
Abstract
Polygenic risk scores (PRS) have ushered in a new era in genetic epidemiology, offering insights into individual predispositions to a wide range of diseases. However, despite recent marked enhancements in predictive power, PRS-based models still need to overcome several hurdles before they can be broadly applied in the clinic. Chiefly, they need to achieve sufficient accuracy, easy interpretability and portability across diverse populations. Leveraging trans-ancestry genome-wide association study (GWAS) meta-analysis, we generated novel, diverse summary statistics for 30 medically-related traits and benchmarked the performance of six existing PRS algorithms using UK Biobank. We built an ensemble model using logistic regression to combine outputs of top-performing algorithms and validated it on the diverse eMERGE and PAGE MEC cohorts. It surpassed current state-of-the-art PRS models, with minimal performance drops in external cohorts, indicating good calibration. To enhance predictive accuracy for clinical application, we incorporated easily-accessible clinical characteristics such as age, gender, ancestry and risk factors, creating disease prediction models intended as prospective diagnostic tests, with easily interpretable positive or negative outcomes. After adding clinical characteristics, 12 out of 30 models surpassed 80% AUC. Further, 25 traits exceeded the diagnostic odds ratio (DOR) of five, and 19 traits exceeded DOR of 10 for all ancestry groups, indicating high predictive value. Our PRS model for coronary artery disease identified 55–80 times more true coronary events than rare pathogenic variant models, reinforcing its clinical potential. The polygenic component modulated the effect of high-risk rare variants, stressing the need to consider all genetic components in clinical settings. These findings show that newly developed PRS-based disease prediction models have sufficient accuracy and portability to warrant consideration of being used in the clinic.
Similar content being viewed by others


Introduction
Polygenic risk scores (PRSs) have emerged as a transformative tool in genetic epidemiology, harnessing the wealth of data generated by genome-wide association studies (GWAS) to predict an individual’s predisposition to complex diseases1 By aggregating numerous genetic variants, each with a small to modest effect, PRS offers an understanding of genetic susceptibility across a spectrum of human diseases, including cardiovascular conditions, psychiatric disorders, and cancers2,3,4,5,6 The utility of PRSs extends beyond mere risk prediction; they hold promise for personalized medicine, where interventions can be tailored based on an individual’s genetic risk profile. PRSs can enhance disease screening strategies, inform clinical decision-making, and potentially guide lifestyle and therapeutic interventions aimed at mitigating disease risk1,3,7,8,9 Despite this potential, the application of PRSs in clinical settings is in its nascent stages, grappling with challenges such as improving the accuracy and interpretability of scores, ensuring equitable performance across diverse populations, and integrating genetic risk information with environmental and lifestyle factors for a comprehensive approach to disease prediction and prevention1,10,11.
Recent advancements in computational methods, alongside the exponential growth in GWAS sample sizes, have markedly enhanced the predictive power of PRSs8,12 However, developing and validating PRS models that are generalizable across different ancestries remains imperative, as current models often exhibit reduced efficacy in non-European populations1,8 Additionally, any PRS used in a clinical setting must demonstrate clinical utility and easy interpretability that will alter patient care decisions based on the results13.
With those aims in mind, we set about to create and validate 30 multi-ancestry PRS models and benchmark our results against two sets of recently released PRS models, described in Thompson et al. (2024)8 and Lennon et al. (2024)14. Thompson et al. evaluated the performance of the UK Biobank PRS Release and demonstrated that those models consistently outperformed a comprehensive array of 76 previously published PRS models. In addition the authors showed that the risk profiles associated with high PRSs rivaled those of known rare pathogenic variants. On the other hand, Lennon et al. (2024)14 focused on the optimization of 10 PRS models for use in the diverse Electronic Medical Records and Genomics (eMERGE) Network and their clinical implementation. We show that further gains in PRS model accuracy are possible with the meta-analyses of even larger and more diverse datasets, and by employing an ensemble PRS model that combines the strengths of individual PRS algorithms. We then build PRS-based disease prediction models by adding demographic and clinical risk factors to our PRS models to maximize their predictive power. We treat this task as a binary classification problem and systematically test different combinations of inputs to make disease predictions that are easily interpretable and would alter physician intervention decisions. We show that the final models are highly predictive and have sufficient accuracy to warrant consideration of being used in a clinical setting.
Methods
Study cohorts
UK biobank cohort
In the UK Biobank project (UKB), 500,000 participants aged 40–69 years were genotyped with a custom Axiom genotyping array, which assayed 825,927 genetic variants15 Genotyping data was further imputed using the Haplotype Reference Consortium and the UK10K + 1KG reference panel, resulting in 96 million variants15. From this imputed dataset, we selected 13.7 million high-quality variants, with a minor allele frequency greater than 0.001 and a Hardy-Weinberg equilibrium P value exceeding 10− 10.
We next chose a subset of individuals used for computing the principal components in the UK Biobank (Field 22020) that were defined as the ‘White British ancestry’ group (UKB Data Field 22006). These individuals, referred to as the White British Unrelated (WBU) subgroup, are a high quality set of unrelated individuals that underwent stringent quality controls including the exclusion of samples with an autosomal missing rate greater than 0.02, mismatches between inferred and self-reported sex, and outliers based on heterozygosity, as detailed in Bycroft et al. (2018)15 We used the GWAS summary statistics generated by Thompson et al. (2024)8 for this WBU UKB subgroup (available at zenodo.org/records/6631952) as the UKB-derived component in our meta-analyses.
