
Abstract
The Nipah virus (NiV), a lethal pathogen from the Paramyxoviridae family, presents a significant global health threat as a result of its high mortality rate and inter-human transmission. This investigation employed in silico methods that were assisted by machine learning to identify small-molecule inhibitors that target the NiV glycoprotein, a critical component of viral entry. Out of the 754 antiviral compounds that were screened using Lipinski’s Rule of Five and DeepPurpose, 333 are identified. Five best hits were identified through molecular docking, each of which exhibited superior binding scores in comparison to the control. This was further refined to three compounds through density functional theory (DFT) analysis, with compound 138,567,123 exhibiting the highest electronic stability (DFT energy: -1976.74 Hartree; HOMO-LUMO gap: 0.83 eV). Its stability was verified by molecular dynamics (MD) simulations, which demonstrated consistent hydrogen bonding and minimal RMSD. Additionally, it possessed the highest docking score (-9.7 kcal/mol) and binding free energy (-24.04 kcal/mol, MM/GBSA). The results underscore ligand 138,567,123 as a promising antiviral candidate for NiV and illustrate the efficacy of machine learning-based in silico drug discovery.
Similar content being viewed by others
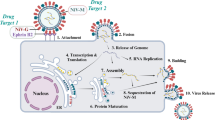

Introduction
Nipah virus (NiV), a member of the Paramyxoviridae family and genus Henipavirus, is one of the most notorious viruses due to its high fatality rate, which ranges from 40–75%1. Paramyxovirus is an enveloped, negative-sense single-stranded RNA virus, classified under the broader group of paramyxoviruses2. These viruses are generally highly infectious, with a host range that includes mammals, fish, birds, and reptiles3,4,5. The reservoir host of NiV is fruit bats of the genus Pteropus, which play a key role in its transmission6. NiV was first identified during an outbreak in Malaysia in 1998, NiV has since caused sporadic outbreaks, particularly in South and Southeast Asia, with devastating impacts on public health and local economies7,8,9. Its ability for human-to-human transmission has drawn significant attention due to its potential to cause severe outbreaks. Infected individuals often experience fever, neurological symptoms like altered sensorium, agitation, and seizures, respiratory issues like laboured breathing and cough, and other symptoms like trembling, muscle spasms, and frothy salivation10,11,12.
A prior work used in silico approaches such as molecular docking, MM-GBSA, ADMET analysis, and MD simulations to identify neem-derived phytochemicals as possible Nipah virus attachment glycoprotein inhibitors13. Quercetin, Cianidanol, and Kaempferol showed high binding affinities and excellent pharmacokinetic characteristics, exceeding the control medication ribavirin. The nanoscale architecture of the Nipah virus fusion protein (NiV-F) on membranes was investigated previously using single-molecule localization microscopy. It was shown that NiV-F generates unique nanoclusters that improve membrane fusion triggering, which are governed by protein interactions and particular mutations, providing new insights into NiV’s fusion activation process14. The previous work examined over 1,500 quinolone-based derivatives for potential inhibitors of both the Nipah virus glycoprotein (NiV-G) and human ephrin-B2. Using docking, ADMET analysis, MD simulations, and MM-GBSA, three top candidate compounds with substantial binding affinities and favorable pharmacokinetic characteristics were discovered as prospective NiV treatment leads15. A previous study computationally evaluated a database of marine natural products to find potential inhibitors of the Nipah virus RNA-dependent RNA polymerase. Deep learning-based docking, MD simulations, and MM/GBSA analysis were used to identify five interesting compounds with stable binding at the RdRp active site, providing possible leads for future antiviral medication development16. Another study used a proteome-wide approach to find possible therapies for all nine Nipah virus proteins. The researchers proposed 4 peptide and 146 small molecule inhibitors targeting multiple viral proteins (G, F, M, N, and P) using homology modeling, peptide design, docking, and MD simulations, significantly expanding the structural data on NiV and providing diverse candidates for antiviral development17. Another prior study employed deep mutational scanning to determine how all potential amino acid changes in the Nipah virus receptor-binding protein (RBP) affect function, receptor binding, and antibody escape18. It discovered crucial conserved areas required for RBP function and mapped escape mutations, offering important insights for vaccine and antibody therapy development.
Treatments against NiV infections have primarily focused on repurposing broad-spectrum antiviral agents and developing monoclonal antibodies. Though Ribavirin is not a proven treatment for NiV, it is particularly being used in a state of emergency to treat acute NiV encephalitis as a first-line treatment strategy. Remdesivir, a broad-spectrum antiviral targeting RNA polymerase, has demonstrated efficacy in animal models and has been considered for emergency use9,19. Favipiravir and Galidesivir, both RNA-dependent RNA polymerase inhibitors, are also being investigated for their potential against NiV20,21. Monoclonal antibodies, such as m102.4, have showed promise in preclinical research and compassionate-use situations, but challenges relating to manufacture, transport, and accessibility have limited their widespread usage22. These limitations highlight the critical need to explore alternative approaches for NiV therapeutics.
Viral entry into the host begins with relatively nonspecific interactions between the virus and attachment factors on the cell surface. The Nipah virus employs two key glycoproteins, the attachment glycoprotein (G) and the fusion glycoprotein (F), for its entry into host cells23,24. The G protein plays an essential role in binding the cellular receptors, such as ephrin-B2 and ephrin-B3, present on endothelial and neuronal cells, facilitating viral attachment and subsequent entry25,26. Given its important involvement in the viral lifecycle, the G protein is a promising target for the development of antiviral therapies27. There are several studies which uses glycoprotein as targets. It was identified that antibodies targeting glycoproteins have disrupted the interaction between the viral G protein and its host receptors and holds as a promise in treating and preventing infections in animal models28,29,30.
Recent advances in computational methodologies have transformed the landscape of drug discovery. In silico approaches enable the identification and optimization of lead compounds from vast chemical libraries, significantly reducing the time and cost associated with traditional experimental workflows31. Small molecule therapeutics play a pivotal role in drug development due to their size, which allows them to interact with biological targets easily32. In-silico techniques have been used to design small-molecule inhibitors that target the G protein with high specificity and binding affinity. These computational tools provide valuable insights into the molecular interactions between the glycoprotein and potential inhibitors, enabling rational design and optimization of drug candidates33,34. Molecular docking studies have been used to identify compounds that fit within the receptor-binding domain of the G protein, while molecular dynamics simulations have provided a detailed understanding of the stability and binding kinetics of these interactions35,36.
This study focuses on the in-silico discovery and optimization of small-molecule inhibitors targeting the Nipah virus glycoprotein. By using computational methods such as docking, molecular dynamics simulations including machine learning techniques such as DeepPurpose, this study aim to identify promising candidates that can serve as the basis for therapeutic development against NiV. Previous study identified the ligands G1-G5 as Nipah virus glycoprotein inhibitors37. These inhibitors were screened and used as control for this present study.
Materials and methods
Protein retrieval and compound library
The crystal structure of the Nipah virus glycoprotein (PDB ID: 2 VSM)38 was obtained from the Protein Data Bank39. The Computed Atlas of Surface Topography of Proteins (CASTp)40 was employed to discover prospective binding sites on the protein surface. The important binding residues revealed from CASTp analysis were subsequently employed to delineate the grid box for molecular docking. The selection of control ligands (G1–G5) was based on previously reported inhibitors of the Nipah virus glycoprotein37. These known binders served as benchmark molecules to validate the docking protocol and assess the relative binding affinity of newly screened compounds. The top-performing control ligand, based on docking score and binding pose quality, was used as a reference for comparing the effectiveness of the candidate molecules identified in this study. These ligands were incorporated to evaluate the binding efficacy of the screened compounds and to function as reference standards during the docking and simulation processes. A collection of 754 antiviral compounds was obtained from the Selleckchem antiviral chemical database (https://www.selleckchem.com/screening/antiviral-compound-library.html) for virtual screening.
