
Abstract
The present study was to investigate potential biomarkers and therapeutic targets for epilepsy by conducting a transcriptomic and proteomic analysis of human brain tissue from patients with epileptic lesions. Brain tissue was collected from the epileptic lesions after surgical resection and surgical removed brain tissue from non-epileptic patients. Using RNA sequencing and iTRAQ-based proteomic analysis, The transcriptomic analysis identified 1,604 DEGs, with 584 upregulated and 1,020 downregulated. The proteomic analysis identified 694 DEPs, with 331 upregulated and 363 downregulated. The combined transcriptomic and proteomic analysis showed that the DEGs and DEPs were mainly enriched in biological processes such as D-aspartate transport, transmembrane transport, cell junctions, vesicle transport, and metabolic processes. Tubulin polymerization promoting protein family member-3 (TPPP3), proprotein convertase subtilisin/kexin type-1 (PCSK1), and dihydropyrimidinase-like 3 (DPYSL3) were significantly altered in the epilepsy patients, and their expression trends were confirmed by the RT-qPCR, WB, and IHC staining results. By integrating transcriptomic and proteomic analyses, we identified genes and proteins expressed differently in epileptic and non-epileptic patients and their associated biological processes. Three key DEPs (TPPP3, PCSK1, and DPYSL3) were identified, indicating their potential significance in the pathological mechanisms of epilepsy.
Similar content being viewed by others


Introduction
Epilepsy is a prevalent neurological disorder affecting 70 million individuals globally. Its primary pathological characteristic is the abnormal discharge of brain neurons, resulting in recurrent seizures1,2. The mechanisms underlying epilepsy are intricate and multifaceted. Despite advances in treatment, 30% of patients continue to experience suboptimal or ineffective therapeutic outcomes3.
Currently, epilepsy treatment primarily relies on anti-seizure drugs (ASMs), which inhibit seizures by modulating neuronal electrical activity. However, prolonged ASMs use can lead to severe side effects, including cognitive impairment, mood disturbances, and organ toxicity4. Additionally, some patients develop drug resistance5. To address these issues, researchers are investigating novel therapies, such as gene therapy and stem cell therapy6.
In recent years, advancements in high-throughput sequencing and proteomics technologies have enabled researchers to increasingly employ multi-omics approaches to investigate the molecular mechanisms of diseases7. It is now possible to analyze gene transcription, the intermediate process that occurs during gene expression and includes steps such as transcriptional and post-transcriptional regulation8. It is also possible to quantitatively analyze proteins, the ultimate effectors of function, and this is important because all physiological and pathological changes directly result from alterations in protein function9.”
While such technologies are being used to elucidate the mechanisms that contribute to the multi-level regulation of gene expression in epilepsy, the genes closely related to the onset of epilepsy and its resistance to drug effects have not yet been identified. Therefore, the aim of the present study was to identify significantly differentially expressed genes (DEGs) and proteins (DEPs) in patients with epilepsy and determine their enriched biological processes. The findings of this study enhance our understanding of the molecular mechanisms of epilepsy and provide new potential targets for future diagnosis and treatment.
Materials and methods
Human brain samples
This study was conducted in accordance with the principles outlined in the Declaration of Helsinki. Written informed consent was obtained from all participants prior to their inclusion in the study. All methods were performed in compliance with relevant guidelines and regulations. Brain tissue was collected from five patients with epilepsy after surgery and five non-epileptic patients who underwent surgery at the Affiliated Hospital of Xuzhou Medical University. This study was approved by the ethics committee of the Affiliated Hospital of Xuzhou Medical University (license number: XYFY2024-KL222-01). For patients diagnosed with epilepsy and requiring surgical intervention, epileptic focus tissue was obtained during craniotomy. After opening the dura mater, intraoperative electrocorticography (ECoG) was employed, with electrodes placed directly on the brain surface to record real-time electrical activity. This technique allowed for the precise identification of regions exhibiting epileptic discharges, thereby guiding the accurate resection of the epileptic focus. Non-epileptic brain tissue was obtained from normal brain tissue excised during brain tumor surgery. These samples were collected from regions adjacent to the tumor that were not infiltrated and were confirmed to be free of epileptic activity and tumor invasion through intraoperative ECoG and postoperative pathological analysis. To ensure complete tumor removal, a standard surgical safety margin was adhered to, involving the excision of a portion of normal brain tissue surrounding the tumor. This approach aligns with clinical guidelines and aims to minimize the risk of recurrence while ensuring the patient’s long-term health. Each patient provided informed consent prior to surgery, agreeing to the use of resected specimens for pathological examination and related medical research. (Inclusion and exclusion criteria are described in Appendix 1.)