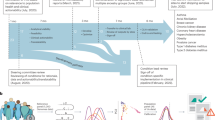
After excluding the WBU subset, we retrieved the 104,604 remaining samples for downstream analysis. This approach, proposed by Thompson et al. (2024)8, increases the representation of non-European ancestries for PRS training, testing and validation. We divided this cohort into a training set of 30,000 individuals and a testing set of 74,604 individuals, to benchmark the performance of each PRS algorithm. Subsequently, we further split the testing set into two sub-cohorts: one of 30,000 participants for retraining our novel ensemble PRS method and developing new risk models that integrate various PRSs and additional demographic factors, and another comprising 44,604 participants for testing. We adopted the methodology outlined by Thompson et al. (2024)8 to infer genetic ancestry in these samples (see Supplementary Methods), categorizing them as Africans (N = 9,501), East Asians (N = 2,931), Europeans (N = 82,318) and South Asians (N = 9,854). This structured approach enabled direct comparisons of our PRS results with those developed by Thompson et al., using the same subsets, thereby ensuring the comparability and robustness of our findings. All experiments and the cohorts and subsets used in this study are detailed in Fig. 1, which shows the schematic of the study design.
eMERGE network cohort
Access to the eMERGE dataset was granted via dbGaP (phs001584.v2.p2). We focused on subsets c1, c4, c5, c6, c7, c8, and c10, which did not require Institutional Review Board approval16 This yielded 84,215 participants with both genetic and phenotypic information, including Blacks or Africans (N = 10,126), Asians (N = 920), Whites (N = 67,316), Latinos (N = 3,303), Native Americans (N = 101) and Pacific Islanders (N = 6). Individuals with unknown self-reported ethnicity were excluded. Due to relatively small numbers, Native Americans were grouped with Latinos, while Pacific Islanders were grouped with Asians. The eMERGE dataset contains over 39 million variants, but it lacks certain SNPs we needed for our analysis. We addressed this by imputing the missing SNPs. The newly released whole genome sequencing data from the UKB (UKB Dragen WGS), available on the UK Biobank Research Analysis platform, was phased with ShapeIt517 and used as the reference panel for imputation. The eMERGE cohort was lifted over to GRCh38 with Picard tools18 and then normalized with BCFtools19. Imputation was performed using Beagle5.420 with default parameters.
PAGE MEC cohort
Access to the PAGE MEC21 cohort was granted via dbGaP (phs000220.v2.p2). We chose individuals genotyped using the MEGA Consortium array over the Metabochip due to the much larger number of variants (1,705,969 vs. 196,725), to ensure reliable imputation and downstream analyses. In total, this yielded 9,098 participants with both genetic and phenotypic information, including Blacks or Africans (N = 3,520), Hawaiians (N = 2,104), Japanese (N = 3,451) and Latinos (N = 23). Individuals with unspecified self-reported ethnicity were excluded from the study, along with Latinos, who were omitted due to their relatively small numbers. Hawaiian and Japanese individuals were combined into an Asian group when necessary, to facilitate performance comparisons with groups from other cohorts. PAGE MEC genotyping data was further imputed in the same way as the eMERGE cohort, to maximize the number of SNPs available for downstream analyses.

Study design schematics. (A) Benchmarking. Six PRS prediction algorithms, SBayesRC, LDpred2, Stacked C + T, lassosum2, PRS-CS and maxCT, were applied to 30 medically-related traits. (B) PRS evaluation. 5 PRS algorithms were combined into an ensemble PRS model. maxCT was excluded since SCT is already a stacked model derived from maxCT. (C) PRS-based disease prediction models. Final risk models were boosted with ancestry, demographic and risk factor information to enhance risk prediction. See main text for details.
Trait selection and definition
We selected 30 clinical traits for this study: age-related macular degeneration, Alzheimer’s disease, asthma, atrial fibrillation, bipolar disorder, bowel cancer, breast cancer, cardiovascular disease, celiac disease, coronary artery disease, Crohn’s disease, epithelial ovarian cancer, hypercholesterolemia (total cholesterol), hypertension, hypertriglyceridemia (triglycerides), ischemic stroke, melanoma, obesity (BMI), osteoporosis, Parkinson’s disease, primary open angle glaucoma, prostate cancer, psoriasis, rheumatoid arthritis, schizophrenia, systemic lupus erythematosus, type 1 diabetes, type 2 diabetes, ulcerative colitis, and venous thromboembolic disease. These diseases are among the most common noncommunicable diseases worldwide. They have been subject to many large and well-powered GWAS that can be used to build robust PRSs. Twenty-seven diseases were adopted from Thompson et al. (2024)8, which enabled direct model comparison. We excluded multiple sclerosis from our study due to a lack of additional summary statistics that showed improvement over random predictions (area under the curve (AUC) not significantly above 50%). Additionally, we incorporated three quantitative health-related traits —body mass index (BMI), total cholesterol and triglyceride levels—which were transformed into binary traits: obesity, hypercholesterolemia and hypertriglyceridemia. We defined hypercholesterolemia and hypertriglyceridemia as values above 200 mg/dL, and obesity as a body mass index above 27.5 for Asian individuals and above 30 for all other ethnicities. All analyses involving epithelial ovarian cancer and breast cancer were restricted to female, while those involving prostate cancer were restricted to male participants. See Supplementary Methods and Table S1 for more information about trait definitions.