Docking (Control)
AutoDock 4.2 was employed to prepare the protein. This involved the removal of crystallographic water molecules, the repair of missing atoms, the addition of polar hydrogens, and the assignment of Kollman charges. The PDBQT format was used to save the constructed structure. The grid box dimensions were defined based on the active site residues identified using the CASTp server, ensuring full coverage of the predicted binding pocket on the Nipah virus glycoprotein. The grid box was centered at coordinates (x = 20.58, y = 69.50, z = −27.51) with dimensions 34 Å × 38 Å × 28 Å and a spacing of 1 Å. These dimensions were selected to encompass not only the core binding site but also surrounding residues that may contribute to ligand interactions, allowing for accurate and comprehensive docking results. The ligands, which included the controls (G1–G5), were prepared and converted into the appropriate format using OpenBabel41. The following parameters were employed to conduct docking simulations: exhaustiveness set to 100, an energy range of 4 kcal/mol, and 20 modes to guarantee comprehensive conformational sampling. 20 configurations were generated for each compound, and the pose with the lowest binding energy was chosen for downstream analysis. The control ligand with the maximum binding affinity was selected as the final reference for comparison with the top hits from the screened antiviral library.
DeepPurpose
Initially, the 754 antiviral compounds were filtered to eliminate null values and duplicates. Lipinski’s Rule of Five (RoF) was implemented to evaluate the remaining compounds’ drug-likeness, with an emphasis on oral bioavailability. According to this rule, a compound is considered more likely to be orally active if it possesses a molecular weight of ≤ 500 Dalton, a LogP (lipophilicity) value of ≤ 5, no more than 5 hydrogen bond donors (-OH and -NH groups), and no more than 10 hydrogen bond acceptors (oxygen and nitrogen atoms). Those compounds which followed the Lipinski’s RoF, was selected and the dataset was saved for further analysis.
The filtered compounds were subsequently assessed using DeepPurpose42, a deep learning-based framework that is specifically designed for the prediction of drug-target interactions (DTIs). This was done to improve the predictive potential of the virtual screening. Traditional scoring functions may neglect the complex nonlinear relationships between molecular structures and protein sequences, which is why DeepPurpose is recommended. DeepPurpose forecasts the interaction probability score for each compound–protein pair by utilizing the amino acid sequence of the Nipah virus glycoprotein and the SMILES strings of the compounds as input. The early-phase drug discovery process is considerably streamlined by this AI-driven approach, which enables the rapid prioritization of compounds with a high likelihood of binding to the target. The compounds with the highest predicted interaction scores, which suggest a greater potential to bind effectively to the target, were selected for subsequent molecular docking and downstream analyses. Pandas was employed to perform data preprocessing and manipulation, and the predictive model was constructed and applied using essential DeepPurpose modules, including utils, dataset, and DTI. The protein sequence was incorporated into a new column designated “Target” in the dataset to guarantee that each compound was assessed against the same target. This integrated screening approach offers a reliable method for identifying high-potential drug candidates prior to the implementation of structure-based validation procedures.
Molecular docking
The SMILES of the top selected compounds were converted to PubChem CIDs using the “PubChem Identifier Exchange Service” (https://pubchem.ncbi.nlm.nih.gov/idexchange/). Finally, the unique CIDs were retrieved after removing duplicates. Further, the CIDs that had 3D-SDF structures were downloaded using the PubChem API, while the remaining 2D-SDF structures were retrieved and then converted to 3D-SDF using Open-babel. These compounds were prepared and docked using the same method as mentioned in the section “Docking(control)”. The top six compounds which showed higher binding energy compared to control was selected for Density functional Theory (DFT) analysis.
Density functional theory (DFT)
Density Functional Theory (DFT) is a widely used ab initio method for the investigation of atoms, molecules, crystals, surfaces, and their interactions43. It is a potent method for the optimization of existing molecules and the discovery of new drugs, and it is essential in quantum chemistry44. The electronic properties of the top-ranked ligands were assessed using a Python-based methodology to conduct DFT calculations in this study. The Python-based Simulations of Chemistry Framework (PySCF) was employed in conjunction with RDKit to calculate the total formal charge and electronic structure of each molecule, which were derived from PDB files45. The B3LYP functional, a hybrid functional that combines the exact Hartree–Fock exchange with the Becke–Lee–Yang–Parr exchange-correlation term46, as well as the 6-31G* basis set47, were employed to conduct geometry optimizations and energy calculations. This combination is widely recognized as a dependable option for organic molecules, as it effectively balances computational cost and accuracy. The electronic stability and reactivity of each compound were evaluated by computing key quantum chemical parameters, including DFT energy, HOMO-LUMO energies, SCF convergence, and dipole moment magnitudes. The ligands with the most favorable electronic profiles were chosen for further investigation through Molecular Dynamics (MD) simulations.
Molecular dynamics (MD) simulation
MD simulations were employed to investigate the dynamic stability and flexibility of the protein–ligand complexes. MD computes the physical movement of atoms and molecules, providing structural integrity and conformational changes that occur in the protein-ligand docked complex48. These simulations were conducted using GROMACS 2021.249 following a thoroughly validated pipeline. The protein topology was generated using the CHARMM36 force field50, while ligand topologies and parameter files were accurately derived using the CGenFF server51. The Particle Mesh Ewald (PME) method52 was used for electrostatics, ensuring accurate long-range interaction calculations. Solvation was performed using the TIP3P water model53 in a cubic simulation box with a 1 Å buffer, and the system was neutralized with Na⁺ and Cl⁻ ions. Energy minimization was carried out using 50,000 steps of the steepest descent algorithm to eliminate steric clashes and bad contacts. LINCS algorithm54 was applied to constrain all bond lengths. Equilibration was performed under NVT conditions for 100 ps and NPT conditions for 1 ns, maintaining a temperature of 310 K and a pressure of 1 atm. The velocity-rescaling thermostat55 and Parrinello-Rahman barostat56 were used to control temperature and pressure, respectively. A 2 fs time step was employed, and coordinates were saved every 10 ps during the 100 ns production run. The post MD analysis was performed on the visual platform called “Analogue” developed by Growdea Technologies57,58 (https://growdeatech.com/Analogue/). Additionally, the conformational changes in the structural level integrity of complexes were analyzed using root mean square deviation (RMSD) and root mean square fluctuation (RMSF) evaluations.
Principle component analysis (PCA)
Principal Component Analysis (PCA) was applied to protein-ligand complexes using the standard configuration in GROMACS. The trajectory was pre-processed through a systematic protocol, including the removal of periodic boundary effects, to prepare it for PCA. The refined trajectory was analyzed with the ‘gmx covar’ function59,60 in GROMACS to compute the covariance matrix, which captures the atomic fluctuations within the protein-ligand complex. The eigenvalues and eigenvectors of the covariance matrix were then computed using the ‘gmx anaeig’ function. Finally, the ‘gmx anaproj’ function was employed to project the trajectory onto the principal component (PC) surfaces, enabling visualization of the trajectory in PC space.
