
Abstract
Rheumatoid arthritis (RA) and cervical cancer (CC) are major global health challenges, yet the molecular connections between these two conditions remain poorly understood. To bridge this gap, our study employs bioinformatics approaches to explore shared genetic pathways and potential biomarkers. We started by identifying differentially expressed genes in RA and CC and then applied WGCNA to detect functionally related gene clusters using gene expression data from the GEO database. Additionally, we constructed protein–protein interaction (PPI) networks and examined the role of the immune microenvironment. To assess the prognostic relevance of key genes in CC, we leveraged survival data from TCGA. Our analysis identified 55 key genes common to RA and CC, with four—CXCL1, CXCL13, ZWINT, and PTTG1—emerging as significant. ROC curve validation confirmed their diagnostic potential, and a model incorporating these genes was associated with poorer prognosis in CC. Among them, CXCL1 stood out as especially crucial. Our findings suggest a potential link between chronic inflammation, immune dysregulation, and chemokine-related pathways in RA patients, which may contribute to an increased susceptibility to CC.
Similar content being viewed by others


Introduction
Rheumatoid arthritis (RA) is a widespread chronic autoimmune disease, affecting approximately 0.5–1% of the global population1. This disorder triggers the generation of numerous inflammatory mediators and autoantibodies, resulting in tissue and organ damage2. The development and exacerbation of inflammation in RA patients are believed to result from a combination of inherited susceptibilities and particular environmental factors3. A key driver of RA progression is angiogenesis, which facilitates the infiltration of leukocytes from the bloodstream into synovial tissues, thereby enhancing inflammation4. Emerging evidence indicates that RA is linked to a higher risk of overall cancer5 and several site-specific malignancies, including lung cancer6, leukemia7 and cervical cancer8.
Cervical cancer (CC) is a significant global health burden, ranking as the third most commonly diagnosed cancer and the fourth leading cause of cancer-related mortality among women9. Persistent human papillomavirus (HPV) infection is widely recognized as the main risk factor for CC10. The oncogenic potential of high-risk HPV types is primarily driven by the early production of two key oncoproteins, E6 and E7, which hijack host cellular mechanisms to create a suitable tumor microenvironment (TME)11. The E6 and E7 oncoproteins can also modulate the expression of immune mediators in host cells12, including cytokines and chemokines, to facilitate the recruitment of immune cells, TME remodeling13, angiogenesis, chronic inflammation14, and proliferation of tumor cells15. Although early administration of the HPV vaccine can prevent CC effectively16, it does not eliminate pre-existing HPV infections17. The clinical management of CC is a significant challenge due to its asymptomatic onset, delayed diagnosis and poor prognosis18. Patients with advanced CC still face limited effective treatment options, with a 5-year survival rate remaining disappointingly low at merely 17%19. This underscores the urgent need to identify novel therapeutic targets that could improve patient outcomes and enhance long-term survival.
RA is a complex autoimmune disorder characterized by chronic inflammation, the underlying mechanisms of which remain incompletely understood20. Similarly, chronic inflammation is a hallmark of HPV infection21. Prolonged inflammation can contribute to DNA damage, genetic instability and an increased risk of mutations, while also promoting the survival and expansion of cancer stem cells22. This pro-inflammatory environment may create conditions that accelerate the progression of CC in RA patients with HPV infection23. The association between RA and cancer has been extensively investigated, with evidence highlighting its significant impact on the quality of life and survival rates for RA patients24. Additionally, the present study found that the majority of RA patients undergo long-term systemic immunosuppressive or corticosteroid therapies25. While these treatments effectively manage RA by reducing inflammation and preserving organ function, they may also increase the risk of cervical dysplasia over time26. A meta-analysis suggests that individuals with RA on long-term immunosuppressive therapies are more susceptible to HPV infection and cervical dysplasia5. Moreover, epidemiological studies consistently show a higher prevalence of CC and cervical intraepithelial neoplasia in RA patients27,28,29. Recent findings from an inverse variance weighted analysis in a two-sample Mendelian randomization (MR) study further support a significant causal link between RA and an increased risk of CC30.
Nevertheless, few studies have delved into the molecular mechanisms linking RA and CC through bioinformatics analysis, and the shared etiology of RA and CC remains obscure. Given the high prevalence of CC among RA patients, coupled with its insidious onset and poor prognosis, the importance of early detection, prompt diagnosis, and efficacious treatment can not be overstated. Recent progress in gene microarray technology has enabled researchers to swiftly evaluate the expression of thousands of genes, deepening our genetic comprehension of disease pathogenesis. This research employs bioinformatics approaches to identify common pathways and key genes in RA and CC, aiming to uncover potential mechanisms, biomarkers, and therapeutic targets. These efforts are intended to enhance RA management and facilitate the early detection and effective management of CC.
Results
Identification of differentially expressed genes
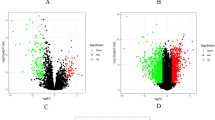
The GSE1919 dataset within the Gene Expression Omnibus (GEO) database encompassed 8760 genes from 5 RA patients and 5 healthy controls. Within this dataset, 840 genes were identified as differentially expressed genes (DEGs) associated with RA, with 408 downregulated and 432 upregulated, as illustrated in the heatmap (Fig. 1a). The GSE77298 dataset included 20,815 genes from 16 RA patients and 7 healthy controls. A total of 1930 genes were recognized as DEGs related to RA, with 819 downregulated and 1111 upregulated, as depicted in the heatmap (Fig. 1b). Similarly, we identified 1238 DEGs related to CC in the GSE9750 dataset, with 604 downregulated and 634 upregulated, as shown in the heatmap (Fig. 1c). In the GSE7803 dataset, we identified 604 DEGs associated with CC, with 276 downregulated and 328 upregulated, as presented in the heatmap (Fig. 1d). The 229 common DEGs associated with RA are shown in the Venn diagram (Fig. 1e) and the 420 common DEGs related to CC are shown in another Venn diagram (Fig. 1f). Ultimately, the Venn diagram (Fig. 1g) highlights 29 shared genes between RA- and CC-related DEGs.