Individuals with missing information were excluded after phenotype definition. This included: (i) those with incomplete, ambiguous or unreliable phenotype assignments (e.g., diabetes decision tree classifications), as well as cases with potentially biased phenotypic estimates (e.g., eMERGE participants on statins), or individuals lacking quantitative measures (e.g., cholesterol levels for hypercholesterolemia); (ii) individuals missing either genotype or phenotype data; and (iii) those missing key covariates in the final risk models (see below ‘Building PRS-based disease prediction models’), including age, sex, ancestry principal components or clinical risk factors (e.g., obesity status). This variability is reported in supplementary tables, which provide the number of cases and controls used for each analysis.
GWAS Meta-Analysis
We generated new genome-wide summary statistics for the 30 health-related traits, using trans-ancestry GWAS meta-analysis. Detailed information about all the studies collected is provided in Table S215,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73. We conducted GWAS meta-analyses using METAL74 employing two specific schemes. For effect size estimates (beta coefficients) and standard errors, we adopted the SCHEME STDERR, which processes effect size estimates and standard errors from each study to compute aggregate effects of each variant. Ensuring uniform units for effect sizes across all included summary statistics was essential for consistency. For P value computation, we implemented the default SCHEME SAMPLESIZE, which adjusts P values and directional effects based on sample size. In this context, we calculated the effective GWAS sample size for binary traits using the formula 4 / (1 / Ncases + 1 / Ncontrols) where Ncases and Ncontrols represent the number of cases and controls in the GWAS, respectively. For quantitative traits, we directly used the total number of samples. This dual-mode approach provided flexibility in our analysis, as the SAMPLESIZE model is capable of estimating corrections for sample overlap, thereby accommodating such corrections in meta-analyses where overlaps are inevitable. Consequently, we sourced our P values from ‘SAMPLESIZE metaanalysis’ and beta coefficients from ‘STDERR metaanalysis’, effectively addressing the complexities of our data.
We sourced summary statistics from publicly available GWAS data, such as the GWAS Catalog75. Additionally, we accessed data from specific project platforms such as the Finngen, BioBank Japan and Global Lipids Genetics Consortium. Access was also obtained from dbGaP for the Million Veterans Program (phs001672.v11.p166) and the summary statistics for melanoma (phs001868.v1.p148). To minimize overfitting, we excluded publicly available GWAS summary statistics that included UKB samples, since these involve the entire UKB cohort or an unknown subset, risking overlap with our validation set of 104,604 individuals. Instead, we exclusively used the clearly defined WBU UKB subset described above (see ‘Study Cohorts - UK Biobank Cohort’) in our meta-analysis, explicitly reserving the 104,604 UKB individuals for validation and downstream analyses. We further excluded studies if they presented fewer than 100,000 variants or were confined to a single chromosome or region. Older studies were removed if they were included in a more recent published release or meta-analysis. From several genetic models presented by Guindo-Martínez et al. (2021)27, we selected the additive model for our analyses. We conducted correlation analyses between studies on nominally significant variants to ensure reliable comparison of effects to verify beta direction consistency. Instances of complete inverse or totally non-correlative beta directions indicated potential labeling errors for effect alleles or allele assignments. We noticed that significantly associated SNPs with allele frequencies close to 50% often displayed such inconsistent beta directions in studies that did not correlate with others, implying potential mislabeling of minor alleles as effect alleles in the GWAS data. We harmonized the summary statistics by annotating SNP IDs from dbSNP version 155. Beta coefficients were calculated from logarithms of odds ratios where needed. When missing, standard errors were derived by dividing the absolute beta coefficients by their corresponding z-scores, which were computed from P values using the inverse of the standard normal cumulative distribution. If a P value was below the threshold of 10− 308, we set it to this value as it represents the lower limit that METAL can process as input. Additionally, we performed liftover from GRCh38 to GRCh37 genome build and implemented other necessary harmonization steps to ensure consistency across datasets.
For the PAGE MEC cohort, we excluded Wojcik et al. (2019)26 from the meta-analysis, as this GWAS included samples from the PAGE study, which may overlap with our PAGE MEC dataset. To prevent potential overfitting, we removed this study from the meta-analyses for hypertension, obesity and type 2 diabetes.
PRS algorithm benchmarking
We benchmarked the following PRS algorithms: PRS-CS76, SBayesRC77, PolyPred78, NPS (non-parametric shrinkage)79, lassosum280, LDpred281, maxCT and Stacked C + T (SCT)22. The UKB training set was used to determine the optimal hyper-parameters for PRS-CS, NPS, PolyPred, lassosum2, LDpred2, and maxCT, in addition to the stacking weights for SCT. SBayesRC was trained using only summary statistics, eliminating the need for individual-level data.
To implement PolyPred, we integrated two complementary predictors—PolyFun-pred and SBayesRC outputs—and applied this model to five traits: asthma, age-related macular degeneration, ulcerative colitis, venous thromboembolism, and cardiovascular disease. However, the performance gains from PolyPred were minimal, improving by only about 0.1% over the results obtained from SBayesRC alone. Additionally, PolyPred was computationally demanding, roughly 75 times more resource-intensive than SBayesRC. This increase in computational load is likely due to its extensive linkage disequilibrium (LD) reference data requirements, where SBayesRC utilizes a 50 GB LD panel, in contrast to PolyPred’s 2.9 TB LD matrices. We also evaluated the NPS software for the same set of traits and encountered similar challenges, including the need for over 3 TB of local storage for dosage matrices, prolonged processing times, and comparatively lower accuracy among the PRS methods we reviewed. The artificially high AUC observed during training is typically indicative of model overfitting. Given these findings, coupled with faster and more accurate performance from other methods, we decided against including PolyPred and NPS in our final benchmarking analysis.