Free energy landscape (FEL)
The analysis of the system’s equilibrium state, characterized by the minima on the Free Energy Landscape (FEL), and its non-equilibrium condition, defined by the barriers on the FEL, serves this objective effectively61. In order to determine the FEL, the following equation was applied:
Where ΔG(X) is the Gibbs free energy, kB is the Boltzmann constant, T is the absolute temperature, X denotes the reaction coordinate, and P(X) is the probability distribution of the system along the reaction coordinate.
Binding free energy
The binding free energy was calculated using the MM/GBSA (Molecular Mechanics Generalized Born Surface Area) method62,63. During the 100 ns simulation, the binding free energy of the complexes was calculated using the GROMACS tool gmx_MM/PBSA. The equation for computing binding free energy is outlined below:
In Eq. (Undefined control sequence \- 3), the variables Gcomplex, Greceptor, and Gligand denote the total free energies of the protein-ligand complex, the unbound receptor, and the unbound ligand in solvent, respectively. Equation (Undefined control sequence \- 3) through (7) provide a more detailed decomposition of the binding free energy (ΔGbinding). Equation (Undefined control sequence \- 3) defines ΔGbinding as the differential of enthalpy change (ΔH) and the product of temperature (T) and entropy change (ΔS). Equation (4) decomposes enthalpy into gas-phase energy (ΔGGAS) and solvation free energy (ΔGSOLV). Additionally, Eq. (5) decomposes ΔGGAS into electrostatic energy (ΔEEL) and van der Waals energy (ΔEVDW). Equation (6) separates the solvation energy into polar (ΔEGB) and nonpolar (ΔESURF) components. Equation (7) delineates the nonpolar solvation energy ΔESURF as the product of surface tension (γ) and solvent-accessible surface area (SASA). The energy components were analyzed and graphically represented to emphasize their contributions to binding affinity. The research highlighted the significance of electrostatic interactions (ΔEEL), van der Waals forces (ΔEVDW), and solvation energies in the stabilization of the protein-ligand complex.
Result and discussion
Protein structure
The three-dimensional crystal structure of the Nipah virus attachment glycoprotein (NiV-G) provided a detailed representation of the protein, essential for subsequent computational analyses. Further, the binding site of NiV-G was predicted using the Computed Atlas of Surface Topography of Proteins (CASTp), which identified key surface cavities and pockets likely to be involved in ligand interactions. The binding pocket is shown in the Fig. 1(a) and the predicted binding pocket residues, are listed in Fig. 1(b). These residues are critical for understanding the protein-ligand interaction. Similary, the binding site of the protein was identified from studies to be GLN559, GLU579, TYR581, and ILE588 which was also included in the binding site residues obtained from the CASTp server37,64.

(a) Surface representation of the protein, with the predicted binding pocket highlighted in pink and the rest of the protein in green. (b) List of residues predicted to form the binding site by CASTp server analysis.
Docking (Control)
The known inhibitors G1, G2, G3, G4, and G5 identified in the study were subjected to molecular docking for the selection of a control ligand. The binding energy of the complex are given in the Table 1. Among this, G5 exhibited the strongest binding affinity, with a top binding energy of −9.3 kcal/mol, indicating consistent high-affinity interactions. In contrast, G1 showed the weakest binding affinity, with a top binding energy of −7.2 kcal/mol and a worst binding energy of −6.5 kcal/mol. Based on the top binding energy, G5 was selected as the control ligand for further analysis.
The docked pose of G5 within the binding pocket of a protein is given in Fig. 2 (a) and its interaction with key residues in the binding site was analysed using the discovery studio(BIOVIA)65 and is given in the Fig. 2 (b). As shown in Fig. 2 (a), the ligand is positioned within this cavity and appears to fit snugly into the pocket, indicating a strong binding interaction. The orientation of the ligand suggests that it is stabilized by specific non-covalent interactions with the surrounding protein residues. Further, the interaction plot (Fig. 2(b)), shows π-donor hydrogen bond formed with TYR508. Similarly, the ligand forms π-π stacked with TRP504 and a π-anion bond with ASP302.

(a) Docked pose of ligand (G5) with protein (b) interaction profile of the ligand with the protein binding site.
Compound library and screening
The antiviral library, containing 754 compounds, was downloaded and processed for analysis. During preprocessing, null values and duplicate entries were removed to ensure data quality. Compounds were then filtered based on their adherence to Lipinski’s Rule of Five, which is used to evaluate drug-likeness and oral bioavailability. Following this filtering process, 498 compounds were identified which follows the criteria of the Lipinski’s RoF. The list of these compounds is given in supplementary sheet (Table S1).
Additionally, the screened compounds were further evaluated using DeepPurpose for Drug-Target Interaction (DTI) analysis. The candidates with the highest probability ratings were selected for additional analysis. The score indicates the probability of interaction between the compound and the target protein, with higher scores signifying an increased likelihood of interaction. The predicted scores of the compounds are given in supplementary sheet (Table S2). Based on the predicted scores, the top-ranking compounds with the strongest predicted interactions compared to the control were selected for further refinement through molecular docking. This process allowed for the identification of the most promising candidates for subsequent analysis. The compounds with the predicted score that had the strongest likelihood of interaction compared to the control were selected. This resulted in 333 compounds which is highted in yellow in the supplementary sheet (Table S2).
Docking (Screened Compounds)
The selected 333 small compounds were docked with the protein and the binding energy were retrieved as shown in supplementary sheet (Table S3). Among the top 333 compounds, there were some compounds which didn’t get docked into the binding pocket of the protein. Out of the 333 ligands subjected to molecular docking, only 270 ligands were successfully docked, likely due to structural factors of the undocked ligands. The ligands which are docked are listed in the supplementary sheet (Table S3) along with the binding affinity. Some of the ligands may have had unfavourable geometric conformations, such as excessive steric bulk, which could have prevented proper accommodation within the binding pocket. Moreover, the ligands which couldn’t get docked might have lacked key functional groups required for stable interactions or had structural features, such as high flexibility, that reduced their likelihood of adopting a favourable binding pose. Additionally, ligands with extremely large or small molecular sizes may not have been compatible with the physical dimensions of the binding site, further contributing to their inability to dock. The results, presented in the table (supplementary sheet Table S3), indicate that compound 5,279,171 exhibits the most favourable average binding energy of −9.43 kcal/mol, as well as the highest top binding score of −10.2 kcal/mol. 5,279,171 exhibited the highest binding affinity, with an average of −9.43 kcal/mol and a peak score of −10.2 kcal/mol. 154,701,550 exhibited a mean binding energy of −9.32 kcal/mol, demonstrating consistent stability with a minimum binding score of −9.0 kcal/mol. 9,956,222 exhibited a similar high binding score of −10.2 kcal/mol, however its average was marginally lower at −9.13 kcal/mol. Compounds 129,130,827 and 138,567,123 demonstrated somewhat lower average binding energies of −8.99 and − 8.78 kcal/mol, respectively.
Further, the binding scores were compared to the control (G5) and the top binding scores which were better than the control was selected for further analysis. The selected ligands along with the binding scores is shown in the Table 2. The ligands 5,279,171 and 9,956,222 shows the lowest binding scores compared to the selected compounds with − 10.2 kcal/mol, indicating highly stable interactions within the binding site.