RA and CC DEGs analysis. (a) A heatmap of RA DEGs analysis results based on the GSE1919 dataset. (b) A heatmap of RA DEGs analysis results based on the GSE77298 dataset. (c) A heatmap of CC DEGs analysis results based on the GSE9750 dataset. (d) A heatmap of CC DEGs analysis results based on the GSE7803 dataset. (e) Identification of 229 overlapping genes between the DEGs of the two RA datasets. (f) Identification of 420 overlapping genes between the DEGs of the two CC datasets. (g) Identification of 29 overlapping genes between the DEGs of RA and CC. RA rheumatoid arthritis, CC cervical cancer, DEGs differentially expressed genes.
Co-expression modules in RA and CC
By conducting Weighted Gene Co-expression Network Analysis (WGCNA) on dataset GSE1919, we identified four gene modules associated with RA development compared to normal samples, each assigned a unique color. To ensure a biologically meaningful scale-free network, we selected the optimal soft-threshold power β for each dataset based on the criteria of scale independence (R2 > 0.85) and mean connectivity approaching zero. The chosen β values were 6, 4, 7, and 6 for GSE1919, GSE77298, GSE9750, and GSE7803, respectively (Fig. 2a,d,g,j). Among these modules, we found that the “magenta” module exhibited a strong positive correlation with RA in the GSE1919 dataset (r = 0.83, P = 3e−3; Fig. 2b,c). In the GSE77298 dataset, we detected six modules, with the “darkred” module showing a significant positive association with RA (r = 0.71, P = 1.4e-4; Fig. 2e,f). Similarly, in the GSE9750 dataset, the “blue” module exhibited a significant positive correlation with CC (r = 0.84, P = 6.8e−19; Fig. 2h,i), while in the GSE7803 dataset, the “lightgreen” module showed a significant positive correlation with CC (r = 0.71, P = 2.4e−6; Fig. 2k,l). Therefore, we considered these two RA-related modules as the key target modules, and the overlapping genes between them were identified as RA-related co-expressed genes (Fig. 2m). Similarly, we designated the two CC-associated modules as the focal modules, with their shared genes considered as CC-related co-expressed genes (Fig. 2n). Ultimately, the Venn diagram (Fig. 2o) illustrates 27 genes that are common between the RA- and CC-related co-expressed gene sets.

Identification of co-expression genes of RA and CC using WGCNA. (a) Analysis of network topology for various soft thresholds (β) in the GSE1919 dataset. The left panel shows the scale-free fit index (scale independence, y-axis) as a function of the soft threshold power (x-axis); the right panel displays the mean connectivity (degree, y-axis) as a function of the soft threshold power (x-axis). Figures (d,g,j) are drawn in the same way. (b) Cluster dendrogram of co-expressed genes in RA based on the GSE1919 dataset. (c) Heatmap of module–trait relationships in the GSE1919 dataset. (d) Analysis of network topology for various soft thresholds (β) in the GSE77298 dataset. (e) Cluster dendrogram of co-expressed genes in RA based on the GSE77298 dataset. (f) Heatmap of module–trait relationships in the GSE77298 dataset. (g) Analysis of network topology for various soft thresholds (β) in the GSE9750 dataset. (h) Cluster dendrogram of co-expressed genes in CC based on the GSE9750 dataset. (i) Heatmap of module–trait relationships in the GSE9750 dataset. (j) Analysis of network topology for various soft thresholds (β) in the GSE7803 dataset. (k) Cluster dendrogram of co-expressed genes in CC based on the GSE7803 dataset. (l) Heatmap of module–trait relationships in the GSE7803 dataset. (m) Identification of 568 overlapping genes between co-expression genes of the two RA datasets. (n) Identification of 578 overlapping genes between co-expression genes of the two CC datasets. (o) Identification of 27 overlapping genes between co-expression genes of RA and CC. RA rheumatoid arthritis, CC cervical cancer, WGCNA weighted gene co-expression network analysis.
Combination of DEGs and co-expressed genes from WGCNA
Genes associated with RA and CC were determined by integrating 29 DEGs and 27 co-expressed genes derived from WGCNA analysis. We then proceeded with 55 genes, among which CXCL1 was the only overlapping gene (Fig. 3a). CXCL1 was upregulated in both RA and CC (Fig. 3b–e).

Identification of key genes of RA and CC. (a) Combination of DEGs and co-expressed genes from WGCNA. (b) The differential expression of CXCL1 in the GSE1919 dataset. (c) The differential expression of CXCL1 in the GSE77298 dataset. (d) The differential expression of CXCL1 in the GSE9750 dataset. (e) The differential expression of CXCL1 in the GSE7803 dataset. RA rheumatoid arthritis, CC cervical cancer, DEGs differentially expressed genes, WGCNA weighted gene co-expression network analysis.
Gene Ontology and Kyoto Encyclopedia of genes and genomes analyses
To explore the molecular mechanisms of the 55 genes, we conducted Gene Ontology (GO) and Kyoto Encyclopedia of genes and genomes (KEGG) enrichment analyses. These analyses identified 246 GO terms, comprising 216 biological processes (BP), 22 molecular functions (MF), and 8 cellular components (CC). In terms of BP (Fig. 4a), the genes were predominantly associated with mitotic nuclear division (GO:0140014), negative regulation of sister chromatid segregation (GO:0033046 and GO:0033048), negative regulation of mitotic sister chromatid separation (GO:2000816), and regulation of T cell migration (GO:2000404). For MF (Fig. 4b), the genes were significantly enriched in fibroblast growth factor binding (GO:0017134), chemokine activity (GO:0008009), growth factor binding (GO:0019838), iron ion binding (GO:0005506), and chemokine receptor binding (GO:0042379). Regarding CC (Fig. 4c), the genes were mainly linked to the endocytic vesicle membrane (GO:0030666), endocytic vesicle (GO:0030139), outer kinetochore (GO:0000940), clathrin-coated endocytic vesicle membrane (GO:0030669), and spindle pole (GO:0000922). KEGG analysis revealed that these genes were involved in the epithelial cell signaling in Helicobacter pylori infection, the chemokine signaling pathway, and the cell cycle (Fig. 4d).

GO analysis and KEGG analysis of the 55 key genes. (a) Circle plot of BP analysis. (b) Circle plot of CC analysis. (c) Circle plot of MF analysis. (d) KEGG analysis. GO Gene Ontology, KEGG Kyoto Encyclopedia of Genes and Genomes, BP biological process, CC cellular component, MF molecular function.
Intersection analysis of protein–protein interaction network and univariate COX regression
To delve deeper into the molecular mechanisms, we utilized Cytoscape software along with the STRING database to construct a protein–protein interaction (PPI) network. The network visualized the interactions among 34 genes (Fig. 5a), and the bar charts showcased the top 12 genes ranked by node degree in Fig. 5b. Notably, the combined interaction score for each gene pair exceeded 0.9. Following this, we carried out a univariate COX regression analysis on the survival data of CC patients derived from The Cancer Genome Atlas (TCGA) datasets to identify key prognostic factors among the 55 pivotal genes (Fig. 5c). Subsequently, we conducted an intersection analysis between the pivotal nodes in the PPI network and the top 17 factors with P < 0.10 from the univariate COX regression. This analysis revealed four common genes—CXCL1, CXCL13, ZWINT and PTTG1—that were identified in both analyses (Fig. 5d). Consequently, these four genes were identified as core genes.