Transcriptome analysis
The transcriptome analysis was performed using a previously reported methodology10. Total RNA was extracted using a TRIzol kit (Thermo Fisher Scientific, USA). The mRNA was fragmented into 200–300 bp fragments, and the first strand of cDNA was synthesized using random primers and reverse transcriptase, with RNA as the template. This was followed by the synthesis of the second strand of cDNA. After a quality inspection was conducted, paired-end sequencing was performed on the Illumina platform. The sequencing data were filtered and aligned with the reference genome data to calculate gene expression and perform differential expression, enrichment, and clustering analyses. Differential expression sequencing (DESeq) was performed to screen for DEGs with a |log2FoldChange| value > 1 and a P-value < 0.05, and gene functions were annotated using the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases. All raw data were deposited into the National Center for Biotechnology Information (NCBI) database as a BioProject (accession no. PRJNA1142196).
Proteome analysis
The samples that were analyzed in the transcriptome analysis were also subjected to a proteome analysis, which was performed as previously described11. Peptides (100 µg) from each sample were labeled using the Thermo Fisher Tandem Mass Tag (TMT) labeling kit. Chromatographic separation was conducted using an Easy nLC 1200 system (Thermo Scientific), and the resultant Liquid Chromatography with tandem mass spectrometry (LC-MS/MS) RAW files were imported into Sequest HT in the Proteome Discoverer application (version 2.4, Thermo Scientific) for database searching. The TMT-labeled proteomic data were qualitatively and quantitatively analyzed using Proteome Discoverer. DEPs, with a |log2FoldChange| value > 1.2 and a P-value < 0.05, were identified. Functional annotation of the DEPs was performed using the GO and KEGG databases. The raw mass spectrometry proteome data have been submitted to the iProx integrated proteome resources center (dataset identifier: IPX0009404000).
mRNA and protein correlation analyses
The mRNA and protein correlation analyses were conducted as previously described12, and significant DEGs and DEPs were identified. A quantitative analysis of these DEGs and DEPs was performed, and a Venn diagram was created to visualize their overlap in the transcriptome and proteome. The correlations between the shared DEGs and DEPs were visualized using a nine-square grid analysis, and a heat map was generated for the expression cluster analysis. Finally, GO and KEGG enrichment analyses were conducted.
Quantitative reverse transcription polymerase chain reaction (qPCR)
The qPCR process used in this study has been previously described13. Total RNA was extracted from brain tissue samples using TRIzol (Thermo) and quantified using a spectrophotometer and 1 µL aliquots of RNA. Each nuclease-free PCR tube contained 2 µg of RNA, 2 µL of 4× AccuRT Reaction Mix, and nuclease-free water to a total volume of 8 µL. After incubating at room temperature for 5 min, 2 µL of 5× AccuRT Reaction Stopper was added, and the solution was mixed. Then, 4 µL of 5× All-In-One RT MasterMix was added, and the mixture was placed in a pre-set PCR machine for cDNA reverse transcription. RNA quantification was based on the 2 − ΔΔCt method. The specific primers used in the PCR are listed in Table S1.
Western blot (WB)
The WB analysis was performed as previously described14. Each brain tissue sample was lysed in radioimmunoprecipitation assay (RIPA) buffer and centrifuged, and the supernatant was used for protein concentration measurement and sample preparation. Samples were subjected to electrophoresis on a pre-prepared gel. After electrophoresis, the gel was transferred to a nitrocellulose (NC) membrane using a transfer system. The membrane was blocked with a quick block solution for 30 min, then incubated with a primary antibody overnight at 4 °C. The following day, the membrane was incubated with a secondary antibody at room temperature for 2 h. After incubation, the membrane was developed using a chemiluminescence instrument (Bio-Rad Inc., USA). The dilutions and suppliers of the primary antibodies used in the WB and immunohistochemical (IHC) staining procedures are listed in Table S2.
Immunohistochemical (IHC) staining
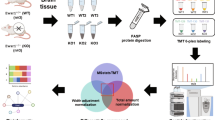
Fixed tissues were washed, dehydrated, cleared, and embedded in paraffin. Sections were cut, mounted, deparaffinized, and rehydrated. IHC staining was then conducted using specific antibodies and chromogens to visualize target antigens. Figure 1 summarizes the various analytical steps performed on the two sample groups.

Overall Approach of the Study. The experimental design and overall approach of this study.
Statistical analysis
The results of the real time (RT)-qPCR and WB analyses, as well as the fragments per kilobase of transcript per million mapped reads (FPKM) values and the relative abundance values, are expressed as mean ± standard error of the mean (SEM). The statistical significance of the differences between the epileptic (EP) and non-epileptic (NEP) groups was determined using Student’s t-test. with the significance level set as p < 0.05. The DEPs were assessed using Student’s t-test. For the DEG analysis, DESeq2 was employed to calculate p-values based on a negative binomial distribution model. Hypothesis testing was conducted using the Wald test or the likelihood ratio test.