The interaction of the selected compounds with the protein is given in the Fig. 3. The ligands 138,567,123 and 129,130,827 shown in Fig. 3(d, e) forms the highest number of hydrogen bonds compared to the other ligands. 138,567,123 forms conventional hydrogen bonds with LYS560 and carbon-hydrogen bonds with GLU505, ASP302, TYR351, and HIS281. Similarly, the ligand 129,130,827 forms conventional hydrogen bonds with residues GLN559, GLY506, and ALA532, followed by carbon-hydrogen bonds with GLU579 and GLU505. The ligand 5,279,171 in Fig. 3(a) shows the least number of hydrogen bonds, TYR508 forms a π-donor hydrogen bond and the residue ASP302 forms a conventional hydrogen bond. Further the ligand 9,956,222 (Fig. 3 (b)) forms carbon hydrogen bonds and conventional hydrogen bonds with the residues GLN530, TRP504 and THR218, ARG236 respectively. Furthermore, the Fig. 3 (c) shows one carbon-hydrogen bond with GLN490 and two conventional hydrogen bonds with ARG236 and ALA532. These complexes were further caried for analysis using density functional theory (DFT).
The top five compounds selected for the active site of the Nipah virus glycoprotein are more thoroughly examined in the bond distance study to determine their binding behavior and stability. Conventional hydrogen bonds often lay within the 3–6 Å range shown in the Table 3, indicating stable and energetically advantageous interactions. Compounds such as 5,279,171 and 9,956,222 made many critical contacts with residues such as Asp302, Trp504, and Asp219, which are required to stabilize the ligand within the binding pocket. Furthermore, π-π stacking and π-anion interactions with residues such as Phe458, Trp504, and Glu579, with bond lengths typically ranging from 4 to 7 Å, increase the conformational stability of the ligand-protein complexes. The existence of halogen and alkyl interactions in compounds like 154,701,550 and 129,130,827 adds another layer of non-covalent stability.

2D interaction plot of selected compounds (a) 5,279,171 (b) 9,956,222 (c) 154,701,550 (d) 138,567,123 (e) 129,130,827.
Density functional theory (DFT)
The DFT (Density Functional Theory) study offers valuable insights into the electronic properties of the ligands, which are crucial for comprehending their reactivity and prospective interactions with the protein during docking. Energy levels of the Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO), SCF (Self-Consistent Field) convergence, DFT energy, and overall energy are essential for assessing the ligand’s reactivity, electronic distribution, and stability. The DFT calculations showed significant variations in the electronic properties of the five ligands. The HOMO-LUMO energy gap is a measure of chemical reactivity and kinetic stability; a smaller gap indicates greater reactivity, while a wider gap denotes greater stability. Among the ligands, compound 9,956,222 had the smallest HOMO-LUMO gap (0.06 eV), indicating better electronic reactivity, which could improve its interaction with the target protein. In contrast, compound 129,130,827 had a bigger gap (0.74 eV), indicating greater stability and possibly lower reactivity. Dipole moments, which represent molecular polarity and potential solubility, varied greatly between compounds, with 9,956,222 having the highest dipole moment (70.11 Debye), indicating a strong polar nature and the potential for improved aqueous solubility. Notably, 138,567,123 and 5,279,171 obtained SCF convergence, which increased the trustworthiness of their computed attributes.
Among the selected compounds, as shown in Tables 4 and 138,567,123 exhibited the most stable structure, with the lowest DFT energy (−1976.74 Hartree), a successful SCF convergence, and a large HOMO-LUMO gap (0.83 eV), suggesting excellent electronic stability. Followed by the ligand 5,279,171, exhibiting a slightly less negative DFT energy (−1308.32 Hartree), successful SCF convergence, and the largest HOMO-LUMO gap (0.94 eV), indicating strong electronic stability even though it has the lowest dipole moment (3.39 Debye). The compounds 129,130,827 and 9,956,222 follow with moderate DFT energies, although the SCF for these compounds did not converge, and 9,956,222 shows a very small HOMO-LUMO gap (0.054 eV) and an extremely high dipole moment (70.11 Debye). Furthermore, 154,701,550 has lower stability and a moderate HOMO-LUMO gap (0.63 eV) but lacks SCF convergence, reducing its reliability. Thus, the top three compounds (138567123, 5279171, and 129130827) which shows lowest DFT energy were selected for further analysis. The selected ligands for MD simulation are highlighted in Table 4.
MD simulation
RMSD
The root mean square deviation (RMSD) is calculated for the 100 ns simulation in order to evaluate the stability of their interaction with protein. The Fig. 4 (a) shows the RMSD of the protein when ligands (129130827, 138567123, 5279171, and control) is bound. Here, the control ligand (G5) exhibits the most stable behaviour, with RMSD values fluctuating around 0.15 nm, indicating minimal structural deviation. Slightly higher RMSD fluctuations are observed for 138,567,123 and 5,279,171, though their behaviour remains relatively stable throughout the simulation. However, 129,130,827 demonstrates the highest and most variable RMSD, suggesting significant structural deviation and potential instability in the protein backbone. Overall, all the compounds showed a stable RMSD for the protein ranging from 0.15 nm to 0.2 nm.
Further, Fig. 4 (b) shows the RMSD of the ligands. Here the control (G5) is observed to exhibit stable behaviour, with RMSD values remaining below 0.5 nm throughout the simulation. When compared to the control, the ligand 138,567,123 shows slightly lower fluctuations, indicating a moderate degree of deviation from the initial ligand position. Further, ligand 5,279,171 shows greater deviations compared to the control with the initial RMSD below 0.5 nm which increased to 2.5 nm around 20–30 ns and reached 1.55 nm at the end of the simulation. In contrast, 129,130,827 demonstrated drastic increases in RMSD values, especially after 70 ns. These deviations are significantly higher than those of the control, reflecting considerable movement or instability of the ligand within the binding pocket.
Overall, all ligands exhibit varying degrees of deviation when compared to the control. Ligand 138,567,123 surpasses the control (G5) in maintaining both protein and ligand stability. However, ligand 5,279,171 displays significant deviations for both the protein and ligand, highlighting its overall instability in comparison to the control. The consistently low RMSD values and reduced fluctuations demonstrate that 138,567,123 supports a more robust structural conformation, making it a valuable candidate.

RMSD trajectories for protein and ligands for the 100 ns simulation (a) RMSD of the protein (b) RMSD of the ligands control, 138,567,123, 5,279,171, and 129,130,827.
RMSF
Figure 5 shows the results of a subsequent analysis of the Root Mean Square Fluctuation (RMSF). The control ligand produces minimal variation, signifying stable areas. Similarly, 138,567,123 and 129,130,827 showed enhances stability, with lesser peaks at high RMSF. Additionally, Ligand 5,279,171 demonstrated numerous peaks with elevated RMSF. Here, 5,279,171 exhibits the most pronounced fluctuations, indicating increased flexibility and potential instability. The residues, ARG258, ASP420, and GLU422 shows more fluctuations which is higher than 0.3 nm. Contrastingly, the compounds 138,567,123 and 129,130,827 shows similar flexibility patterns but with slightly lower RMSF values than 5,279,171. In this case, 138,567,123 and 129,130,827 did not exhibit any residues with RMSF > 0.3 nm. contrast, the Control remains the most rigid overall, with only slight fluctuations at the residue LEU207, and ASN326 higher than 0.3 nm. The terminal residues exhibit consistently high RMSF across all compounds, despite the diverse effects of the ligands on other regions of the protein. These results suggest that 5,279,171 is the most flexible, whereas 138,567,123 and 129,130,827 display more stable conformations, with the Control showing minimal conformational changes throughout the simulation.

Root means square Fluctuation (RMSF) of protein when ligands (control, 138567123, 5279171, and 129130827) are bound.