PPI network and univariate COX. (a) Interaction network constructed with the nodes with interaction confidence value > 0.9. (b) The top 12 genes ordered by the number of nodes. (c) Univariate COX regression analysis with the 55 key genes, listing the top significant factors with P value < 0.10. (d) Venn plot showing the common genes shared by leading nodes in PPI and top significant factors in univariate COX. PPI protein–protein interaction.
Receiver operating characteristic curve analysis of core genes
We constructed the receiver operating characteristic (ROC) curves for the core genes to assess their diagnostic utility. The findings indicated that these markers possessed strong diagnostic value for disease classification. Both in RA (Fig. 6a) and CC (Fig. 6b), the predictive performance was commendable, with all area under the curve (AUC) values exceeding 0.70.

ROC curves of the four common core genes in RA and CC. (a) ROC curves of CXCL1, CXCL13, ZWINT and PTTG1 in the RA datasets, respectively. (b) ROC curves of CXCL1, CXCL13, ZWINT and PTTG1 in the CC datasets, respectively. RA rheumatoid arthritis, CC cervical cancer, ROC receiver operating characteristic.
Prognostic model construction of core genes
Subsequently, we conducted a prognostic evaluation of CXCL1, CXCL13, ZWINT and PTTG1 in CC patients, and developed a predictive model based on these four genes utilizing LASSO Cox regression analysis. The risk score was calculated using the following formula:
The 95% confidence intervals (CI) for the coefficients of the four genes are as follows: CXCL1: 0.168 (95% CI 0.092 to 0.244), CXCL13: − 0.113 (95% CI − 0.198 to − 0.028), ZWINT: − 0.354 (95% CI − 0.441 to − 0.267) and PTTG1: − 0.310 (95% CI − 0.397 to − 0.223).
Patients were categorized into high- and low-risk groups based on their risk scores. Figure 7a displays the distribution of scores, survival status, and expression levels of CXCL1, CXCL13, ZWINT and PTTG1. An increase in the risk score was associated with a higher mortality risk and shorter overall survival (OS) for CC patients. Subsequently, we conducted survival analysis. The median OS was 213.6 months for the low-risk group and 53.9 months for the high-risk group. The high-risk group exhibited poorer OS compared to the low-risk group (P value = 0.0012, HR 2.24, Fig. 7b). Moreover, the AUCs for the 1-, 5-, and 10-year ROC curves were 0.66 (95% CI 0.53–0.79), 0.73 (95% CI 0.63–0.83), and 0.73 (95% CI 0.59–0.87), respectively (Fig. 7c), indicating moderate to high predictive accuracy.

Prognostic model construction. (a) Risk scores, survival time, and survival status in CC patients in the TCGA dataset. Top: scatterplot of risk scores from low to high; middle: scatterplot distribution of survival time and survival status corresponding to risk scores of different samples; bottom: heat map of gene expression in the prognostic model. (b) Kaplan–Meier curves for high-risk patients and low-risk patients. (c) ROC curves for 1, 5, and 10 years for this risk model. CC cervical cancer, TCGA The Cancer Genome Atlas, ROC receiver operating characteristic.
CIBERSORT analysis of immune cell correlations
Furthermore, we conducted an immune cell correlation analysis using CIBERSORT. The analysis revealed significant differences in the proportions of 10 out of 22 immune cell types in RA, such as regulatory T cells, resting NK cells, activated NK cells, macrophages M0, macrophages M1, resting dendritic cells, follicular helper T cells, plasma cells, activated mast cells and memory B cells (Fig. 8a). In CC, significant differences were observed in the proportions of 9 out of 22 immune cell types, such as memory B cells, CD8 T cells, resting CD4 memory T cells, resting NK cells, macrophages M0, macrophages M1, resting dendritic cells, activated dendritic cells and activated mast cells (Fig. 8b). A notable commonality between the two diseases was the increased proportion of macrophages M0 and M1 and the decreased proportion of resting dendritic cells. In RA, the core gene expression levels were predominantly linked to the proportions of plasma cells, regulatory T cells and resting dendritic cells (Fig. 8c). In CC, the core gene expression levels were associated with the proportions of resting CD4 memory T cells, macrophages M0, macrophages M1 and resting dendritic cells (Fig. 8d). We also found that in RA, CXCL1 correlated positively with plasma cells and negatively with Tregs and resting dendritic cells. In CC, CXCL1 showed similar correlations, with a positive link to plasma cells and negative links to resting dendritic cells and Tregs (though the latter was not statistically significant). Additionally, CXCL1 correlated positively with activated CD4 memory T cells and M0 and M1 macrophages in CC.

CIBERSORT analysis of immune cell correlations. (a) Comparison of 22 immune cells in samples with Con and RA. (b) Comparison of 22 immune cells in samples with Con and CC. (c) Spearman correlation analysis of the four core genes and 22 immune cells in RA. (d) Spearman correlation analysis of the four core genes and 22 immune cells in CC. RA rheumatoid arthritis, CC cervical cancer. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Association between the risk score and tumor immune microenvironment
Additionally, we calculated the StromalScore, ImmuneScore and ESTIMATEScore for CC patients in the TCGA dataset, categorizing them into high- and low-risk groups based on the ESTIMATE algorithm. Consistent with our earlier findings, the ImmuneScore was considerably lower in the high-risk group compared to the low-risk group (P value = 0.02, Fig. 9a). Moreover, the TumorPurity was higher in the high-risk group than the low-risk group, and the difference was statistically significant (P value = 0.04, Fig. 9b).

Immune microenvironment analysis in CC patients in the TCGA dataset with high and low risk scores. (a) Comparison of ESTIMATE Score, Stromal score and Immune score between high and low risk score groups. (b) Comparison of Tumor Purity between high and low risk score groups. CC cervical cancer, TCGA The Cancer Genome Atlas.
CXCL1 is the most important common gene
Using the Gene Expression Profiling Interactive Analysis (GEPIA), Genotype-Tissue Expression (GTEx) and TCGA datasets of CC, we verified the upregulation of core genes in CC patients, as shown in Fig. 10a. However, only the high expression of CXCL1 was associated with shorter OS in CC patients (P value = 0.033, HR 1.70, Fig. 10b). Since CXCL1 was identified as a significant gene when intersecting the genes of both diseases and has clear predictive value for the prognosis of CC patients compared to other genes, CXCL1 is considered the most important common gene in our study.

