial cells, reduce oxidative stress damage, and thus delay the development of atherosclerotic lesions to a certain extent40. Research suggests that excessive autophagy can lead to endothelial damage or promote the formation of small blood vessels, both of which can worsen atherosclerosis and even compromise the stability of atherosclerotic plaques. This can lead to myocardial fibrosis and ultimately impair the function of heart muscle cells41,42. Conversely, insufficient autophagy can lead to the accumulation of necrotic cells due to impaired clearance, which can exacerbate atherosclerotic changes. However, appropriate autophagy can enhance angiogenesis and provide blood flow to alleviate myocardial ischemia caused by atherosclerosis43. Our findings demonstrate that inhibiting GAL8DNA methylation weakens autophagy compared to the atherosclerotic group, while concurrently strengthening the inflammatory effect. However, this effect is less pronounced than directly inhibiting the MAPK/mTOR pathway. Therefore, we speculated that MAPK/mTOR activity was inhibited after inhibition of GAL8 DNA methylation, which might regulate downstream events by affecting the expression of MAPK and mTOR in this pathway, resulting in the reduction of autophagosomes and damage to vascular endothelial cells. The direct use of MAPK/mTOR pathway inhibitors further aggravates this phenomenon, resulting in increased atherosclerotic lesions. To further investigate the impact of inhibiting GAL8 gene methylation and the MAPK/mTOR pathway on atherosclerosis, we established a foam cell model using Ox-LDL-induced human-derived monocytes. We found that directly silencing GAL8 expression with siRNA significantly reduced GAL8 expression in the siGAL8 group compared to both the AS and DC05 groups. Compared to the DC05 group, the siGAL8 group exhibited a relative weakening of autophagy and intracellular lipid deposition, alongside downregulation of inflammatory factors. Compared with DC05 group, fat deposition in UO126 group was further increased. Therefore, we ulteriorly concluded that when the autophagy process is inhibited, and aggravate the inflammatory response. there maybe have a problem in the autophagosome lysosome fusion, and the cell’s ability to decompose lipids is reduced. At this time, low-density lipoprotein cholesterol is easy to deposit in the blood vessel wall and is not easy to clear, thus promoting the formation of atherosclerotic plaque. Besides, Studies have confirmed that transcription factors are sensitive to DNA methylation44,45. Therefore, whether autophagy and inflammatory effects may be mediated by specific transcription factors in atherosclerosis will be further explored in our follow-up studies.
In summary, we speculate that inhibition of GAL8 DNA methylation may aggravate atherosclerosis by reducing autophagy, increasing inflammation, leading to lipid deposition and endothelial cell damage. The results are consistent with those of Vazquez MM et al.46. GAL8 DNA methylation plays a unique role in regulating autophagy and inflammation in atherosclerosis, which brings new ideas for future targeted therapy. In the treatment of atherosclerosis, the DNA methylation level of GAL8 maybe targeted to regulate, and more accurate and effective therapeutic drugs or means can be developed by affecting the initiation and progression of autophagy and inflammation. It is expected to break through the limitations of existing treatments, providing patients with better treatment options, and may be an important direction for future medical research and clinical application. Elucidating the precise mechanisms underlying this process will be the next step of our research, as depicted in Fig. 7.

Regulatory map of GAL8 DNA methylation on autophagy and inflammation.
Limitations
Our study investigated the role of GAL8 DNA methylation in atherosclerosis. DNA methylation is a complex process catalyzed by DNA methyltransferases (DNMTs), includingDNMT1, DNMT2, DNMT3a, DNMT3b, and DNMT3L. Among these, DNMT1 plays a primary role in maintaining stable DNA methylation patterns. In this study, we did not directly inhibit GAL8 DNA methylation. Instead, we employed DC05 to inhibit the activity of DNMT1, which indirectly altered the overall DNA methylation landscape. This approach may affect the methylation status of all genes undergoing methylation, not just GAL8. Therefore, we cannot definitively exclude the influence of methylation changes in other genes on atherosclerotic lesions. However, our results do demonstrate that DC05 treatment led to changes in GAL8 DNA methylation. The molecular mechanism has only been preliminarily discussed, and the complete signaling pathway and interaction network have not been thoroughly analyzed, which may neglect other factors that may be regulated simultaneously. In addition, we lack long-term dynamic observations. We will do further research in the follow-up study.
Data availability
The datasets generated and analysed during the current study are available in the [NCBI] repository, [LINKNUMBER TO DATASETS: https://www.ncbi.nlm.nih.gov/nuccore/NM_006499.5].
Change history
01 July 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41598-025-08357-9
References
World Health Organization. Global health estimates: leading causes of death. 2021. https://www.who.int/data/gho/data/themes/mortality. Accessed 01 Mar 2021.
Robert, J. et al. Sudden cardiac death caused by coronary heart disease. Circulation 125(8), 1043–1052 (2012).