Conformation changes
The initial (0 ns) and final (100 ns) trajectories of the protein-ligand complex are extracted and given in Fig. 6. It reveals the conformational changes that occurred during the molecular dynamics simulation. These alterations signify the ligand’s interaction with the protein binding site and the occurrence of substantial structural rearrangements. Figure 6 (a) of the control ligand shows that the initial trajectory is positioned deeply within the binding pocket, suggesting initial strong binding interactions. In the final trajectory, the ligand remains bound in a similar position but exhibits a slight shift and rotation. This indicates that the ligand has adjusted its orientation to form more favorable interactions within the binding pocket. As contrary to this, the ligand 138,567,123 (Fig. 6(b)) initially occupies the binding pocket with a compact orientation, fitting tightly within the cavity but the ligand undergoes a significant positional shift, indicating partial rearrangement within the binding site. This might suggest that the initial pose was less stable, and the ligand found a more favourable binding conformation over time. Minor adjustments in the protein surface are observed, possibly reflecting local flexibility around the binding pocket to accommodate the ligand’s movement. The ligand 5,279,171 (Fig. 6(c)) starts in a deeply buried position within the binding pocket, showing strong initial interactions. In the final trajectory, it shifts toward the edge of the binding pocket, suggesting a partial loss of binding interactions. This could indicate weaker binding stability or the inability of the ligand to remain tightly bound during the simulation. Lastly, the ligand 129,130,827 (Fig. 6 (d)), initially positioned centrally within the binding pocket, appeared to establish significant contacts with the protein surface. In the final trajectory this remains largely within the binding pocket but shifts slightly and adopts a different orientation. This indicates that while the ligand retained its binding, it underwent minor adjustments to optimize its interactions with the protein. In conclusion, 138,567,123 and 129130827showed enhanced stability compared to the other systems, as evidenced by the ligand’s significant yet purposeful conformational adjustment within the binding pocket. While the ligand reoriented itself during the simulation, it remained securely bound and transitioned into a more energetically favourable pose. This indicates that the ligand might have achieved a stable configuration by optimizing its interactions with the protein. The minimal structural adjustments observed in the protein further support the notion that the binding pocket effectively accommodates the ligand without destabilization.

Conformational changes of the protein-ligand complex at different time points during molecular dynamics simulation. (a, c, e, g) represent the structures at 0 ns, (b, d, f, h) show the corresponding structures at 100 ns. (a, b) control, (c, d) 138,567,123, (e, f) 5,279,171, and (g, h) 129,130,827.
Hydrogen bonds
The formation of hydrogen bonds (H-bonds) between proteins and ligands plays a crucial role in stabilizing their interactions. These chemical bonds are crucial for ligand recognition and complex stability because they provide specificity, binding affinity, and induce conformational changes. The Fig. 7 shows fluctuations in the number of hydrogen bonds, which ranges from 0 to 5, over a time span of 100 ns. The control (Fig. 7 (a)) exhibits frequent hydrogen bond formation but maintains a limited number of hydrogen bonds, mostly between 0 and 2 throughout the simulation. The distribution of hydrogen bond interactions is irregular, with fluctuations observed over time, indicating transient interactions rather than sustained bonding. Further, 138,567,123 shows an increased number of hydrogen bonds compared to the control, with values fluctuating primarily between 1 and 2, occasionally reaching 3. The interactions are more sustained and frequent throughout the simulation, suggesting stronger and more consistent hydrogen bonding. Additionally, 129,130,827 shows moderate hydrogen bond interactions, with values typically fluctuating between 1, 2 and 3 occasionally reaching 4. This hydrogen bond interactions are more frequent and sustained, particularly in the latter half of the simulation, suggesting a strengthening of the ligand-protein interaction over time. Figure 7 (c) showed that 5,279,171 exhibits the highest number of hydrogen bonds among other ligands, frequently reaching values of 3 or more and occasionally as high as 5 bonds. The interactions are relatively frequent but not as consistent or high in number as system 138,567,123 or 129,130,827.
In conclusion, 138,567,123 exhibits the strongest hydrogen bonding, with the consistency of interactions. It also forms stronger and more reliable interactions with the protein, potentially enhancing ligand stability and binding efficiency. It sustained hydrogen bonding, surpassing the control in terms of stability and binding potential.

Hydrogen bond formed between glycoprotein and ligands during 100 ns of simulation. (a) control, (b) 138,567,123, (c) 5,279,171, and (d) 129,130,827.
Interaction analysis
Further, the protein-ligand interactions at the 100 ns time point of the simulation is obtained and given in Fig. 8, with the ligand and surrounding amino acids involved in various types of molecular interactions. The control ligand forms stable yet straightforward interactions, including hydrogen bonds and π-interactions with critical residues such as GLU506, PHE458, TRP504, PRO441, and LEU221, indicating a basic level of binding stability. Ligand 138,567,123 demonstrates the most extensive and varied interaction profile, characterised by strong hydrogen bonds with LYS560 and an alkyl bond with GLU504, indicating robust and stable binding. Ligand 5,279,171 establishes alkyl connections with ARG402 and ILE401, alongside π-anion interactions with ARG372, indicating a dynamic yet stable binding configuration that may provide flexibility inside the binding pocket. Ligand 129,130,827 exhibits a more intricate interaction profile, featuring hydrogen bonds with CYS240, carbon-hydrogen bonds with ASN586 and SER239, a halogen interaction with ASP582, and a π-π T-shaped interaction with GLY238. This indicates a moderate degree of stability, with both conventional and non-conventional interactions contributing to its binding. Ligand 138,567,123 has the most diversified and stable interactions, followed by ligand 5,279,171, which displays a more dynamic binding profile, whilst ligand 129,130,827 shows moderate stability with a range of unique interaction types.
The bond distances were listed in the Table 5. The control compound exhibited moderate stability, as evidenced by its robust π–π interaction with Phe458 (5.87 Å) and its conventional hydrogen bond with Gly506 (3.90 Å). Additionally, it maintained multiple alkyl interactions. Compound 138,567,123 maintained a stable and persistent binding conformation by forming two strong hydrogen bonds with Lys560 (4.46 and 3.93 Å) and retaining a π–π interaction with Trp504 (6.95 Å). Compound 5,279,171 exhibited a unique π–cation interaction with Arg372 (7.86 Å) and significant alkyl contacts with Arg402 and Ile401, which suggests that it has a favorable hydrophobic anchoring. A complex network of interactions was demonstrated by compound 129,130,827, which included a conventional H-bond with Cys240 and multiple carbon H-bonds with Ser239 and Asn586. In addition, it established a π–π interaction with Gly238 and a halogen bond with Asp582, which indicates that it is in a conformationally favorable and well-stabilized state following MD. The selected compounds’ potential as promising Nipah virus glycoprotein inhibitors is further supported by these results, which affirm their structural integrity and binding persistence over the simulation time.

Molecular interactions of the ligands (a) control, (b) 138,567,123, (c) 5,279,171, and (d) 129,130,827 with surrounding amino acid residue of the protein at the 100 ns time point of the simulation.