Analysis of core genes in the TCGA and GTEx datasets. (a) The expression differences of core genes between CC samples and Con samples in the TCGA and GTEx datasets. (b) Analysis of survival time differences between high and low expression groups of core genes in the TCGA dataset. TCGA The Cancer Genome Atlas, GTEx genotype-tissue expression, CC cervical cancer, Con control.
Biological processes associated with CXCL1 and CXCL1-related signaling pathways
After identifying CXCL1 as the paramount shared gene in CC, we explored the mechanisms behind its role utilizing the GeneMANIA database. We generated the PPI network for CXCL1 and pinpointed related proteins (Fig. 11a). Subsequently, we conducted GO and KEGG functional enrichment analyses on these genes, revealing that CXCL1 is predominantly linked to the chemokine signaling pathway (GO:0042379), leukocyte chemotaxis (GO:0008009), neutrophil chemotaxis (GO:0030674), myeloid leukocyte migration (GO:0097529), and inflammatory responses (GO:0006954) (Fig. 11b), as well as interactions between viral proteins and cytokines, the chemokine signaling pathway, and cytokine-cytokine receptor interactions (Fig. 11c).

Analysis of CXCL1-related signaling pathways. (a) PPI network for CXCL1 and identified proteins associated with CXCL1. (b) GO functional enrichment analysis of CXCL1 and associated genes. (c) KEGG functional enrichment analysis of CXCL1 and associated genes. PPI protein–protein interaction, GO Gene Ontology, KEGG Kyoto Encyclopedia of Genes and Genomes.
Pancancer analysis of CXCL1
To assess the broad significance of CXCL1, we examined its expression levels across various tumor types using the GEPIA platform (Fig. 12a). Our findings revealed that of 33 cancer types analyzed, 12 including CESC (CC) exhibited significantly elevated CXCL1 expression. Kaplan–Meier (K-M) survival analysis, comparing the high CXCL1 expression group (4750 patients) with the low CXCL1 expression group (4748 patients), demonstrated that elevated CXCL1 expression correlated with reduced OS in a pan-cancer context (P value < 0.0001, HR 1.80, Fig. 12b). Furthermore, we investigated the relationship between CXCL1 expression levels and OS in various types of cancer. The results showed that tumors with red borders indicate a positive correlation between CXCL1 expression and OS, while blue borders indicate a negative correlation (Fig. 12c).

Generalization value of CXCL1 across cancer. (a) Comparison of CXCL1 mRNA expression between cancer and paracancerous tissues across cancers. (b) The association between CXCL1 expression and OS in the pan-cancer cohort. (c) Associations between CXCL1 expression and OS across human cancers. OS overall survival, CESC cervical squamous cell carcinoma and endocervical adenocarcinoma.
Analysis of drug responsiveness regarding CXCL1
In the final phase of our research, we investigated the possible correlation between drug responsiveness and CXCL1 expression by leveraging the CellMiner™ database (https://discover.nci.nih.gov/cellminer/home.do), an online platform with genomic and pharmacological tools. Significantly, CXCL1 expression exhibited a predominantly inverse correlation with drug sensitivity, particularly showing a strong negative association (|cor| > 0.4, Spearman’s test) with the sensitivity to EMD-534085, TAK-960, Tamoxifen, Daporinad, and others (P value < 0.01, Fig. 13a). However, CXCL1 showed a positive correlation (cor > 0, Spearman’s test) with the sensitivity to two drugs, (−)-Nutlin-3 and TAS-6417, though the correlation was relatively weak (P value < 0.05, Fig. 13b).