Results
Transcriptomic analysis
To determine whether there were any differences in gene expression, we conducted an RNA sequencing (RNA-seq) analysis of the brain tissue samples from the EP and NEP groups. The principal component analysis (PCA) showed that there were significant transcriptional differences between the EP and NEP groups (Fig. 2A) and that there was clear separation and directionality between the groups, based on gene expression similarities. A subsequent comparison of the RNA-seq results revealed 1,604 DEGs, with 584 upregulated and 1,020 downregulated in the EP group (Fig. 2B, C). The heat map of the gene expression profiles indicated that most of the samples were divided into two clusters (Fig. 2D).

Analysis of mRNA Expression Differences Between the Epilepsy Group (EP) and the Non-Epilepsy Group (NEP). (A) Principal component analysis (PCA) of RNA sequencing data. Points represent biological replicates. (B) Bar chart displaying genes differentially expressed between the groups. (C) Volcano plot illustrating the relative abundances of the transcripts; the vertical dashed lines represent a 2-fold expression difference threshold, and the horizontal dashed line represents a P-value threshold of 0.05. The red dots indicate the upregulated genes in the EP group, the blue dots indicate the downregulated genes, and the gray dots represent non-significant differentially expressed genes (DEGs). (D) Heat map of the gene expression across the different groups. (E) GO enrichment analysis results, showing the top 10 most significantly enriched GO terms with the smallest P-values for each category. (F) KEGG enrichment analysis results for the DEGs, measured by rich factor, FDR value, and the number of genes enriched in each pathway. The top 20 most significantly enriched KEGG pathways with the smallest FDR values are presented.
To analyze the biological functions of the DEGs, we performed GO annotation and KEGG pathway enrichment analysis. As shown in Fig. 2E, most of the DEGs were primarily associated with the following GO terms in the biological process category: plasma membrane, cell periphery, intrinsic component of plasma membrane, integral component of plasma membrane, and extracellular matrix. In the cellular component category, many of the DEGs were associated with the following terms: extracellular matrix structural constituent, calcium ion binding, G protein-coupled peptide receptor activity, peptide receptor activity, and signaling receptor binding. In the molecular function category, the DEGs were mainly related to the following terms: anatomical structure development, multicellular organismal process, developmental process, system development, and biological adhesion. The KEGG pathway enrichment analysis of the DEGs indicated that these genes were primarily involved in the transforming growth factor-beta (TGF-beta) signaling pathway, the advanced glycation end products and receptor for advanced glycation end products (AGE-RAGE) signaling pathway, pathways in cancer, the Hippo signaling pathway, viral protein interactions with cytokines and cytokine receptors, the cyclic adenosine monophosphate (cAMP) signaling pathway, and neuroactive ligand–receptor interactions(Fig. 2F).
Proteomic analysis
Proteomic technologies can be used to study the large-scale structure and function of proteins in complex biological samples. In this study, we initially assessed the clustering of the groups via PCA (Fig. 3A). The results indicated that there was clear directionality between the two groups and showed that there were significant differences in protein expression between the groups. As shown in Figs. 3B and 694 DEPs were identified, with 331 upregulated and 363 downregulated. Additionally, a volcano plot was generated to display the distribution of the DEPs (Fig. 3C). The heat map generated to examine the protein expression profiles revealed two distinct clusters among the samples (Fig. 3D).

Analysis of Protein Expression Differences between the EP and NEP Groups. (A) PCA of proteomic data. Points represent biological replicates. (B) Bar chart displaying the proteins differentially expressed between the groups. (C) Volcano plot illustrating the relative abundance of the proteins. (D) Heat map of the protein expression across different groups. (E) Bubble plot for the GO functional enrichment analysis. (F) Bubble plot for the KEGG pathway enrichment analysis of the differentially expressed proteins (EP vs. NEP, top 20). The x-axis represents the negative logarithm of the P-value, the circle color represents the rich factor, and the circle size represents the count.