Principal component analysis
The Fig. 9 shows the principal component analysis (PCA). The control (Fig. 9 (a)) exhibits a broad distribution with two distinct clusters separated along eigenvector 1. This suggests that the control system transitions between two major conformational states during the simulation, with moderate sampling of intermediate states. Alternatively, ligand 138,567,123 leads to a more diffuse distribution of data, particularly along eigenvector 1, suggesting a higher degree of conformational variability and systemic flexibility. Stabilising the protein-ligand complex into two independent conformational states for ligand 5,279,171, as seen in the two lobes, suggests dynamic equilibrium between both states. At the same time, ligand 129,130,827 causes mild alterations, showing a longer and less dense cluster in comparison to the control. This suggests it has an effect on protein dynamics, but a smaller one than 138,567,123 or 5,279,171. In general, the findings demonstrate that the protein’s conformational behaviour is modulated differently by each ligand, with the most pronounced split into discrete states observed with ligand 5,279,171.

Principal component analysis (PCA) of the protein-ligand complex (a) control, (b) 138,567,123, (c) 5,279,171, and (d) 129,130,827.
Free energy landscape (FEL)
The control (Fig. 10 (a)) shows a well-defined low-energy basins which are separated by a moderate energy barrier. This suggests that the control has four primary stable conformational states with some degree of interconversion. The energy barrier between the basins is not extremely high, indicating moderate flexibility in conformational transitions. The ligand 138,567,123 shows a single dominant low-energy basin Fig. 10 (b), with only a few surrounding high-energy regions. This indicates that 138,567,123 primarily stabilizes one conformation, with minimal sampling of alternative states. The energy barrier between conformations is relatively low, suggesting high stability and reduced flexibility. In the Fig. 10 (c), multiple low-energy basins are present, similar to the control system, indicating higher conformational diversity. The basins are separated by moderate energy barriers, which suggests a balance between stability and flexibility. Additionally, the ligand 129,130,827 Fig. 10 (d) has a single, deep low-energy basin which dominates the landscape, with higher energy regions encompassing the conformational space. The deep basin indicates a highly stable conformation, but the surrounding high-energy regions suggest significant energy barriers for transitions to other states.
The compounds 138,567,123 and 129,130,827 show strong stabilization of their respective conformations, with 138,567,123 being more flexible due to lower energy barriers and 129,130,827 being more rigid due to high barriers. The control and 5,279,171 exhibit higher conformational diversity, with moderate barriers allowing transitions between multiple stable states. These FELs provide a clear understanding of the energy landscape, with 138,567,123 emerging as a balanced system offering both stability and flexibility.

Free Energy Landscape (FEL) of the protein-ligand complexes at 100 ns simulation (a) control, (b) 138,567,123, (c) 5,279,171, and (d) 129,130,827.
Binding free energy
In addition to FEL, MM/GBSA energy for the last 20 ns of the simulation was also computed for the complex as given in Table 6. Table 6 shows the energetic components contributing to the protein-ligand binding energy, divided into van der Waals (VDWAALS), electrostatic (EEL), polar solvation (EGB), and non-polar solvation (ESURF) energies, along with their total contributions.
The total binding free energy, ΔTOTAL, represents the overall stability of the protein-ligand complex, incorporating all the individual energetic contributions, including van der Waals interactions (VDWAALS), electrostatic interactions (EEL), polar solvation (EGB), and non-polar solvation energies (ESURF). As shown in table, 138,567,123 demonstrates the most favourable total binding free energy of −24.04 kcal/mol and a standard deviation of ± 3.29, indicating a strong and stable interaction between the ligand and the protein. Further, the control shows a total binding free energy of −21.64 kcal/mol ± 2.57, which is also favourable but less than 138,567,123. The compounds 129,130,827 shows a total binding free energy of −20.17 kcal/mol ± 3.78 exhibiting moderately strong binding. Finally, the compound 5,279,171 has the weakest binding affinity, with a total binding free energy of −14.74 kcal/mol ± 3.42. Overall, the total binding free energy analysis highlights 138,567,123 as the most stable complex, with control and 129,130,827 showing moderately strong binding, while 5,279,171 exhibits the least favourable binding energy.
Discussion
This study highlights the potential of computational tools in the discovery and optimization of small-molecule inhibitors targeting the Nipah virus glycoprotein (NiV-G). The use of molecular docking, density functional theory (DFT), and molecular dynamics (MD) simulations facilitated the identification of lead compounds with promising binding affinity and stability, underscoring the efficiency of in silico methodologies in antiviral drug discovery.
Our computational analysis identified two highly promising ligands, 5,279,171 and 9,956,222, each exhibiting exceptional projected binding affinities of − 10.2 kcal/mol against the Nipah virus glycoprotein (NiV-G). The other three compounds 154,701,550, 138,567,123 and 129,130,827 also showed strong binding affinities in the range − 9.9 kcal/mol to −9.4 kcal/mol. These values significantly exceed the binding energies documented in many computational studies, underscoring substantial progress in the discovery of effective inhibitors.
Compared to previous investigations, ligand 211,559 from the BBB + Asinex library exhibited the highest binding affinity at − 9.2 kcal/mol66, while ligands ND_nw_193 and ND_nw_93 from another prior study displayed affinities of − 8.8 and − 8.3 kcal/mol, respectively64. Our identified ligands clearly outperform these compounds, emphasizing the robustness and effectiveness of our computational screening approach. This advancement is further emphasized by the fact that phytoconstituents previously studied for Nipah inhibition, including 6-Deoxy-L-Mannopyranose (− 6.65 kcal/mol), Vanillic acid (− 6.31 kcal/mol), and the standard antiviral Ribavirin (− 5.52 kcal/mol), exhibited significantly lower predicted binding energies67. The considerable disparity between these phytoconstituents and our identified ligands underscores the importance of our findings.
This study’s identified ligands showed interactions with critical residues that were also found in previous studies. Specifically, the interaction between the protein and the ligands involved residues His281, Tyr351, Tyr508, Gln490, and Tyr506, consistent with previous findings68. Additionally, residues Gln559 and Gln579 identified in earlier studies were also observed in this study64,37. Another prior study reported interactions involving critical residues Arg236, Ala532, and Lys560, which were similarly detected in our current analysis66,67. This evident improvement in anticipated inhibitory action rectifies prior shortcomings in computational analyses of Nipah virus glycoprotein and considerably propels the domain of computational drug research against viral infections.
Among the screened compounds, ligand 138,567,123 consistently outperformed the control ligand (G5) across multiple analyses. 138,567,123 showed sustained hydrogen bonding and strong interactions with key residues within the NiV-G binding pocket. These results were further validated by MD simulations69, which demonstrated minimal root mean square deviation (RMSD) and fluctuation (RMSF) values for 138,567,123, indicating a stable and robust protein-ligand complex. Additionally, principal component analysis (PCA) and free energy landscape (FEL) studies confirmed the ligand’s ability to stabilize the protein while maintaining structural flexibility. The DFT calculations provided insights into the electronic properties of the compounds. A large HOMO-LUMO gap and favourable DFT energy for 138,567,123 highlighted its electronic stability and potential reactivity. These properties translated into the most favourable binding free energy (−24.04 kcal/mol) among all selected ligands, as calculated using the MM/GBSA method. Comparatively, the control ligand showed moderately strong binding, while ligands 5,279,171 exhibited lower stability and binding efficiency, as evidenced by higher RMSD values and weaker energetic contributions.
These results reinforce the feasibility of targeting NiV-G with small molecules as a therapeutic strategy against Nipah virus infections. By focussing on the receptor-binding domain, this method directly reports a vital step in the viral entry process, providing a new channel for therapeutic intervention. The integration of AI-based screening approaches with conventional computational methodologies facilitated the identification of potential drug candidates, effectively accelerating the drug discovery process.