Drug sensitivity analysis of CXCL1. (a) Negative associations. (b) Positive associations.
Discussion
RA, marked by synovial inflammation and joint erosion, is a global health issue31. Chronic inflammation is central in the progression of RA32. While RA itself is not directly life-threatening, the associated complications can significantly affect quality of life and mortality. CC is a common cancer, the third most frequent and a leading cause of mortality in women9. Although persistent HPV infection is identified as the primary risk factor of CC, the detailed process of cervical carcinogenesis remains not fully understood33. Recent studies have elucidated the connection between RA and CC through epidemiological studies27,28, MR analyses30,34 and comprehensive reviews23,24 with preliminary clinical and foundational research suggesting that RA patients have a higher risk of developing CC. However, the underlying molecular processes of this connection have not been extensively explored. To our knowledge, this is one of the first studies to employ a comprehensive bioinformatics approach to explore potential molecular mechanisms linking RA and CC.
In our research, we explored two gene expression datasets for RA and two for CC. These datasets were selected due to their frequent citations, recognition and representativeness. We performed differential gene expression analyses for each RA and CC dataset. The intersection yielded 29 common DEGs. From the WGCNA analysis, 27 co-expressed genes were obtained. Notably, we identified 55 key genes that are implicated in both RA and CC. In the GO analysis, these genes were enriched in processes such as cell cycle regulation, cell division and immune responses. They were also associated with cellular components like the nucleus and cytoplasm, as well as molecular functions like enzyme and signal molecule activity. These processes are essential for maintaining cellular homeostasis but may contribute to disease when dysregulated. Furthermore, the KEGG analysis emphasizes their involvement in pathways like the cell cycle and chemokine signaling. The cell cycle pathway is central to cancer development, as its dysregulation often leads to uncontrolled cell proliferation35. The chemokine signaling pathway is crucial for immune cell migration and inflammation, which contribute to chronic inflammatory diseases and tumor progression36. Additionally, enrichment in the pathway “epithelial cell signaling in Helicobacter pylori infection” suggests a role in host–pathogen interactions. Overall, these findings indicate that the 55 key genes may be involved in pathways critical for both normal cellular functions and disease processes, particularly inflammation and cancer.
Subsequently, a PPI network revealed 12 hub genes, and TCGA survival data identified 17 survival-associated genes, narrowing down to four core genes: CXCL1, CXCL13, ZWINT and PTTG1, all of which are upregulated in both diseases. The diagnostic significance of core genes was validated with ROC curves, and a prognostic model including these genes showed an evident correlation with poor prognosis of CC. Additionally, using TCGA and GTEx data, we found that within the CC patient group, four core genes are highly expressed, yet only CXCL1 shows a significant correlation with patient survival, with elevated levels suggesting a worse prognosis. These results suggest that these four genes, particularly CXCL1, may serve as novel biomarkers for RA and CC.
Chemokine (C-X-C motif) ligand 1 (CXCL1), a key chemokine, was first recognized for its autocrine stimulation of melanoma cell growth37. CXCL1, via CXCR2, recruits neutrophils to tumors, contributing to a pro-oncogenic TME38. Another crucial role of CXCL1 is the stimulation of angiogenesis39, influenced by CXCR2 expression on endothelial cells. CXCL1 may also impact cancer-associated fibroblasts40, leading to their senescence and transformation into cells that support tumorigenesis. CXCL1 has been observed with elevated expression in CC41. It enhances the proliferation and inhibits apoptosis of CC cells42,43. The effects of CXCL1/CXCR2 in CC cells were explored through in vitro experiments42,43. The influence of HPV oncoproteins such as E6 and E7 in carcinogenesis involves complex pathways that adjust chemokine expression15,44. The CXCL family is one of the principal chemokine families, and it is established that the abnormal expression of the CXCL family in cancers and inflammation can act as potential biomarkers, targets and indicators45. Additionally, CXCL1 contributed to chemoresistance and radioresistance46. Therefore, targeting CXCL1/CXCR2 could improve the efficacy of current anticancer therapies, especially for doxorubicin47, paclitaxel47 and oxaliplatin48. Our analysis of the PPI network, coupled with GO and KEGG enrichment studies, confirmed CXCL1’s direct connections to numerous chemokines and receptors, and its critical role in chemokine pathways, leukocyte chemotaxis, inflammation and cytokine action. Strikingly, CXCL1’s involvement in viral-cytokine interactions may shed light on the HPV-CC nexus. Additionally, CXCL1’s overexpression in various cancers often signals a graver prognosis. Chemokine ligand 13 (CXCL13), also known as B-lymphocyte chemoattractant, plays a significant role in various cellular functions, including migration, invasion, motility, proliferation and apoptosis49. While primarily chemotactic to B cells, CXCL13 also influences the migration of macrophages50. The CXCL13/CXCR5 axis is also involved in the recruitment of suppressive immune cells, which can enhance survival and invasion of tumors51. Zeste White 10-interacting kinetochore protein (ZWINT), a centromeric complex component, plays a role in cell growth and is linked to chromosomal instability in cancer52. Previously, ZWINT was linked to the emergence and progression of various malignant tumors53. Western blot analysis confirmed significantly elevated ZWINT expression in CC cells54. Pituitary tumor-transforming gene 1 (PTTG1) is involved in cell cycle regulation and cell proliferation55. Moreover, the elevated expression of PTTG1 is associated with the disruption of sister chromatid segregation and the interference with DNA repair mechanisms by causing chromosomal instability56,57.
Using the CIBERSORT algorithm, our findings suggest that the four core genes may be associated with an increase in macrophages M0 and M1. Macrophages, through pro-inflammatory cytokines and chemokines, create a persistent inflammatory environment that can promote tumorigenesis58. Within the macrophage classification, M0 denotes the resting state, while M1 represents the activated state with significant pro-inflammatory properties and M2 assumes an anti-inflammatory role59. M1 is closely linked to inflammatory responses and the initiation and progression of tumors, making them potential targets for anti-tumor therapy60. We also found that in RA and CC patients, CXCL1 shows similar correlations with immune cells, potentially promoting inflammatory responses in both conditions. Furthermore, analysis utilizing the ESTIMATE algorithm revealed that the high-risk group exhibited a decreased ImmuneScore and an increased TumorPurity, implying that tumors within this cohort may be classified as cold tumors, which are known to respond poorly to immunotherapy61. Notably, in CC patients, the suppressive TME, measured by the expression of associated genes, correlates with a higher risk and worse prognosis. These observations align with previous research on the four core genes, suggesting their potential role in the pathogenesis of RA and CC. However, further functional studies are needed to establish causality.
Our findings suggest a potential link between chronic inflammation, immune dysregulation, and chemokine-related pathways in RA patients, which may contribute to an increased susceptibility to CC. During the long-term management of RA, patients need to be vigilant about the risk of developing CC. However, there are no specific guidelines recommending tailored screening programs for RA patients62,63. To identify cervical abnormalities early and enhance the prognosis of CC, female RA patients should be considered for HPV vaccination64 and regularly screened for CC65. Addressing chronic inflammation in RA and CC treatments may mitigate CC risk in RA patients, especially those with HPV23.
Targeted therapy plays a crucial role in the treatment of advanced cervical cancer, with an increasing number of therapeutic targets being identified66. Our research indicates that CXCL1 is a promising therapeutic target in CC, representing our first discovery in this area. However, CXCL1 correlates inversely with the responsiveness to a range of drugs and positively only with (−)-Nutlin-3 and TAS6417 temporarily. This indicates that effective drugs targeting CXCL1 require further exploration.
In conclusion, this research suggests that 55 pivotal genes potentially play a role in the mechanisms underlying the co-occurrence of RA and CC. Four diagnostic markers not only modulate immune cells but are also potential therapeutic targets for RA with comorbid CC. CXCL1 and chemokine-related pathways may play a significant role in RA and CC, warranting further investigation to elucidate their precise mechanisms and therapeutic potential.
However, the study has some limitations. Initially, we do not possess case data from individuals who suffer from both RA and CC, which limits validation capabilities. More critically, since all data originate from publicly accessible databases, our findings require further validation from future experimental and clinical research. Additionally, a major limitation of this study arises from potential confounding factors related to patient heterogeneity. Gene expression profiles in RA and CC patients may be influenced by clinical variables such as disease severity, comorbidities and therapeutic interventions. To address this, future investigations should prioritize stratified analyses to clarify the effects of distinct health states on transcriptional signatures. Moreover, further exploration of the molecular pathways and their significance is still needed, and we will continue to work in this area.
Methods
Data download and processing
We retrieved two gene expression profiling datasets for RA (GSE1919 and GSE77298) and two for CC (GSE9750 and GSE7803) from the GEO repository (https://www.ncbi.nlm.nih.gov/geo/). Detailed dataset information is accessible through the GEO database link. We also extracted gene mutation, clinical, and transcriptome data for CC patients from the TCGA repository (https://portal.gdc.cancer.gov/). We performed background correction and normalization on all raw datasets, converting probe identifiers to their corresponding gene symbols for further examination. All data processing and analytical work were carried out in RStudio (version 2024.09.0) (https://www.rstudio.com/). The GSE1919 dataset (GPL91 platform) encompasses transcriptional profiles of synovial tissues from 5 RA patients and 5 healthy controls. The RA dataset GSE77298 (GPL570 platform) includes 23 synovial fluid samples derived from 16 RA patients and 7 healthy controls. The GSE9750 dataset (GPL96 platform) is composed of 42 tumor samples and 24 samples of normal cervical epithelium. The GSE7803 dataset (GPL96 platform) contains 10 normal cervical epithelium samples and 24 invasive squamous cell carcinomas of the cervix samples. We categorized the samples into RA groups, CC groups, and normal control (Con) groups based on their origins. A streamlined process of our study is illustrated in Fig. 14.