For maxCT, tuning parameters included a squared correlation (r²) threshold for clumping and a base size for the clumping window, with default settings specified in Privé et al., (2019)22. Additionally, a series of 50 thresholds on P values were applied, ranging from the least to the most significant in the final meta-analyzed summary statistics, and evenly distributed on a log-log scale. LDpred2’s tuning involved selecting the proportion of causal SNPs from a sequence of 21 logarithmically spaced values ranging from 10− 5 to 1, and inferring the per-SNP heritability chosen from {0.3, 0.7, 1, 1.4} times the total heritability, as estimated by LD score regression. We enabled the ‘sparse’ option to truncate minor effects to zero, which effectively doubled the number of models considered for training by incorporating both sparse and non-sparse models. lassosum2’s tuning parameters included the lasso penalty lambda (L1-regularization), selected from 30 values logarithmically spaced between lambda0 – the maximum value where all coefficients are zero, derived from the maximum absolute standardized beta coefficient – and 1% of lambda0, along with a delta (L2-regularization) parameter for the LD matrix chosen from {0.001,0.01,0.1,1}. We developed the PRS-CS PRS model using default settings for the gamma-gamma prior parameters (a = 1 and b = 0.5) calculated on a per-chromosome basis. We did not specify the global shrinkage parameter phi, allowing PRS-CS to learn phi from the data through a fully Bayesian approach, which is advisable in the context of large GWAS sample sizes such as ours75 SBayesRC was executed using its default settings, enabling the software to determine the optimal estimates for heritability and the count of non-zero effect variants. Additionally, we adjusted the eigen variance cutoffs for model tuning in response to software-generated warnings. The original threshold set of (0.995, 0.99, 0.95, 0.9) was expanded to (0.995, 0.9, 0.8, 0.7, 0.6) to accommodate findings that suggested the optimal tuning parameter was near the minimum threshold, thus necessitating lower cutoff values.
Both LDpred2 and lassosum2 were implemented using the functions snp_ldpred2_grid and snp_lassosum2 from the R package bigsnpr, respectively. We adopted an LD radius of 3 cM to approximate local LD patterns, which assumes that variants further away than this distance are not correlated. Moreover, genetic markers were further restricted to the HapMap3 panel, following recommendations by Privé et al. (2021)81 Conversely, maxCT and SCT were executed using corresponding functions from the same R package (snp_grid_clumping and snp_grid_PRS, and snp_grid_stacking, respectively). However, for these analyses, we limited the set of genetic variants to those achieving a significance level of P < 0.1 in the meta-analysis. PRS-CS scores were also based on HapMap3 sites, as precomputed by the PRS-CS authors. In this case, we opted for data from the UKB as our reference panel for LD after it showed improved results in preliminary testing across several traits compared to those using the 1000 Genomes Project. PRS-CS is a Python based command line tool (available here: github.com/getian107/PRScs). Final PRSs were computed from the SNP weights output using the PLINK --score command. We utilized SBayesRC (available at github.com/zhilizheng/SBayesRC), applying the LD reference supplied by the authors from the UKB. For our analysis, we employed two different sets of SNPs: the HapMap3 set and a larger set consisting of 7 million SNPs, both incorporating functional genomic annotations to enhance polygenic prediction. During an initial testing phase across various traits, the 7 million SNP panel demonstrated superior performance compared to the HapMap3 set. Thus, we selected the 7 million SNP set for benchmarking and further analyses.
The models with the highest prediction accuracy on the training set were selected based on the area under the receiver operating characteristic curve (AUC). The predictive performance of these final models was then evaluated on the independent testing set. Unlike diseases encoded as binary outcomes (case/control), optimal values for the quantitative traits – body mass index, total cholesterol, and triglyceride levels – were initially determined by training models to maximize the predictive R² between observed and predicted traits. Subsequently, these traits were converted to binary outcomes to facilitate AUC calculation and comparison with the testing samples.
Multi-algorithm ensemble PRSs
We developed new ensemble PRS models for each disease by integrating scores previously obtained from SBayesRC, PRS-CS, SCT, LDpred2 and lassosum2. Ensemble models were retrained using logistic regression, employing the second training set to optimize the integration of these scores. This strategy proved to be an effective method of leveraging a larger cohort for training without the need to directly retrain the more computationally intensive and time-consuming PRS algorithms described above, which had been previously calibrated within a smaller, yet adequate cohort, thereby streamlining the overall process.
We tested various training setups to assess the stability of the ensemble model coefficients, ranging from a simpler logistic regression without hyperparameter tuning or cross-validation to more complex models like Naive Bayes and Light Gradient Boosting Machine. As results remained virtually unchanged across models (data not shown), reinforcing the stability of our approach, we selected logistic regression with minimal tuning, which provided a slight performance gain over basic logistic regression while maintaining reasonable computational efficiency.
We optimized the logistic regression models using the glmnet method for binary classification, configured with 5-fold cross-validation to enhance reliability. The models were implemented in R using the caret package. Model training was controlled through the trainControl function, set to compute class probabilities and evaluate model performance based on the AUC metric. Hyperparameters were finely tuned using a grid search across a range of values for alpha (0 to 1, in 11 steps) and lambda (10− 4 to 101, in 50 logarithmic steps), which regulate the balance between L1 and L2 regularization. This approach allowed us to systematically explore the parameter space to maximize the AUC metric, ensuring optimal discrimination of binary (case/control) outcomes.