Subsequently, we performed a GO annotation and KEGG pathway enrichment analysis of the DEPs. As shown in Fig. 3E, the majority of the DEPs were primarily associated with the following GO annotation terms in the biological process category: protein–DNA complex assembly, protein–DNA complex subunit organization, nucleosome assembly, chromosome condensation, and negative regulation of DNA recombination. In the cellular component category, a large number of the DEPs were associated with the following terms: cell junction, cell projection, nucleosome, plasma membrane-bounded cell projection, and cell periphery. Within the molecular function category, the DEPs were primarily associated with the following terms: nucleosomal DNA binding, ATPase-coupled cation transmembrane transporter activity, nucleosome binding, immune receptor activity, and chemokine binding. The KEGG pathway enrichment analysis of the DEPs (Fig. 3F) revealed associations with the synaptic vesicle cycle, metabolic pathways, endocrine- and other factor-regulated calcium reabsorption, neurodegeneration pathways, the peroxisome proliferator-activated receptor (PPAR) signaling pathway, glutamatergic synapses, extracellular matrix (ECM)–receptor interactions, and arginine and proline metabolism.
Correlation between the transcriptome and proteome
To evaluate the complementarity between the mRNA and protein levels, we analyzed the correlation between the DEGs and DEPs. From the Venn diagram shown in Fig. 4A, we identified 606 significant DEPs and 1,596 significant DEGs, and 89 molecules exhibited differential expression at both the mRNA and protein levels. To further explore the correlations between the transcriptomic and proteomic data, we plotted correlation diagrams of the shared DEGs/DEPs that exhibited the same or opposite trends (Fig. 4B and C). This analysis showed that there were molecules with a strong positive correlation between the proteomic and transcriptomic data, with a correlation coefficient (R) of 0.86, indicating consistency at both the transcriptional and translational levels (Fig. 4B). Conversely, we identified molecules with a strong negative correlation (R = -0.97), which suggested that potential complex feedback mechanisms or post-translational regulation caused discrepancies between the transcription- and translation-level data (Fig. 4C).

Correlation between Transcriptome and Proteome. (A) Venn diagram comparing the significant differentially expressed genes/proteins (DEGs/DEPs) in the proteome and transcriptome. (B) Correlation plot of shared DEGs/DEPs with the same trend. Red indicates commonly upregulated genes/proteins, and green indicates commonly downregulated genes/proteins. The x-axis shows the fold change in the proteome, and the y-axis shows the fold change in the transcriptome, both on a log2 scale. (C) Correlation plot of shared DEGs/DEPs with opposite trends. The blue dots and gray dots represent the genes/proteins with opposite regulatory trends. The x-axis shows the fold change in the proteome, and the y-axis shows the fold change in the transcriptome, both on a log2 scale. (D) Clustering heat map of the DEGs/DEPs PCSK1, TPPP3, and DPYSL3. (E) GO functional enrichment analysis. (F) Pathway enrichment analysis of the significant DEGs/DEPs in the proteome and transcriptome. The x-axis represents the pathway names, and the y-axis represents the omics data. The color intensity of the dots indicates the significance level, and the size of the dots represents the number of enriched genes/proteins (count).
Our analysis revealed that the significant DEGs/DEPs were prominently enriched in several GO functional categories, such as D-aspartate transport, transmembrane transport, cell junctions, vesicle transport, metabolic processes, extracellular region, secretory granule lumen, cytoplasmic vesicle lumen, extracellular space, response to xenobiotic stimulus, and central nervous system development. The significant enrichment in D-aspartate transport, transmembrane transport, cell junctions, and vesicle transport suggests that these are pivotal functions of the DEGs/DEPs. Additionally, nervous system-related functions (e.g., central nervous system development and astrocyte projection) were significantly enriched, indicating that these genes/proteins may play crucial roles in the function of the nervous system or the development of diseases of the nervous system (Fig. 4E). To explore the functional distribution of these genes/proteins in biological pathways, we performed a pathway enrichment analysis with the proteomic and transcriptomic data. The results showed that there was significant enrichment—at both the proteomic and transcriptomic levels—in various biological pathways, including viral protein interactions with cytokines and cytokine receptors, T helper (Th)1 and Th2 cell differentiation, the calcium signaling pathway, axon guidance, the regulation of the actin cytoskeleton, pathways of neurodegeneration (multiple diseases), the synaptic vesicle cycle, and ECM–receptor interactions (Fig. 4F).
As shown in Fig. 4D, three of the identified DEGs are genes predominantly expressed in brain tissue or the nervous system that may play significant roles in neurological function and have not been previously reported in epilepsy. These genes are PCSK1, TPPP3, and DPYSL3.
Verification of DEGs by RT-qPCR and of deps by WB and IHC staining
Based on the transcriptomic and proteomic data, we identified several significant DEGs and DEPs. To further verify their biological significance, we selected three representative genes for validation: PCSK1, TPPP3, and DPYSL3. These genes exhibited significant differential expression in the omics data. DPYSL3 is primarily expressed in the nervous system and is crucial for nervous system development, particularly for axon growth and neuronal guidance15. PCSK1, a PI3K family member, is widely expressed in the brain and is essential in the neuroendocrine system for converting inactive prohormones and neuropeptides into active forms and thus for regulating metabolism, body weight, and energy balance16,17. TPPP3, a microtubule-associated protein, is highly expressed in the brain and is involved in microtubule dynamics and the formation and maintenance of neuronal processes18,19.