Although this study offers a comprehensive computational framework for the identification of potential inhibitors of the Nipah virus glycoprotein, it is important to recognize several limitations. In silico methods, such as molecular docking, MM-GBSA, molecular dynamics simulations, and DFT analyses, are the primary foundation of all results. Despite their robustness and widespread acceptance, these methods are predictive in nature. These methods are unable to completely capture the intricacy of biological environments, including immune responses, enzymatic metabolism, and potential off-target effects. In addition, the confirmation of the actual efficacy, bioavailability, and safety of the proposed compounds is restricted by the absence of in vitro or in vivo experimental validation. Consequently, these compounds require additional experimental studies to validate their findings and progress toward clinical development, despite the fact that the computational data offer robust initial evidence and a valuable starting point.
Future work should prioritize the experimental validation of these findings, particularly the in vitro and in vivo evaluation of ligand 138,567,123. Further optimization of this compound, along with its analogues, could enhance its pharmacokinetic and pharmacodynamic properties. This study provides a strong foundation for the development of targeted antiviral therapies and highlights the transformative potential of computational approaches in addressing global health challenges like the Nipah virus.
Conclusion
This study demonstrates the effective application of computational methods to identify potential small-molecule inhibitors that target the Nipah virus glycoprotein (NiV-G). Among the compounds that were screened, ligand 138,567,123 demonstrated favourable electronic stability, dynamic behaviour, and binding affinity, suggesting that it has the potential for further investigation. Although these preliminary in silico findings provide valuable preliminary insights, they must be validated through experimental studies to corroborate their therapeutic relevance. Overall, the study emphasizes the importance of computational screening in the early stages of antiviral drug discovery and establishes a foundation for future experimental-based investigations.
Data availability
Data is provided within the manuscript or supplementary information files.
References
Hughes, J. M., Wilson, M. E., Luby, S. P., Gurley, E. S. & Hossain, M. J. Transmission of human infection with Nipah virus. Clin. Infect. Dis. 49, 1743–1748 (2009).
Amarasinghe, G. K. et al. Taxonomy of the order mononegavirales: update 2019. Arch. Virol. 164, 1967–1980 (2019).
Paramyxoviruses in reptiles. A review - ScienceDirect. https://www.sciencedirect.com/science/article/pii/S0378113513001971
Thibault, P. A., Watkinson, R. E., Moreira-Soto, A., Drexler, J. F. & Lee, B. Chapter One - Zoonotic Potential of Emerging Paramyxoviruses: Knowns and Unknowns. in Advances in Virus Research (eds. Kielian, M., Mettenleiter, T. C. & Roossinck, M. J.) vol. 98 1–55Academic Press, (2017).
Choi, E. J., Ortega, V. & Aguilar, H. C. Feline morbillivirus, a new paramyxovirus possibly associated with feline kidney disease. Viruses 12, 501 (2020).
TRANSMISSION OF HUMAN INFECTION WITH NIPAH VIRUS - Improving Food Safety Through a One Health Approach. - NCBI Bookshelf. https://www.ncbi.nlm.nih.gov/books/NBK114486/
Chua, K. B. Nipah virus outbreak in Malaysia. J. Clin. Virol. 26, 265–275 (2003).
Chua, K. B. et al. Nipah virus: A recently emergent deadly paramyxovirus. Science 288, 1432–1435 (2000).
Johnson, K., Vu, M. & Freiberg, A. N. Recent advances in combating Nipah virus. Fac. Rev. 10, 74 (2021).
Mrcp, K. S. T., Tan, C. T. & Mrcp, K. J. G. Epidemiological aspects of Nipah virus infection. (1999).
Wong, K. T., Shieh, W. J., Zaki, S. R. & Tan, C. T. Nipah virus infection, an emerging paramyxoviral zoonosis. Springer Semin Immunopathol. 24, 215–228 (2002).
Clinical Presentation of Nipah Virus Infection in. Bangladesh | Clinical Infectious Diseases | Oxford Academic. https://academic.oup.com/cid/article-abstract/46/7/977/292786
Saha, O. et al. Antiviral activity, Pharmacoinformatics, molecular docking, and dynamics studies of Azadirachta indica against Nipah virus by targeting envelope glycoprotein: emerging strategies for developing antiviral treatment. Bioinform Biol. Insights. 18, 11779322241264145 (2024).
Wang, Q. et al. The nanoscale organization of the Nipah virus fusion protein informs new membrane fusion mechanisms. Preprint Https://doi. https://doi.org/10.7554/eLife.97017.2 (2024).
Yang, S. & Kar, S. Computer-assisted identification of potential quinolone derivatives targeting Nipah virus glycoprotein attachment with human cell surface receptor ephrin-B2: multistep virtual screening. Comput. Biol. Med. 163, 107240 (2023).
Abduljalil, J. M., Elfiky, A. A., Sayed, E. S. T. A. & AlKhazindar, M. M. Computational identification of drug-like marine natural products as potential RNA polymerase inhibitors against Nipah virus. Comput. Biol. Chem. 104, 107850 (2023).
Sen, N. et al. Predicting and designing therapeutics against the Nipah virus. PLoS Negl. Trop. Dis. 13, e0007419 (2019).
Larsen, B. B. et al. Functional and antigenic landscape of the Nipah virus receptor binding protein. Preprint At. https://doi.org/10.1101/2024.04.17.589977 (2024).
Aljofan, M., Lo, M. K., Rota, P. A., Michalski, W. P. & Mungall, B. A. Off label antiviral therapeutics for henipaviruses: new light through old windows. J. Antivir Antiretrovir. 2 https://doi.org/10.4172/jaa.1000014 (2010).
Dawes, B. E. et al. Favipiravir (T-705) protects against Nipah virus infection in the hamster model. Sci. Rep. 8, 7604 (2018).
Julander, J. G. et al. An update on the progress of Galidesivir (BCX4430), a broad-spectrum antiviral. Antiviral Res. 195, 105180 (2021).
Tit-oon, P. et al. Prediction of the binding interface between monoclonal antibody m102.4 and Nipah attachment glycoprotein using structure-guided Alanine scanning and computational Docking. Sci. Rep. 10, 18256 (2020).
Byrne, P. O. et al. Structural basis for antibody recognition of vulnerable epitopes on Nipah virus F protein. Nat. Commun. 14, 1494 (2023).
Viral Entry into Host Cells | SpringerLink. https://link.springer.com/book/10.1007/978-1-4614-7651-1#page=110
Bowden, T. A. et al. Crystal structure and carbohydrate analysis of Nipah virus attachment glycoprotein: a template for antiviral and vaccine design. J. Virol. 82, 11628–11636 (2008).
Single Amino Acid. Changes in the Nipah and Hendra virus attachment glycoproteins distinguish EphrinB2 from EphrinB3 usage |. J. Virol. https://journals.asm.org/doi/full/https://doi.org/10.1128/jvi.00999-07
Ortega, V. et al. Novel roles of the Nipah virus attachment glycoprotein and its mobility in early and late membrane fusion steps. mBio 13, e03222–e03221 (2022).
Dang, H. V. et al. Broadly neutralizing antibody cocktails targeting Nipah virus and Hendra virus fusion glycoproteins. Nat. Struct. Mol. Biol. 28, 426–434 (2021).
Exceptionally Potent Cross-Reactive Neutralization of Nipah. and Hendra Viruses by a Human Monoclonal Antibody | The Journal of Infectious Diseases | Oxford Academic. https://academic.oup.com/jid/article/197/6/846/919925
Xu, K. et al. Crystal structure of the Hendra virus attachment G glycoprotein bound to a potent Cross-Reactive neutralizing human monoclonal antibody. PLoS Pathog. 9, e1003684 (2013).