After model training, the final ensemble model weights were extracted from the best-performing logistic regression model at the optimal alpha and lambda values. To ensure comparability across PRS algorithms, coefficients were adjusted by multiplying them with the standard deviation of each PRS predictor, correcting for variance differences. The adjusted contributions were then normalized to sum to 100%, reflecting each PRS algorithm’s relative importance in the model. Importantly, the weights were data-driven, with the elastic net framework optimizing each PRS method’s contribution based on cross-validation performance.
We compared our ensemble PRS models to those of Thompson et al. (2024)8 and Lennon et al. (2024)14, as examples of cutting-edge models with potential medical actionability and clinical utility. Thompson et al.’s PRS profiles in UKB were retrieved from the UK Biobank Research Analysis platform, while Lennon et al.’s PRS models were accessed from: github.com/broadinstitute/eMERGE-implemented-PRS-models-Lennon-et-al. Thompson et al. provided PRSs exclusively for UKB samples, limiting our comparison to the UKB dataset. Lennon et al. provided PRS models for 10 diseases, eight of which overlapped with ours. Since these models generalized beyond the UKB, we applied them to eMERGE.
Building PRS-based disease prediction models
To improve the predictive accuracy of our PRS models, we first incorporated the first four principal components (PCs) into the logistic regression model. Principal component analysis (PCA) was run on variants that were pruned to ensure linkage equilibrium. We removed strand-ambiguous SNPs A/T and G/C, then filtered the SNP dataset to exclude those with a minor allele frequency (MAF) below 2%, those failing the Hardy-Weinberg equilibrium test with a P value below 1 × 10− 6 and those with over 5% missing data. Then we applied genotype pruning using a window size of 1000 kb, a step size of 50 SNPs, and an r2 threshold of 0.2, which allowed us to capture broad ancestry patterns through PCA.
Next, we integrated ancestry estimates from Orchestra82, a method for local ancestry inference. We aggregated the results from all genomics regions into vectors that quantify the percentage of each ancestry assessed for every individual. This approach allowed us to incorporate a series of columns into our model – each corresponding to a different ancestry, enabling more precise genetic profiling per individual. Orchestra results were provided at level 2, which offers a regional granularity, intermediate between continental and population-specific granularity.
Further, we also incorporated age, sex and other clinical characteristics, easily gatherable in a clinical setting. Age was calculated as the age at the earliest recorded diagnosis. We chose characteristics that are known risk factors for each medically-related trait. Known risk factors were retrieved by doing a literature search, and those that matched one of our 30 traits were added to the model (Table S3). For example, obesity (BMI), hypertension, type 1 and type 2 diabetes were used as risk factors in the cardiovascular disease model.
We employed the previously-described logistic regression model, using grid-search techniques to fine-tune the regularization parameters, thereby ensuring the model’s generalizability.
PRS-based models vs. Rare variants
We used whole exome sequencing data from the European portion of UK Biobank to identify carriers with significant mutations in genes associated with coronary artery disease, breast cancer and bowel cancer. This allowed us to use cumulative incidence plots for carriers within the full UKB cohort, maximizing sample size and statistical power due to the low frequency of these mutations, while minimizing confounding factors related to ancestry between the overall cohort and our UKB testing group, where PRS distribution percentiles were calculated.
We started with a list from Fahed et al. (2020) to pinpoint mutations in genes associated with familial hypercholesterolemia (APOB, LDLR, PCSK9), breast cancer (BRCA1, BRCA2), and colorectal cancer (Lynch syndrome genes: MSH2, MSH6, MLH1, PMS2)2 We expanded this list by including additional mutations identified as likely pathogenic or pathogenic in the ClinVar database. For familial hypercholesterolemia, we incorporated FH variants from Chora et al. (2018)83, which adhered to diagnostic guidelines set by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology84. We also added more ClinVar variants for the APOB gene (search: apob[gene] and Hypercholesterolemia). For breast cancer, we added mutations in the ATM and PALB2 genes, classified as likely pathogenic and pathogenic under the category of familial breast cancer (X[gene] and familial cancer of breast). Additionally, we included the CHEK2 1100delC mutation, known for its significant association with breast cancer risk.
We determined the PRS percentile that matched the risk associated with having a pathogenic mutation. This percentile would indicate that individuals above this threshold have a comparable overall risk level to mutation carriers. To ascertain the accuracy of our findings, we calculated confidence intervals using a binomial test.
Diagnostic odds ratios of PRS-based disease prediction models
We calculated the diagnostic odds ratio (DOR)85 for each PRS-based disease prediction model. To achieve sufficient sample sizes for certain traits, we aggregated East Asians and South Asians from the UKB into a broader ‘Asian’ category. If non-European populations did not meet this threshold, we combined all non-European groups (African and Asian) into a ‘Non-European’ category for the analysis. In addition, we defined high risk groups as the top 1%, 2%, 3%, 5%, 10%, or 20% of the population, and looked for the largest high risk group for which the DOR exceeded the pre-set threshold of 10. We assessed the impact of varying PRS percentile cutoffs on odds ratio calculations for defining high-risk individuals (e.g., top X% of the PRS distribution vs. the remaining population), finding that these variations did not alter the interpretation of the results or the conclusions drawn from our analyses (data not shown).