Van den Hoogen, I. J. et al. What atherosclerosis findings can CT see in sudden coronary death: Plaque rupture versus plaque erosion. J. Cardiovasc. Comput. Tomogr. 14(3), 214–218 (2020).
Golsa, J. et al. Coronary artery disease and atherosclerotic risk factors in a population-based study of sudden death. J. Gen. Intern. Med. https://doi.org/10.1007/s11606-019-05486-6 (2020).
Ridker, P. M. et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377(12), 1119–1131 (2017).
Desiderio, A. et al. DNA methylation in cardiovascular disease and heart failure: novel prediction models?. Clin. Epigenet. 16(1), 115 (2024).
Philibert, R. et al. Validation of an integrated genetic-epigenetic test for the assessment of coronary heart disease. J. Am. Heart Assoc. 20(12), e030934 (2023).
Li, M. et al. Genome-wide DNA methylation analysis of peripheral blood cells derived from patients with first-episode schizophrenia in the Chinese Han population[J]. Mol. Psychiatry 26(8), 4475–4485 (2021).
You, C. et al. A cell-type deconvolution meta-analysis of whole blood EWAS reveals lineage-specific smoking-associated DNA methylation changes. Nat. Commun. 11(1), 4779 (2020).
Cai, L. et al. Epigenetic alterations are associated with tumor mutation burden in non-small cell lung cancer. J. Immunother. Cancer. 7(1), 198 (2019).
Schnitzler, G. R. et al. Convergence of coronary artery disease genes onto endothelial cell programs. Nature 626(8000), 799–807 (2024).
Wong, Q. H. et al. Molecular landscape of IDH-mutant primary astrocytoma grade IV/glioblastomas. Mod. Pathol. 34(7), 1245–1260 (2021).
Pu, Y. et al. Transcriptome and differential methylation integration analysis identified important differential methylation annotation genes and functional epigenetic modules related to vitiligo. Front. Immunol. 12, 587440 (2021).
Zhu, W. et al. Deoxyribonucleic acid methylation signatures in sperm deoxyribonucleic acid fragmentation. Fertil. Steril. 116(5), 1297–1307 (2021).
Jiang, D. et al. DNA methylation and hydroxymethylation are associated with the degree of coronary atherosclerosis in elderly patients with coronary heart disease. Life Sci. 224, 241–248 (2019).
Krolevets, M. et al. DNA methylation and cardiovascular disease in humans: a systematic review and database of known CpG methylation sites. Clin. Epigenet. 15(1), 56 (2023).
Agha, G. et al. Blood leukocyte DNA methylation predicts risk of future myocardial infarction and coronary heart disease. Circulation 140(8), 645–657 (2019).
Navas-Acien, A. et al. Blood DNA methylation and incident coronary heart disease: evidence from the strong heart study. JAMA Cardiol. 6(11), 1237–1246 (2021).
Xia, Y., Brewer, A. & Bell, J. T. DNA methylation signatures of incident coronary heart disease: findings from epigenome-wide association studies. Clin. Epigenet. 13(1), 186 (2021).
Xia, B. et al. Association of GAL-8 promoter methylation levels with coronary plaque inflammation. Int. J. Cardiol. 19, 131782 (2024).
Li, F. et al. Blockade of CXCR4 promotes macrophage autophagy through the PI3K/AKT/mTOR pathway to alleviate coronary heart disease. Int. J. Cardiol. 392, 131303 (2023).
Zhou, H. et al. Artemisinin and procyanidins loaded multifunctional nanocomplexes alleviate atherosclerosis via simultaneously modulating lipid influx and cholesterol efflux. J. Control Release. 341, 828–843 (2022).
Romero-Becerra, R. et al. MKK6 deficiency promotes cardiac dysfunction through MKK3-p38γ/δ-mTOR hyperactivation. Elife. 11, e75250 (2022).
Traughber, C. A. et al. Disulfiram reduces atherosclerosis and enhances efferocytosis, autophagy, and atheroprotective gut microbiota in hyperlipidemic mice. J. Am. Heart Assoc. 13(8), e033881 (2024).
Dong, Z. et al. Myocardial infarction drives trained immunity of monocytes, accelerating atherosclerosis. Eur. Heart J. 45(9), 669–684 (2024).
Bieniasz-Krzywiec, P. et al. Podoplanin-expressing macrophages promote lymphangiogenesis and lymphoinvasion in breast cancer. Cell Metab. 30(5), 917–936 (2019).
Diskin, S. et al. GALectin-8 promotes cytoskeletal rearrangement in trabecular meshwork cells through activation of Rho signaling. PLoS One 7(9), e44400 (2012).
Xie, B. et al. Induction of autophagy and suppression of type I IFN secretion by CSFV. Autophagy 17(4), 925–947 (2021).