The simplified research workflow.
Analysis of DEGs
We employed the limma package (version 3.60.6) in R for normalizing and correcting all microarray-based gene expression profiling data and for gene name annotation. To confirm accuracy and counteract batch effects, the two RA gene expression datasets (GSE1919 and GSE77298), which included 12 healthy control samples and 21 RA samples, were analyzed separately to identify DEGs. The overlapping genes from these datasets were considered as the DEGs for RA. The same preprocessing was applied to the two CC gene expression datasets (GSE9750 and GSE7803). A threshold of |log2FC| > 1.0 and P value < 0.05 was used to detect DEGs in RA or CC patients. The threshold was chosen to balance sensitivity and specificity, allowing for the identification of biologically significant genes while minimizing false positives. The common DEGs between RA and CC were identified by intersecting the DEGs from both diseases.
Construction of co-expressed gene modules
Based on the DEGs identified in RA and CC, we proceeded to conduct WGCNA to uncover genes that are co-expressed in the transcriptome and common to both diseases. Utilizing the WGCNA package (version 1.73) in R, we established unsigned co-expression networks. The process commenced with hierarchical clustering analysis of the samples through the flashClust program in R to identify and exclude outliers. A “soft” thresholding power (β) determined by the “pickSoftThreshold” algorithm in WGCNA was applied to create a scale-free network in accordance with the scale-free topology criterion. A dynamic tree-cutting method was then used to generate a topological overlap matrix (TOM) based on the adjacency matrix to detect gene modules. We calculated gene significance (GS) and module membership (MM) to link modules with clinical features. Finally, we constructed the eigengene network. Two RA gene expression profiling datasets and two CC gene expression profiling datasets were analyzed respectively. Taking into account the number of genes and the degree of correlation, we finally selected the target gene modules. The intersection genes obtained from the two RA datasets were taken as the co-expressed genes of RA. For the CC datasets, the same analytical method was applied. The intersection of co-expressed genes between RA and CC was determined to identify common co-expressed genes for both diseases.
Finally, the genes associated with RA and CC were identified by combining DEGs and co-expressed genes from WGCNA.
Functional enrichment analysis of core genes
The main goals of this study were to discover the mechanisms behind the co-occurrence of RA and CC and to elucidate the molecular mechanisms behind the key disease genes. We utilized GO (https://geneontology.org/) and KEGG67,68,69 (https://www.kegg.jp/) pathway enrichment analyses to pinpoint distinctive biological and functional properties. Carried out with the clusterProfiler package (version 4.12.6) in R, significance was set at a P value of < 0.05.
PPI network construction and univariate COX regression
In our study, the STRING database (https://string-db.org/) was utilized to construct a PPI network for key genes in RA with comorbid CC. For building the network, the settings were as follows: organism, Homo sapiens, combined score threshold, 0.4. The “ClueGO” and “MCODE” plugins in Cytoscape (version 3.10.1) (https://cytoscape.org/) were used to build PPI networks, identifying protein relationships that may be crucial for screening core genes with significant roles. Additionally, we performed univariate COX regression analysis for CC using survival data from TCGA, selecting genes with P values < 0.10 as significant. Genes screened in PPI networks were intersected with those identified from univariate COX regression to determine the core genes in RA with comorbid CC.
Prognostic modeling
The sensitivity and accuracy of the core genes were validated using ROC curves with the pROC package (version 1.18.5). The SVA package (version 3.54.0) was applied to correct for batch effects. Core genes linked to CC prognosis were determined, and a risk score was formulated for each participant based on the regression coefficients of the genes within the signature and their respective expression levels. The formula for the risk score is presented below:
The 95% CI for the coefficients were estimated using the bootstrap method. Patients were divided into high-risk and low-risk cohorts based on the median risk score, followed by K-M analysis for OS and subsequent log-rank testing. In this process, we utilized the ggrisk package (version 1.3) and the survminer package (version 0.5.0). The predictive accuracy of this prognostic model was assessed using time-dependent ROC analysis. The time-dependent ROC curve was plotted using the survivalROC function from the survivalROC package (version 1.0.3.1).
Assessment of the immune landscape
To investigate the immune environment of RA and CC, CIBERSORT and ESTIMATE algorithms were implemented using the CIBERSORT package (version 0.1.0) and the estimate package (version 1.0.13), respectively. CIBERSORT, a deconvolution algorithm, was used to calculate the proportions of 22 immune cell types in each RA or CC patient and normal control, with the total fractions summed for each sample. The reference gene set for the CIBERSORT algorithm is LM22 (22 immune cell types and 547 genes), using raw expression values that are internally linearly normalized. The SVA package (version 3.54.0) was applied to correct for batch effects in this process. ESTIMATE, a method for determining the fractions of stromal and immune cells based on gene expression signatures in tumor samples, was applied to evaluate TME of each CC patient in the TCGA database, including StromalScore, ImmuneScore, EstimateScore, and TumorPurity. The reference gene set for the ESTIMATE algorithm is the immune/stromal gene set (141 genes each), with data being pre-normalized (TPM). We compared StromalScore, ImmuneScore, EstimateScore and TumorPurity across high-risk and low-risk cohorts.
Further analysis of CXCL1
Since the commonality between DEGs and co-expressed genes from WGCNA analysis yielded only one gene, CXCL1, which is among the core genes and significantly associated with OS in CC after validation in the GEPIA2 (http://gepia2.cancer-pku.cn/), we proceeded to conduct further analysis on CXCL1.
Biological mechanisms linked to CXCL1 and pathways related to CXCL1 signaling