Sadeghi, S. S. & Keyvanpour, M. R. An analytical review of computational drug repurposing. IEEE/ACM Trans. Comput. Biol. Bioinf. 18, 472–488 (2021).
Makurvet, F. D. Biologics vs. small molecules: drug costs and patient access. Med. Drug Discovery. 9, 100075 (2021).
Shah, M., Patel, M., Shah, M., Patel, M. & Prajapati, M. Computational transformation in drug discovery: A comprehensive study on molecular docking and quantitative structure activity relationship (QSAR). Intell. Pharm. 2(5), 589–595. https://www.sciencedirect.com/science/article/pii/S2949866X24000340 (2024).
Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis (Eds. Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R. & Yamanishi, K.). Cambridge University Press (Cambridge, UK, 2007).
Singh, M. P. et al. Computational approaches to designing antiviral drugs against COVID-19: A comprehensive review. Curr. Pharm. Des. 29, 2601–2617 (2023).
Mongia, A. et al. A computational approach to aid clinicians in selecting anti-viral drugs for COVID-19 trials. Sci. Rep. 11, 9047. https://www.nature.com/articles/s41598-021-88153-3 (2021).
Kalbhor, M. S., Bhowmick, S., Alanazi, A. M., Patil, P. C. & Islam, M. A. Multi-step molecular Docking and dynamics simulation-based screening of large antiviral specific chemical libraries for identification of Nipah virus glycoprotein inhibitors. Biophys. Chem. 270, 106537 (2021).
Bowden, T. A. et al. Structural basis of Nipah and Hendra virus attachment to their cell-surface receptor ephrin-B2. Nat. Struct. Mol. Biol. 15, 567–572 (2008).
Burley, S. K. et al. Protein Data Bank (PDB): The Single Global Macromolecular Structure Archive. in Protein Crystallography: Methods and Protocols (eds. Wlodawer, A., Dauter, Z. & Jaskolski, M.) 627–641 (Springer, New York, NY, 2017). https://doi.org/10.1007/978-1-4939-7000-1_26.
Tian, W., Chen, C., Lei, X., Zhao, J. & Liang, J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 46, W363–W367 (2018).
O’Boyle, N. M. et al. Open babel: an open chemical toolbox. J. Cheminform. 3, 33 (2011).
Huang, K. et al. DeepPurpose: a deep learning library for drug–target interaction prediction. Bioinformatics 36, 5545–5547 (2021).
Argaman, N. & Makov, G. Density functional theory: an introduction. Am. J. Phys. 68, 69–79 (2000).
Rozhenko, A. B. Density functional theory calculations of Enzyme–Inhibitor interactions in medicinal chemistry and drug design. In Application of Computational Techniques in Pharmacy and Medicine (eds Gorb, L. et al.) 207–240 (Springer Netherlands, 2014). https://doi.org/10.1007/978-94-017-9257-8_7.
Sun, Q. et al. PySCF: the Python-based simulations of chemistry framework. WIREs Comput. Mol. Sci. 8, e1340 (2018).
Tirado-Rives, J. & Jorgensen, W. L. Performance of B3LYP density functional methods for a large set of organic molecules. J. Chem. Theory Comput. 4, 297–306 (2008).
Nedelec, J. M. & Hench, L. L. Effect of basis set and of electronic correlation on Ab initio calculations on silica rings. J. Non-cryst. Solids. 277, 106–113 (2000).
Kumar, Y., Singh, H. & Patel, C. N. Silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular Docking and dynamics simulation based drug-repurposing. J. Infect. Public Health. 13, 1210–1223 (2020).
Bauer, P., Hess, B. & Lindahl, E. GROMACS 2022.4 Manual. (2022). https://doi.org/10.5281/ZENODO.7323409
Huang, J. & MacKerell, A. D. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 34, 2135–2145 (2013).
Vanommeslaeghe, K. et al. CHARMM general force field (CGenFF): A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 31, 671–690 (2010).
Petersen, H. G. Accuracy and efficiency of the particle mesh Ewald method. J. Chem. Phys. 103, 3668–3679 (1995).
Boonstra, S., Onck, P. R. & van der Giessen, E. CHARMM TIP3P water model suppresses peptide folding by solvating the unfolded state. J. Phys. Chem. B. 120, 3692–3698 (2016).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Martoňák, R., Laio, A. & Parrinello, M. Predicting crystal structures: the Parrinello-Rahman method revisited. Phys. Rev. Lett. 90, 075503 (2003).
Sim(Ana). Analogue Release 2024, Growdea Technologies Pvt. Lt. v1.1. (2024).
Trajecta(Ana). Analogue Release 2024, Growdea Technologies Pvt. Lt. v1.1. (2024).
Berendsen, H. J. C., van der Spoel, D. & van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56 (1995).
Hess, B., Kutzner, C., van der Spoel, D. & Lindahl, E. GROMACS 4: algorithms for highly efficient, Load-Balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 (2008).
Sgarbossa, A. Natural biomolecules and protein aggregation: emerging strategies against amyloidogenesis. Int. J. Mol. Sci. 13, 17121–17137 (2012).
Valdés-Tresanco, M. S., Valdés-Tresanco, M. E., Valiente, P. A., Moreno, E. & gmx_MMPBSA A new tool to perform End-State free energy calculations with GROMACS. J. Chem. Theory Comput. 17, 6281–6291 (2021).
Miller, B. R. I. et al. MMPBSA.py: an efficient program for End-State free energy calculations. J. Chem. Theory Comput. 8, 3314–3321 (2012).
Randhawa, V., Pathania, S. & Kumar, M. Computational identification of potential multitarget inhibitors of Nipah virus by molecular Docking and molecular dynamics. Microorganisms 10, 1181 (2022).
Sharma, S., Sharma, A. & Gupta, U. Molecular Docking studies on the Anti-Fungal activity of Allium Sativum (Garlic) against mucormycosis (Black Fungus) by BIOVIA discovery Studio Visualizer 21.1.0. Preprint at (2021). https://doi.org/10.21203/rs.3.rs-888192/v1
Naeem, I. et al. In Silico identification of potential drug-like molecules against G glycoprotein of Nipah virus by molecular docking, DFT studies, and molecular dynamic simulation. J. Biomol. Struct. Dynamics. 41, 7104–7118 (2023).
Geetha, D. et al. G SITE INHIBITORS FROM VARIOUS MEDICINAL PLANTS J. Popl Ther. Clin. Pharmacol. 32, 576–589 (2025).
Ropón-Palacios, G. et al. Integrated computational biophysics approach for drug discovery against Nipah virus. Biochem. Biophys. Res. Commun. 745, 151140 (2025).
Borjian Boroujeni, M., Shahbazi Dastjerdeh, M., Shokrgozar, M., Rahimi, H. & Omidinia, E. Computational driven molecular dynamics simulation of keratinocyte growth factor behavior at different pH conditions. Inf. Med. Unlocked. 23, 100514 (2021).
Author information
Authors and Affiliations
Contributions
Jawaher A.Abdulhakim, wrote the manuscrip as single autrhor.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Abdulhakim, J.A. Machine learning assisted in Silico discovery and optimization of small molecule inhibitors targeting the Nipah virus glycoprotein. Sci Rep 15, 16067 (2025). https://doi.org/10.1038/s41598-025-01243-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-01243-4