Statistical analysis
PRS performance was primarily evaluated using AUC as the main metric, ranging from 0.5 (no discrimination) to 1 (perfect discrimination). To estimate AUC and its 95% confidence interval (CI), we sampled 10,000 bootstrap replicates of the testing set and computed AUC for each iteration. The final AUC value corresponds to the mean of these replicates, with CIs derived from the 2.5% and 97.5% quantiles, as implemented in the AUCBoot function from the bigstatsr R package. Additional evaluation metrics included odds ratios (ORs), comparing individuals in the top PRS percentile (high-risk) against the remainder of the population, calculated via logistic regression (glm in R, binomial family). We also computed OR per Standard Deviation (OR per SD) of PRS, as in Thompson et al. (2024), where PRS profiles were normalized and standardized (z-score transformation) before fitting a logistic regression model. Given that our ensemble PRS model is trained via logistic regression, which outputs probabilities rather than a direct risk distribution, we applied a rank-based inverse normal transformation to enforce a normal distribution while preserving rank order. The exponentiated regression coefficient from the logistic model provides the OR per SD of PRS. AUC comparisons between models were performed within each phenotype using DeLong’s test (roc.test function from pROC), applying False Discovery Rate (FDR) correction (p.adjust function) for multiple testing across the 30 traits. Moreover, we assessed overall model performance by counting how often each model outperformed another across traits and tested for statistical significance using a sign test, which assumes no systematic advantage for either method (binom.test in R, null hypothesis: p = 0.5).
Additional statistical analyses included: (i) pairwise correlations between PRS profiles obtained from different algorithms, calculated using Pearson correlation coefficients (cor function in R) and averaged across traits, followed by hierarchical clustering (hclust with Euclidean distance and complete linkage) to generate a dendrogram; (ii) cumulative incidence graphs, estimated as the proportion of individuals diagnosed with a specific trait by a given age, with 95% CIs derived using binomial tests at each data point; and (iii) post-hoc power analysis, where observed AUC in the testing cohort was converted to Cohen’s using ≈ 2 Φ⁻¹(AUC) approximation and power was estimated via pwr.t2n.test from the R package pwr at α = 0.05, confirming that most analyses had power values close to 1. All statistical analyses were performed in R (version 3.6.3; hthttp://www.R-project.org), with visualizations generated using ggplot2 and cowplot. All P values were two-sided, with statistical significance set at P < 0.05.
Results
Trans-ethnic GWAS Meta-analysis and the benchmarking of PRS algorithms
Leveraging trans-ancestry GWAS meta-analyses, we generated novel genome-wide summary statistics for 30 medically-related traits. The sample sizes and contributions of different studies to our meta-analyses are summarized in Figure S1 and Table S2. For binary traits, the median number of cases and controls across all studies was 42,768 and 821,539, respectively. For quantitative traits, the median sample size was 1,205,118 individuals. Compared to the UK Biobank (UKB) alone, the meta-analyses have increased the number of cases for the binary traits 6.4 times and the number of samples for quantitative traits 7 times. Key contributors to the meta-analyses of binary traits included the FinnGen project (26.5% of cases), the UKB (15.73%) and the BioBank Japan (4.9%). Major contributors to the meta-analysis of quantitative traits included the Global Lipids Genetics Consortium (67.8% of the samples), the UKB (14.4%), the FinnGen project (11.8%) and the BioBank Japan (4.7%).
We trained six PRS algorithms: PRS-CS76, SBayesRC77, lassosum280, LDpred281, maxCT and Stacked C + T (SCT)22 to benchmark their performance and scrutinize each method’s strengths and weaknesses. A subset of UKB individuals was used as the cohort for training and testing (see Methods, Fig. 1A). Although we initially also trained PolyPred78 and NPS (non-parametric shrinkage)79, these were excluded from the final benchmarking because their performance did not enhance the results compared to the other methods, in addition to having longer computational training times (data not shown). Table S4 summarizes each method’s tuning strategy, computational demand, strengths and limitations. PRS performance was evaluated using the area under the receiver operating characteristic curve (AUC), with the best results for each trait ranging from 83.4% (type 1 diabetes) to 55.9% (epithelial ovarian cancer), and a median of 68.1% (Fig. 2A; Table S5). SBayesRC outperformed the other algorithms for 21 out of 30 medical traits, 11 of which were statistically significant. In contrast, SCT and LDpred2 were superior in three traits each, with two of the SCT traits reaching statistical significance. Lassosum2 and PRS-CS performed the best in two and one trait, respectively (Fig. 2A and B). When we relaxed our criterion to identify phenotypes where two algorithms significantly outperformed the other four PRS algorithms (rather than just one method surpassing all others), we identified six additional traits where SBayesRC was one of the two top-performing algorithms. We note that SCT performed well in cardiovascular traits, such as coronary artery disease, atrial fibrillation and hypertriglyceridemia, while LDpred2 and lassosum2 showed stronger performance in neuropsychiatric and gastrointestinal conditions, including Alzheimer’s disease, bipolar disorder, schizophrenia, Crohn’s disease and bowel cancer.
When we checked the pairwise correlations of PRS models between algorithms, we found high correlation between lassosum2 and LDpred2, as well as between LDpred2 and the other two Bayesian regression-based PRS algorithms (PRS-CS and SBayesRC), whereas correlations with SCT were less pronounced (Fig. 2C). Therefore, we hypothesized that integrating insights from different algorithms would lead to enhanced predictive accuracy.