After pinpointing CXCL1 as the paramount gene in CC, we delved into the mechanisms underlying CXCL1’s role utilizing the GeneMANIA database (http://genemania.org/), an interactive online tool designed for identifying proteins correlated with particular genes and gene ensembles. We mapped the PPI network for CXCL1 and uncovered associated proteins. Following this, we conducted GO and KEGG67,68,69 enrichment analyses on the identified genes.
Pancancer analysis of CXCL1
The pancancer expression and survival analysis of CXCL1 was conducted using the GEPIA. We utilized TISIDB to analyze the relationships between CXCL1 expression and OS in pancancer, generating K-M curves using TCGA cohort data.
Statistical analysis
All analytical procedures and graphical representations were carried out using R software (version 4.4.1) (https://cran.r-project.org/). Data organization was primarily carried out using the tidyverse package (version 2.0.0), and result visualization was partly accomplished with the ggplot2 package (version 3.5.1). Unless otherwise specified, the threshold for significance was established at α = 0.05, with a P value less than 0.05 deemed statistically significant.
Data availability
This study utilized publicly available datasets accessible from the following sources: the GEO database (https://www.ncbi.nlm.nih.gov/geo/) and the TCGA database (https://portal.gdc.cancer.gov/).
References
Liu, L. et al. Secreted protein acidic and rich in cysteine mediated biomimetic delivery of methotrexate by albumin-based nanomedicines for rheumatoid arthritis therapy. ACS Nano 13, 5036–5048 (2019).
Deane, K. D. & Holers, V. M. Rheumatoid arthritis pathogenesis, prediction, and prevention: An emerging paradigm shift. Arthritis Rheumatol. 73, 181–193 (2021).
van Oosterhout, M. et al. Differences in synovial tissue infiltrates between anti-cyclic citrullinated peptide-positive rheumatoid arthritis and anti-cyclic citrullinated peptide-negative rheumatoid arthritis. Arthritis Rheum. 58, 53–60 (2008).
MacDonald, I. J. et al. Implications of angiogenesis involvement in arthritis. Int. J. Mol. Sci. 19, 1 (2018).
Simon, T. A., Thompson, A., Gandhi, K. K., Hochberg, M. C. & Suissa, S. Incidence of malignancy in adult patients with rheumatoid arthritis: A meta-analysis. Arthritis Res. Ther. 17, 212 (2015).
Wu, X. et al. Rheumatoid arthritis and risk of lung cancer: Meta-analysis and Mendelian randomization study. Semin. Arthritis Rheum. 51, 565–575 (2021).
Luo, X. et al. The risk of leukemia in patients with rheumatoid arthritis: A systematic review and meta-analysis. Clin. Rheumatol. 40, 1283–1289 (2021).
Kim, S. C. et al. Risk of high-grade cervical dysplasia and cervical cancer in women with systemic inflammatory diseases: A population-based cohort study. Ann. Rheum. Dis. 74, 1360–1367 (2015).
Yu, X., Li, Z., Bai, R. & Tang, F. Transcriptional factor 3 binds to Sirtuin 1 to activate the Wnt/β-catenin signaling in cervical cancer. Bioengineered 13, 12516–12531 (2022).
Kusakabe, M., Taguchi, A., Sone, K., Mori, M. & Osuga, Y. Carcinogenesis and management of Human Papillomavirus-associated cervical cancer. Int. J. Clin. Oncol. 28, 965–974 (2023).
Pal, A. & Kundu, R. Human Papillomavirus E6 and E7: The cervical cancer hallmarks and targets for therapy. Front. Microbiol. 10, 3116 (2019).
Dust, K. et al. Human Papillomavirus 16 E6 and E7 oncoproteins alter the abundance of proteins associated with DNA damage response, immune signaling and epidermal differentiation. Viruses 14, 1 (2022).
Tomaić, V. Functional roles of E6 and E7 oncoproteins in HPV-induced malignancies at diverse anatomical sites. Cancers 8, 1 (2016).
Iuliano, M. et al. Human Papillomavirus E6 and E7 oncoproteins affect the cell microenvironment by classical secretion and extracellular vesicles delivery of inflammatory mediators. Cytokine 106, 182–189 (2018).
Castro-Amaya, A. M. et al. E6/E7 from Beta-2-HPVs 122, 38b, and 107 possess transforming properties in a fibroblast model in vitro. Exp. Cell Res. 414, 113088 (2022).
Oyouni, A. A. A. Human Papillomavirus in cancer: Infection, disease transmission, and progress in vaccines. J. Infect. Public Health 16, 626–631 (2023).
Hancock, G., Hellner, K. & Dorrell, L. Therapeutic HPV vaccines. Best Pract. Res. Clin. Obstet. Gynaecol. 47, 59–72 (2018).
Yu, S., Li, X., Zhang, J. & Wu, S. Development of a novel immune infiltration-based gene signature to predict prognosis and immunotherapy response of patients with cervical cancer. Front. Immunol. 12, 709493 (2021).
Adiga, D., Eswaran, S., Pandey, D., Sharan, K. & Kabekkodu, S. P. Molecular landscape of recurrent cervical cancer. Crit. Rev. Oncol. Hematol. 157, 103178 (2021).
De Cock, D. & Hyrich, K. Malignancy and rheumatoid arthritis: Epidemiology, risk factors and management. Best Pract. Res. Clin Rheumatol. 32, 869–886 (2018).
Fernandes, J. V. et al. Link between chronic inflammation and Human Papillomavirus-induced carcinogenesis (Review). Oncol. Lett. 9, 1015–1026 (2015).
Zhao, H. et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther. 6, 263 (2021).
Gusakov, K., Kalinkovich, A., Ashkenazi, S. & Livshits, G. Nature of the association between rheumatoid arthritis and cervical cancer and its potential therapeutic implications. Nutrients 16, 1 (2024).
Morand, S. et al. Molecular mechanisms underlying rheumatoid arthritis and cancer development and treatment. Future Oncol. 16, 483–495 (2020).
Belal, A. et al. Therapeutic potential of Zeolites/Vitamin B12 nanocomposite on complete Freund’s adjuvant-induced arthritis as a bone disorder: In vivo study and bio-molecular investigations. Pharmaceuticals 16, 285 (2023).
Moscicki, A. B. et al. Guidelines for cervical cancer screening in immunosuppressed women without HIV infection. J Low Genit Tract Dis. 23, 87–101 (2019).
Yang, T. O., Floud, S. & Reeves, G. K. Rheumatoid arthritis and cancer risk in the million women study. Int. J. Epidemiol. 53, 1 (2024).
Beydon, M. et al. Risk of cancer for patients with rheumatoid arthritis versus general population: A national claims database cohort study. Lancet Reg. Health Eur. 35, 1 (2023).
Foster, E. et al. Increased risk of cervical dysplasia in females with autoimmune conditions-Results from an Australia database linkage study. PLoS ONE 15, e234813 (2020).
Xu, M. et al. Exploring the causal association between rheumatoid arthritis and the risk of cervical cancer: A two-sample Mendelian randomization study. Arthritis Res. Ther. 26, 35 (2024).
Smolen, J. S., Aletaha, D. & McInnes, I. B. Rheumatoid arthritis. Lancet 388, 2023–2038 (2016).
Weyand, C. M. & Goronzy, J. J. The immunology of rheumatoid arthritis. Nat. Immunol. 22, 10–18 (2021).