To validate the expression of PCSK1, TPPP3, and DPYSL3 in the EP and NEP groups, we employed RT-qPCR, WB, and IHC staining techniques. The RNA-seq transcriptomic data indicated that there were significant differences in the expression of these genes between the EP and NEP groups. Specifically, compared with the NEP group, the FPKM values and relative abundance of TPPP3 and DPYSL3 were significantly decreased in the EP group (Figs. 6A and C and 7A and C), while the FPKM value of PCSK1 was significantly increased (Figs. 5A, C). The RT-qPCR results confirmed the trends observed in the expression of all three genes (Figs. 5B and 6B, and 7B). Additionally, the WB and IHC staining results validated the iTRAQ-based proteomic data, demonstrating consistency with the observed transcriptomic trends. The proteomic findings for PCSK1, TPPP3, and DPYSL3 were corroborated by the WB and IHC staining results, as shown in Figs. 5D–F and 6D–F, and 7D–F, respectively.

Verification of PCSK1 Expression in the Transcriptome and Proteome. (A) FPKM values of PCSK1 in the RNA sequencing transcriptome data. (B) Quantitative analysis of the relative expression of PCSK1 mRNA. (C) Relative abundance of PCSK1 in the iTRAQ proteomic data. (D) Representative western blot bands of PCSK1. (E) Quantitative analysis of PCSK1 protein expression. (F) Representative immunohistochemical staining image of PCSK1. Data are presented as mean ± SEM, n = 3–5/group. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, EP versus NEP.

Verification of TPPP3 Expression in the Transcriptome and Proteome. (A) FPKM values of TPPP3 in the RNA sequencing transcriptome data. (B) Quantitative analysis of the relative expression of TPPP3 mRNA. (C) Relative abundance of TPPP3 in the iTRAQ proteomic data. (D) Representative western blot bands of TPPP3. (E) Quantitative analysis of TPPP3 protein expression. (F) Representative immunohistochemical staining image of TPPP3. Data are presented as mean ± SEM, n = 3–5/group. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, EP versus NEP.

Verification of DPYSL3 Expression in the Transcriptome and Proteome. (A) FPKM values of DPYSL3 in the RNA sequencing transcriptome data. (B) Quantitative analysis of the relative expression of DPYSL3 mRNA. (C) Relative abundance of DPYSL3 in the iTRAQ proteomic data. (D) Representative western blot bands of DPYSL2. (E) Quantitative analysis of DPYSL3 protein expression. (F) Representative immunohistochemical staining image of DPYSL3. Data are presented as mean ± SEM, n = 3–5/group. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, EP versus NEP.
Discussion
This study focused on the transcriptomic and proteomic analysis of brain tissue samples from epilepsy patients and non-epileptic controls. When we integrated the data, we identified 89 significant DEGs/DEPs. Further analysis revealed that these genes/proteins are associated with specific biological functions and pathways and potentially contribute to the pathological mechanisms of epilepsy.
In the transcriptomic analysis, we identified 1,604 genes that were differentially expressed in the EP group compared to the NEP group; 584 were upregulated and 1,020 were downregulated. The proteomic analysis revealed 694 DEPs, with 331 upregulated and 363 downregulated. We then conducted an integrative analysis of the transcriptomic and proteomic data, and the observed changes in gene and protein expression were strongly correlated. The DEGs/DEPs were found to contribute to multiple biological functions and to be critical in various cellular processes, metabolic pathways, and signal transduction.
Our functional enrichment analysis revealed that the genes/proteins differentially expressed in the EP group are primarily involved in biological processes such as D-aspartate transport, transmembrane transport, cell junctions, vesicle transport, and metabolic processes. Studies have indicated that D-aspartate and transmembrane transport processes are closely associated with neurotransmitter metabolism and transport20,21, suggesting that these processes may influence neural activity by modulating neurotransmitter levels during epileptic seizures. The processes of cell junctions and vesicle transport are crucial for intercellular communication and material exchange22,23, potentially impacting neuronal connectivity and function during the pathological progression of epilepsy. The presence of significantly altered metabolic pathways in the EP group suggests that energy metabolism plays a critical role in epilepsy pathogenesis. DEGs/DEPs were also found to be substantially involved in several critical signaling and disease-related pathways, predominantly those related to neurodegenerative diseases24, such as Parkinson’s disease and Alzheimer’s disease. The enrichment of these pathways suggests potential common molecular mechanisms between epilepsy and neurodegenerative diseases. DEGS/DEPs were found to be involved in inflammation- and immune-related pathways, such as the AGE-RAGE signaling pathway and Th17 cell differentiation25, indicating that inflammatory and immune responses may significantly contribute to the pathogenesis of epilepsy. Finally, given that DEGS/DEPs involved in metabolism and signaling pathways, such as the Hippo signaling pathway and cAMP signaling pathway26,27,28, were detected, cell proliferation, survival, and metabolic regulation processes may play crucial roles in epilepsy pathogenesis.