Benchmarking of six PRS prediction algorithms applied to 30 medically-related traits. (A) Prediction accuracy using area under the curve (AUC) as a performance metric in the testing set of UKB. Conditions marked with an asterisk indicate those where SBayesRC or SCT significantly outperformed the other PRS algorithms. Conditions identified with a dagger represent a relaxed criterion where two algorithms significantly exceed the performance of the others. Error bars present the mean and 95% confidence interval of 10,000 non-parametric bootstrap replicates. DeLong’s test was used to compare the significant difference between AUC results across methods. Performance for breast and epithelial ovarian cancer was calculated using only female, and prostate cancer using only male individuals. (B) Prediction results summarized per algorithm. Each dot represents one trait. The numbers at the top of the plot denote the count of traits where each PRS method demonstrated optimal performance, as indicated by highlighted dots. (C) Mean Pearson correlation coefficient (r) for PRS across the 30 traits between pairs of algorithms, along with a dendrogram depicting hierarchical clustering.
Ensemble PRSs and their performance in UK biobank, eMERGE and PAGE MEC cohorts
Building on the idea of integrating outputs from multiple PRS algorithms, we retrained an ensemble PRS model using logistic regression, incorporating the scores previously obtained from SBayesRC, SCT, LDpred2, PRS-CS and lassosum2 (see Fig. 1B, Methods). We did not include maxCT, since SCT is already a stacked version derived from different clumping plus thresholding models. We assessed the performance of the newly constructed ensemble models by comparing them to SBayesRC, which was identified as the top-performing individual PRS algorithm in our prior evaluations. Additionally, we contrasted the results derived from meta-analyzed summary statistics with those obtained solely using the UKB WBU GWAS component for PRS training with SBayesRC. These findings were benchmarked against the PRSs reported by Thompson et al. (2024)8 and Lennon et al. (2024)14.
Our ensemble model significantly outperformed SBayesRC in the UKB testing cohort, leveraging both UKB-only and meta-analysed summary statistics (Fig. 3A and C; Table S6). The ensemble method exceeded the SBayesRC algorithm for 23 out of 30 traits (P = 0.005, sign test). Moreover, our results proved superior to those reported by Thompson et al. (2024) for 26 traits (P = 6 × 10− 5, sign test), demonstrating significant improvements in AUC for 22 of these traits (DeLong’s test for AUC, P < 0.05), with median AUC improvement of 5.07% (Table S7). The only exception was Parkinson’s disease, where Thompson et al. achieved significantly better results, albeit with a minor difference (P = 0.03, DeLong’s test). PRS analyses based only on summary statistics from the UKB WBU set yielded the poorest performance relative to other strategies (median UKB-only AUC 62.9% vs. 66.9% with meta-analysis and 68.8% with the ensemble method). However, results exclusively based on the UKB data showed marginally better outcomes for celiac disease. This negligible improvement may be attributable to random variation within the analysis.
To additionally validate the performance of our ensemble PRS models, we applied them to the diverse cohorts of eMERGE and PAGE MEC. The UKB WBU summary statistics exhibited the poorest outcomes compared to other approaches for both eMERGE and PAGE MEC, followed by better performances achieved through meta-analysis with SBayesRC and our ensemble models (Figs. 3C and S2; Table S6). Our ensemble models significantly outperformed those by Lennon et al. (2024) for six out of eight traits (DeLong’s test for AUC, P < 0.05), achieving a median improvement in AUC of 1.97% (Fig. 3B; Table S7). However, the significant signal for asthma in eMERGE was lost after FDR adjustment (P = 0.058).
To further evaluate PRS performance, we employed two additional metrics: odds ratio (OR) per standard deviation (SD) of PRS and a comparison between individuals in the top 20% of the PRS distribution, identified as high-risk, versus the remaining population. The results were consistent with our primary findings (Figures S3 and S4; Table S7). Specifically, our approach outperformed Thompson et al. (2024) for 26 out of 30 traits using OR per SD, and for 25 out of 30 traits using OR at the top 20%. Similarly, our PRS models surpassed Lennon et al. (2024) for seven out of eight traits using OR per SD and for eight out of eight using OR at the top 20%.

Predictive performance of PRS models in the UK Biobank, eMERGE and PAGE MEC cohorts. (A) Predictive performance of SBayesRC with UKB-only summary statistics (grey), SBayesRC with meta-analysis summary statistics (light green), and the ensemble PRS with meta-analysis summary statistics (dark green). Prediction accuracy was measured using area under the curve (AUC). Error bars indicate 95% confidence intervals of 10,000 non-parametric bootstrap replicates. Results were compared against AUC obtained with PRSs from Thompson et al. (2024) (pink line and shaded area as 95% CI). Green asterisk indicates traits where our ensemble method performed significantly better. Pink asterisk indicates a significantly better result for Thompson et al. DeLong’s test was used to compare the significant difference between AUC results. Performance for breast and epithelial ovarian cancer was calculated using only female, and prostate cancer using only male individuals. (B) Similar to A), PRS models were compared to Lennon et al. (2024) models in the eMERGE cohort. Significant signal in eMERGE for asthma is lost after FDR adjustment (P = 0.058). (C) Risk prediction results summarized per PRS study and cohort.
Subsequently, we analyzed the performance across different ancestries in UKB, eMERGE and PAGE MEC. The analysis was limited to populations exhibiting more than 50 cases of the trait, to ensure precise estimates. Our findings were comparable to those of Thompson et al. (2024) and Lennon et al. (2024) (Figure S5). As expected, we observed a decline in PRS accuracy for non-European ancestries. For instance, Latinos in eMERGE had a median decrease of 2.4% AUC, while Africans experienced the largest reductions of 4.7% in eMERGE and 7.6% in UKB (Fig. 4; Table S8). The smallest drops were observed in East Asians in UKB, with a 1.7% median decrease in AUC, and Asians from eMERGE, who exhibited a decrease of 1.4%, potentially reflecting the significant representation of Japanese samples from the BioBank Japan in our meta-analysis.