Sun, Q. et al. A novel miRNA identified in GRSF1 complex drives the metastasis via the PIK3R3/AKT/NF-κB and TIMP3/MMP9 pathways in cervical cancer cells. Cell Death Dis. 10, 636 (2019).
Yuan, S. et al. Rheumatoid arthritis and risk of site-specific cancers: Mendelian randomization study in European and East Asian populations. Arthritis Res. Ther. 24, 270 (2022).
Malumbres, M. & Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 9, 153–166 (2009).
Raman, D., Sobolik-Delmaire, T. & Richmond, A. Chemokines in health and disease. Exp. Cell Res. 317, 575–589 (2011).
Richmond, A., Lawson, D. H., Nixon, D. W. & Chawla, R. K. Characterization of autostimulatory and transforming growth factors from human melanoma cells. Cancer Res. 45, 6390–6394 (1985).
Bellomo, G. et al. Chemotherapy-induced infiltration of neutrophils promotes pancreatic cancer metastasis via Gas6/AXL signalling axis. Gut 71, 2284–2299 (2022).
Wu, Z., Neufeld, H., Torlakovic, E. & Xiao, W. Uev1A-Ubc13 promotes colorectal cancer metastasis through regulating CXCL1 expression via NF-кB activation. Oncotarget 9, 15952–15967 (2018).
Cai, L. et al. Adiponectin induces CXCL1 secretion from cancer cells and promotes tumor angiogenesis by inducing stromal fibroblast senescence. Mol. Carcinog. 55, 1796–1806 (2016).
Kong, W. et al. Analysis of therapeutic targets and prognostic biomarkers of CXC chemokines in cervical cancer microenvironment. Cancer Cell Int. 21, 399 (2021).
Man, X. et al. High expression level of CXCL1/GROα is linked to advanced stage and worse survival in uterine cervical cancer and facilitates tumor cell malignant processes. BMC Cancer 22, 712 (2022).
Sun, J. & Yuan, J. Chemokine (C-X-C motif) ligand 1/chemokine (C-X-C motif) receptor 2 autocrine loop contributes to cellular proliferation, migration and apoptosis in cervical cancer. Bioengineered 13, 7579–7591 (2022).
Israr, M., DeVoti, J. A., Papayannakos, C. J. & Bonagura, V. R. Role of chemokines in HPV-induced cancers. Semin. Cancer Biol. 87, 170–183 (2022).
Zhou, C., Gao, Y., Ding, P., Wu, T. & Ji, G. The role of CXCL family members in different diseases. Cell Death Discov. 9, 212 (2023).
Xu, H. et al. CXCR2 promotes breast cancer metastasis and chemoresistance via suppression of AKT1 and activation of COX2. Cancer Lett. 412, 69–80 (2018).
Sharma, B., Varney, M. L., Saxena, S., Wu, L. & Singh, R. K. Induction of CXCR2 ligands, stem cell-like phenotype, and metastasis in chemotherapy-resistant breast cancer cells. Cancer Lett. 372, 192–200 (2016).
de Porras, V. R. et al. Curcumin mediates oxaliplatin-acquired resistance reversion in colorectal cancer cell lines through modulation of CXC-Chemokine/NF-κB signalling pathway. Sci. Rep. 6, 24675 (2016).
Hussain, M. et al. CXCL13/CXCR5 signaling axis in cancer. Life Sci. 227, 175–186 (2019).
Gunn, M. D. et al. A B-cell-homing chemokine made in lymphoid follicles activates Burkitt’s lymphoma receptor-1. Nature 391, 799–803 (1998).
Baniyash, M. Myeloid-derived suppressor cells as intruders and targets: Clinical implications in cancer therapy. Cancer Immunol. Immunother. 65, 857–867 (2016).
Peng, F. et al. ZWINT is the next potential target for lung cancer therapy. J. Cancer Res. Clin. Oncol. 145, 661–673 (2019).
Famulski, J. K., Vos, L., Sun, X. & Chan, G. Stable hZW10 kinetochore residency, mediated by hZwint-1 interaction, is essential for the mitotic checkpoint. J. Cell Biol. 180, 507–520 (2008).
Ma, Z., Cai, Y. & Tian, C. ZWINT promotes the proliferation, migration, and invasion of cervical cancer cells by regulating the p53/p21 signaling pathway. Chin. J. Physiol. 66, 372–378 (2023).
Wang, L. & Liu, X. Multi-omics analysis of the oncogenic value of pituitary tumor-transforming gene 1 (PTTG1) in human cancers. Front. Biosci. 29, 87 (2024).
Lai, Y., Xin, D., Bai, J., Mao, Z. & Na, Y. The important anti-apoptotic role and its regulation mechanism of PTTG1 in UV-induced apoptosis. J. Biochem. Mol. Biol. 40, 966–972 (2007).
Read, M. L. et al. Elevated PTTG and PBF predicts poor patient outcome and modulates DNA damage response genes in thyroid cancer. Oncogene 36, 5296–5308 (2017).
Livshits, G. & Kalinkovich, A. Hierarchical, imbalanced pro-inflammatory cytokine networks govern the pathogenesis of chronic arthropathies. Osteoarthr. Cartil. 26, 7–17 (2018).
Sedighzadeh, S. S., Khoshbin, A. P., Razi, S., Keshavarz-Fathi, M. & Rezaei, N. A narrative review of tumor-associated macrophages in lung cancer: Regulation of macrophage polarization and therapeutic implications. Transl. Lung Cancer Res. 10, 1889–1916 (2021).
Sica, A. & Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 122, 787–795 (2012).
Kao, K. C., Vilbois, S., Tsai, C. H. & Ho, P. C. Metabolic communication in the tumour-immune microenvironment. Nat. Cell Biol. 24, 1574–1583 (2022).
Auguste, P. Cervical cancer screening: Updated guidelines from the American Cancer Society. Am. Fam. Phys. 104, 314–315 (2021).
Fraenkel, L. et al. 2021 American College of Rheumatology Guideline for the treatment of rheumatoid arthritis. Arthritis Care Res. (Hoboken) 73, 924–939 (2021).
Kim, S. C., Feldman, S. & Moscicki, A. B. Risk of human papillomavirus infection in women with rheumatic disease: Cervical cancer screening and prevention. Rheumatology (Oxford) 57, v26–v33 (2018).
Voelker, R. A. Cervical cancer screening. JAMA 330, 2030 (2023).
Watkins, D. E. et al. Advances in targeted therapy for the treatment of cervical cancer. J. Clin. Med. 12, 5992 (2023).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: Biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677 (2025).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Acknowledgements
The authors extend special thanks to all researchers involved in this study. In particular, we express our gratitude to Wei Luo from the Sun Yat-sen University Cancer Center for his support in writing the article.
Author information
Authors and Affiliations
Contributions
S-SQ was responsible for data collection and sorting, computational modeling, and analysis and wrote the research paper. Y-WY provided the design and general guidance of the research framework. YB and L-HX provided research guidance for writing the research paper and assisted with the revision of the article. DG participated in data analysis. CS assisted with the collection of reference literature. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Su, S., Yang, B., Liu, H. et al. Four genes shared between rheumatoid arthritis and cervical cancer are associated with cervical cancer prognosis. Sci Rep 15, 15884 (2025). https://doi.org/10.1038/s41598-025-01177-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-01177-x