To further validate the vital roles that the DEGs/DEPs play in biological functions related to the nervous system, we focused on those predominantly expressed in brain tissue or the nervous system. We subsequently identified genes/proteins not previously reported in epilepsy and selected three representative genes—PCSK1, TPPP3, and DPYSL3—for further validation.
PCSK1 is a member of the protein convertase family and plays a crucial role in the cleavage and activation of precursor proteins, including various hormones and neuropeptides, such as insulin, glucocorticoids, and pro-opiomelanocortin (POMC)29. The end products generated by PCSK1-mediated reactions are widely involved in energy balance, glucose metabolism, and blood pressure regulation. In humans, loss-of-function mutations in the PCSK1 gene cause monogenic obesity, impaired glucose tolerance, hypertension, cardiac remodeling, and microvascular damage30. PCSK1 expression has also been reported in human cancer and tumor cell lines31. While there have been no reports published on the role of PCSK1 in epilepsy to date, it has been shown to play a crucial role in neuroendocrine transformation and neuropeptide processing29. Thus, abnormal expression or function of PCSK1 may lead to nervous system dysfunction. Our results support this hypothesis, as we found that PCSK1 was significantly upregulated in the EP group. Although there is currently no direct evidence linking PCSK1 to epilepsy, its crucial role in nervous system processes suggests that it may contribute to the pathophysiology of epilepsy.
TPPP3, a member of the TPPP family, regulates microtubule dynamics and exhibits unique microtubule-bundling activity32. Studies have shown that knocking down TPPP3 can inhibit cell proliferation, induce apoptosis, and suppress tumor growth33,34. Research also indicates that TPPP3 plays a critical role in female embryo implantation35. Recently, Zhang et al. reported that TPPP3 is essential for both central and peripheral nerve regeneration33. Furthermore, it has been suggested that TPPP3 promotes axon regeneration18. Hence, the role that TPPP3 plays in the development of neurological diseases has garnered increasing attention. In this study, we found that TPPP3 expression was significantly lower in the EP group. Although there is currently no direct evidence linking TPPP3 to epilepsy, its critical role in nervous system processes suggests it may be involved in the pathophysiology of epilepsy.
DPYSL3, also known as collapsing response mediator protein 4 (CRMP4), is a cytoplasmic phosphoprotein highly expressed in the nervous system36. The protein product of the DPYSL3 gene, located on chromosome 5q32, is crucial for many aspects of neurogenesis, including neuronal differentiation, neurite outgrowth, and axonal guidance37. Studies have shown that knocking down DPYSL3 in prostate cancer cells enhances their invasion and migration abilities37, while overexpression of DPYSL3 in advanced neuroblastoma improves the clinical prognosis38. It has also been shown that elevated levels of DPYSL3 in amyotrophic lateral sclerosis (ALS) can impact motor neurons39. CRMP family members are known to be involved in the formation of neural networks, and CRMP4 knockout mice exhibit symptoms such as impaired social activities and abnormal sensory sensitivity40. Overall, DPYSL3 plays an indispensable role in the nervous system. In this study, we found that DPYSL3 expression was significantly lower in the EP group. Although there is currently limited research directly linking DPYSL3 to epilepsy, its crucial role in neurodevelopment and signal transduction processes suggests that its dysfunction may affect neuronal excitability and susceptibility to seizures.
Conclusion
In this study, we comprehensively explored the molecular mechanisms that underlie epileptic seizures by conducting a multi-omics analysis. The rationale for selecting the PCSK1, TPPP3, and DPYSL3 genes as targets is based on their crucial roles in nervous system processes. While there is no direct evidence linking them to epilepsy at present, their involvement in key neurological functions such as neurogenesis, synaptic plasticity, and neurotransmitter processing makes them potential contributors to the pathophysiology of epilepsy. Furthermore, these genes showed significant differential expression in our omics data, suggesting that they may play a role in the disease process, which led us to choose them for validation. The results revealed that patients with epilepsy had significantly different gene and protein expression profiles compared to patients without epilepsy, and multiple key genes and pathways were identified, including PCSK1, TPPP3, and DPYSL3. These genes are implicated in neurotransmitter imbalance, abnormal neuronal excitability, inflammatory responses, and cell signaling. These findings provide new insights into the pathophysiological mechanisms of epilepsy as well as potential biomarkers and therapeutic targets. Specifically, PCSK1, TPPP3, and DPYSL3 may serve as key diagnostic biomarkers or treatment targets for epilepsy in the future. The further experiments that validate the association between PCSK1 and/or TPPP3 and seizures or epilepsy by using an epilepsy animal model will help understand the pathogenesis of epilepsy and strengthen the impact of the present research.