Ding, J. et al. trilobatin alleviates cognitive deficits and pathologies in an alzheimer’s disease mouse model. Oxid. Med. Cell. Longev. 2021, 3298400 (2021).
Erdmann, J. et al. A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc. Res. 114, 1241–1257 (2018).
Palou-Márquez, G. et al. DNA methylation and gene expression integration in cardiovascular disease. Clin. Epigenet. 13(1), 75 (2021).
Troncoso, M. F. et al. Galectin-8: a matricellular lectin with key roles in angiogenesis. Glycobiology 24(10), 907–914 (2014).
Si, Y. et al. Crystallization of galectin-8 linker reveals intricate relationship between the n-terminal tail and the linker. Int. J. Mol. Sci. 17(12), 2088 (2016).
Zhang, Q. et al. Berberine represses human gastric cancer cell growth in vitro and in vivo by inducing cytostatic autophagy via inhibition of MAPK/mTOR/p70S6K and Akt signaling pathways. Biomed. Pharmacother. 128, 110245 (2020).
Qian, Y. et al. Tanshinone IIA regulates mapk/mtor signal-mediated autophagy to alleviate atherosclerosis through the miR-214-3p/ATG16L1 axis. Int. Heart J. 64(5), 945–954 (2023).
Huang, W. et al. Triggering receptor expressed on myeloid cells 2 protects dopaminergic neurons by promoting autophagy in the inflammatory pathogenesis of parkinson’s disease. Front. Neurosci. 18(15), 745815 (2021).
Uddin, M. D. M. et al. Clonal hematopoiesis of indeterminate potential, DNA methylation, and risk for coronary artery disease. Nat. Commun. 13(1), 5350 (2022).
Xu, S. et al. Oxidative stress gene expression, DNA methylation, and gut microbiota interaction trigger Crohn’s disease: a multi-omics Mendelian randomization study. BMC Med. 21(1), 179 (2023).
Qiao, L. et al. Deficient chaperone-mediated autophagy promotes inflammation and atherosclerosis. Circ. Res. 129(12), 1141–1157 (2021).
Cheng, C. et al. NPRC deletion mitigated atherosclerosis by inhibiting oxidative stress, inflammation and apoptosis in ApoE knockout mice. Sig. Transduct. Target Ther. 8(1), 290 (2023).
Qu, W. et al. Long noncoding RNA Gpr137b-ps promotes advanced atherosclerosis via the regulation of autophagy in macrophages. Arterioscler. Thromb. Vasc. Biol. 43(11), e468–e489 (2023).
Hu, J. et al. Autophagy modulates mesenchymal-to-endothelial transition via p53. Aging (Albany NY). 13(12), 22112–22121 (2020).
Xiao, P. et al. RTN4/Nogo-A-S1PR2 negatively regulates angiogenesis and secondary neural repair through enhancing vascular autophagy in the thalamus after cerebral cortical infarction. Autophagy. 18(11), 2711–2730 (2022).
Yin, Y. et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science https://doi.org/10.1126/science.aaj2239 (2017).
Héberlé, É. & Bardet, A. F. Sensitivity of transcription factors to DNA methylation. Essays Biochem. 63(6), 727–741 (2019).
Vazquez, M. M. et al. Nitro-oleic acid, a ligand of CD36, reduces cholesterol accumulation by modulating oxidized-LDL uptake and cholesterol efflux in RAW264.7 macrophages. Redox Biol. 36, 101591. https://doi.org/10.1016/j.redox.2020.101591 (2020).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 82460335) and the Gui zhou Provincial Science and Technology Program (Qiankehe Foundation-ZK[2022] General 357).
Author information
Authors and Affiliations
Contributions
Bing Xia,Yan-lin Lu wrote the main manuscript text and Conducted most of the experiments and data processing. Jin Peng , Jing-wei Liang, Fang-qin Li, Jiu-yang Ding, Chang-wu Wan, Cui-yun Le and Jia-lin Dai participated in animal modeling experiments and drawing. Jie Wang and Cui-yun Le provided a fund. Bing Guo,Jiang Huang provide ideas, supervise and manage the research team, and revise manuscripts to verify data, etc.
Corresponding authors
Ethics declarations
Competing interests
Bing Xia, Yan-lin Lu contributed equally to this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of the Article contained an error in Figure 6. Due to an operational error, images in Figure 6K intended for the PMA group were erroneously duplicated and placed in the position designated for the DC05 group. The original data confirms that the DC05 group sample possesses its own distinct image. Similarly, an error occurred during layer manipulation in Figure 6M, where the LC3/P62 image belonging to the si-NC group was incorrectly copied to the position representing the si-GAL8 group.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xia, B., Lu, Yl., Peng, J. et al. Galactin-8 DNA methylation mediates macrophage autophagy through the MAPK/mTOR pathway to alleviate atherosclerosis. Sci Rep 15, 603 (2025). https://doi.org/10.1038/s41598-024-85036-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-85036-1