Data availability
The transcriptome dataset generated in this study has been deposited in the National Center for Biotechnology Information (NCBI) database as a BioProject (accession no. PRJNA1142196), and is accessible at https://www.ncbi.nlm.nih.gov/sra/PRJNA1142196. The proteome data are stored in the iProx integrated proteome resources center (dataset identifier: IPX0009404000), and can be accessed at https://www.iprox.cn/page/project.html? id=IPX0009404000.
References
Shaikh, T. G. et al. The role of angiotensin receptor blockers in treating epilepsy: a review. Neurol. Sci. 45 (4), 1437–1445 (2024).
Kanner, A. M. & Bicchi, M. M. Antiseizure medications for adults with epilepsy: A review. JAMA 327 (13), 1269–1281 (2022).
Manole, A. et al. State of the Art and challenges in Epilepsy—A narrative review. J. Personalized Med., 13(4). (2023).
García-Rodríguez, C. et al. Contribution of non-selective Membrane Channels and Receptors in Epilepsy231 (Pharmacology & Therapeutics, 2022).
Jaseja, H., Goyal, M. & Mishra, P. Drug-Resistant epilepsy and obstructive sleep apnea: exploring a link between the two. World Neurosurg. 146, 210–214 (2021).
Higurashi, N., Broccoli, V. & Hirose, S. Genetics and Gene Therapy in Dravet Syndrome131 (Epilepsy & Behavior, 2022).
Zhang, L. et al. Liver transcriptomic and proteomic analyses provide new insight into the pathogenesis of liver fibrosis in mice. Genomics 115 (6), 110738 (2023).
Yu, L. et al. MTR4 drives liver tumorigenesis by promoting cancer metabolic switch through alternative splicing. Nat. Commun. 11 (1), 708 (2020).
Liang, Y. et al. Research progress of reduced amino acid alphabets in protein analysis and prediction. Comput. Struct. Biotechnol. J. 20, 3503–3510 (2022).
Ji, S. et al. Integrative transcriptome and proteome analyses elucidate the mechanism of Lens-Induced myopia in mice. Invest. Ophthalmol. Vis. Sci. 64 (13), 15 (2023).
Dou, T. et al. A combined transcriptomic and proteomic approach to reveal the effect of Mogroside V on OVA-Induced pulmonary inflammation in mice. Front. Immunol. 13, 800143 (2022).
Ma, Y. et al. Integrative transcriptomics and proteomics analysis provide a deep insight into bovine viral diarrhea Virus-Host interactions during BVDV infection. Front. Immunol. 13, 862828 (2022).
Zhou, C. et al. Nitro-oleic acid-mediated blood-brain barrier protection reduces ischemic brain injury. Exp. Neurol. 346, 113861 (2021).
Zhou, C. et al. Gain-of-function of progesterone receptor membrane component 2 ameliorates ischemic brain injury. CNS Neurosci. Ther. 29 (6), 1585–1601 (2023).
Kowara, R. et al. Co-localization and interaction of DPYSL3 and GAP43 in primary cortical neurons. Biochem. Biophys. Res. Commun. 363 (1), 190–193 (2007).
Zhu, X. et al. Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proc. Natl. Acad. Sci. 99 (16), 10293–10298 (2002).
Choquet, H., Stijnen, P., Creemers, J. W. M., Mbikay, M. & Seidah, N. G. Genetic and Functional Characterization of PCSK1, in Proprotein Convertases, and Humana: Totowa, NJ. 247–253. (2011).
Olah, J. et al. Modulatory Role of TPPP3 in Microtubule Organization and Its Impact on Alpha-Synuclein Pathology. Cells, 11(19). (2022).
Huang, R. et al. MicroRNA-133b negatively regulates zebrafish single Mauthner-Cell axon regeneration through targeting tppp3 in vivo. Front. Mol. Neurosci. 10, 375 (2017).
Ota, N., Shi, T. & Sweedler, J. V. D-Aspartate acts as a signaling molecule in nervous and neuroendocrine systems. Amino Acids. 43 (5), 1873–1886 (2012).
Herring, B. E. et al. Is Aspartate Excitatory Neurotransmitter? J. Neurosci., 35(28): 10168–10171. (2015).
van Niel, G. et al. Challenges and directions in studying cell–cell communication by extracellular vesicles. Nat. Rev. Mol. Cell Biol. 23 (5), 369–382 (2022).
Kuo, I. Y. et al. Recent advances in conventional and unconventional vesicular secretion pathways in the tumor microenvironment. J. Biomed. Sci. 29 (1), 56 (2022).
Sanghai, N. & Tranmer, G. K. Biochemical and molecular pathways in neurodegenerative diseases: an integrated view. Cells, 12(18). (2023).
Soman, S. et al. A multicellular signal transduction network of AGE/RAGE signaling. J. Cell. Commun. Signal. 7 (1), 19–23 (2013).
Lee, D. Global and local missions of cAMP signaling in neural plasticity, learning, and memory. Front. Pharmacol. 6, 161 (2015).
Zhang, H. et al. Complex roles of cAMP-PKA-CREB signaling in cancer. Exp. Hematol. Oncol. 9 (1), 32 (2020).
Wu, Z. & Guan, K. L. Hippo signaling in embryogenesis and development. Trends Biochem. Sci. 46 (1), 51–63 (2021).
Stijnen, P. et al. PCSK1 mutations and human endocrinopathies: from obesity to Gastrointestinal disorders. Endocr. Rev. 37 (4), 347–371 (2016).
Ramos-Molina, B., Martin, M. G. & Lindberg, I. PCSK1 variants and human obesity. Prog Mol. Biol. Transl Sci. 140, 47–74 (2016).
Chou, C. L. et al. PCSK1 overexpression in rectal Cancer correlates with poor response to preoperative chemoradiotherapy and prognosis. Onco Targets Ther. 13, 3141–3150 (2020).
Shen, A. et al. TPPP3 inhibits the proliferation, invasion and migration of endometrial carcinoma targeted with miR-1827. Clin. Exp. Pharmacol. Physiol. 48 (6), 890–901 (2021).
Ren, Q. et al. Silence of TPPP3 suppresses cell proliferation, invasion and migration via inactivating NF-kappaB/COX2 signal pathway in breast cancer cell. Cell. Biochem. Funct. 38 (6), 773–781 (2020).
Ye, K. et al. Knockdown of tubulin polymerization promoting protein family member 3 inhibits cell proliferation and invasion in human colorectal cancer. J. Cancer. 8 (10), 1750–1758 (2017).
Shukla, V. et al. Uterine TPPP3 plays important role in embryo implantation via modulation of β-catenin†. Biol. Reprod. 99 (5), 982–999 (2018).
Liang, P. I. et al. Upregulation of dihydropyrimidinase-like 3 (DPYSL3) protein predicts poor prognosis in urothelial carcinoma. BMC Cancer. 23 (1), 599 (2023).
Desprez, F. et al. Contribution of the dihydropyrimidinase-like proteins family in synaptic physiology and in neurodevelopmental disorders. Front. Neurosci. 17, 1154446 (2023).
Tan, F. et al. Dihydropyrimidinase-like protein 3 expression is negatively regulated by MYCN and associated with clinical outcome in neuroblastoma. Cancer Sci. 104 (12), 1586–1592 (2013).
Maimon, R. et al. A CRMP4-dependent retrograde axon‐to‐soma death signal in amyotrophic lateral sclerosis. EMBO J., 40(17). (2021).
Ohtani-Kaneko, R. Crmp4-KO mice as an animal model for investigating certain phenotypes of autism spectrum disorders. Int. J. Mol. Sci., 20(10). (2019).
Acknowledgements
We thank Shanghai Bioprofile Biotechnology Co., Ltd for their support in data analysis.
Funding
This work was supported by the National Nature Science Foundation of China to FH (82171420).
Author information
Authors and Affiliations
Contributions
Taiyang Zhu, Yan Zhou, and Lei Zhang participated in the experimental design, and wrote the paper. Xiaoyu Sun, Fanyu Shen, Hui Zhou and Wanyan Ni analyzed the sequencing datasets, Lingwen Kong, Hai Tang, Qihua Xiao and Sha Liu performed the bioinformatics analyses. Huimin Gao, Guoliang Jin and Xiao Jia conducted the basic experiments. Fang Hua supervised the project and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
This study was approved by the ethics committee of the Affiliated Hospital of Xuzhou Medical University (license number: XYFY2024-KL222-01). Each patient provided informed consent prior to surgery, agreeing to the use of resected specimens for pathological examination and related medical research.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, T., Zhou, Y., Zhang, L. et al. A transcriptomic and proteomic analysis and comparison of human brain tissue from patients with and without epilepsy. Sci Rep 15, 16369 (2025). https://doi.org/10.1038/s41598-025-00986-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-00986-4