Furthermore, we evaluated the impact of applying our ensemble PRS models, developed on UKB data, to other cohorts. Comparing performance by ancestry for each trait in UKB vs. eMERGE or PAGE MEC, we found expected performance drops, however none exceeded 4% median reduction—specifically 3.6% for Asians and 3.2% for Europeans. No drop for Africans was observed (Fig. 4).
When we checked the contribution of each PRS algorithm to our ensemble framework, SBayesRC emerged as the most influential component, accounting for 43.7% of the ensemble weight on average. This was followed by SCT with 17.3%, and LDpred2 with 15.7%. PRS-CS and lassosum2 had comparatively lower weights of 12.5% and 10.8%, respectively. Notably, the allocation of weights closely aligned with the performance rankings of the algorithms observed in our preliminary benchmarking assessments (Figure S6; Table S9).

Drop in predictive performance of PRS models across ancestries and cohorts. (A) Relative percentage change in performance (AUC) in non-European groups compared to Europeans. (B) Relative percentage change in performance (AUC) in eMERGE and PAGE MEC compared to accuracy obtained in the UK Biobank per ancestry group. Only ancestry groups with at least 50 cases for each clinical condition were included into the analysis.
PRS-based disease prediction models
We proceeded to enhance our ensemble PRS models by incorporating additional information, starting with ancestry, which led to a marginal improvement in overall accuracy (Fig. 5A and B; Table S10). Specifically, adding only the PCs to the baseline (PRS-alone) model enhanced performance for 24 out of 30 traits analyzed, with a median AUC improvement of 0.3%. The addition of more granular ancestry information resulted in further accuracy gains for 22 traits, with an additional median AUC enhancement of 0.1% beyond PCs alone. Collectively, ancestry information improved accuracy estimates for 26 traits, a significant albeit subtle enhancement (P = 0.00006, sign test).
While creating predictive risk models solely based on genetic information is a good way to test and benchmark various PRS models, in a real world clinical setting, the addition of non-genetic information to the final model is needed to increase performance. With this in mind, we incorporated other easily obtained clinical characteristics – sex, age, and known risk factors (Table S3) – into a final logistic regression model that would serve as a prospective diagnostic test, with easily interpretable positive or negative value, typical of other diagnostic tests used in a clinical setting. We obtained improvements in a large subset of the traits tested, with 12 models surpassing the 80% AUC accuracy range. Including age and sex to the previous model improved performance for all but one trait, with a median AUC improvement of 8.4%. The addition of risk factors (where available) added a modest extra 1.3% on average to the final performance. Specifically, 16 out of 21 conditions with defined risk factors improved their accuracy mark (P = 0.03, sign test).
When we look at the contributions of individual features to the full risk models, PRS proved to be the predominant factor, contributing roughly one-third of the predictive weight on average (Fig. 5C). Although the addition of ancestry information to the models did not substantially enhance performance over PRS alone, up to 22% of the model’s weight was attributable to ancestry, suggesting that our PRS model may have partially captured some ancestry effects. Age was second only to genetics, accounting for an average of 25% of the model’s weight. Gender and other risk factors were less influential, contributing 8.8% and 10.6% respectively.
Significant variability was evident across traits (Figure S7; Table S11). For instance, several conditions which predominantly manifest later in life showed a higher weighting for age, such as Alzheimer’s disease (74% of total weight), age-related macular degeneration (71%), Parkinson’s disease (64%), and prostate cancer (51%). Conditions where there is a known gender bias had a higher weighting for gender, e.g. systemic lupus erythematosus (37% of total weight), osteoporosis (32%), total cholesterol (29%), rheumatoid arthritis (28%), and coronary artery disease (20%). Cardiovascular-related conditions such as hypertension and coronary artery disease, along with other conditions like venous thromboembolism, atrial fibrillation, and ischemic stroke, displayed a higher dependency on risk factors such as body mass index, pre-existing hypertension and prior cardiovascular events. Type 2 diabetes was impacted by body mass index.

Predictive performance of risk models that incorporate ancestry, age, sex and risk factor information. (A) Prediction accuracy was measured using AUC in the UKB testing set. Bars indicate 95% confidence intervals of 10,000 non-parametric bootstrap replicates. Models shown include performance with: ensemble PRS alone; the addition of ancestry PCs, ancestry with Orchestra for local ancestry deconvolution in addition to PCs; sex and age in addition to ancestry, and relevant risk factors in addition to all other information. (B) Prediction results summarized per risk model where each dot represents a trait. If a trait lacked relevant risk factors, the accuracy of the final comprehensive model equaled that of the preceding model, which included only prs, ancestry, age and sex. (C) Average weights per feature in the final logistic regression model across 30 studied traits.
Several clinical conditions demonstrated a substantial reliance on ancestry. Examples include Crohn’s disease (93% total weight), celiac disease (60%), and type 1 diabetes (47%), which are known to be more prevalent in Europeans; ischemic stroke (37%) and schizophrenia (31%), which exhibit higher rates in African-Americans; or type 2 diabetes (30%), which has a higher prevalence in non-Europeans. Melanoma, bipolar disorder, and breast cancer were the diseases most affected by genetics (PRS) in our models. Particularly, the melanoma risk model was almost entirely based on PRS, achieving an accuracy of nearly 80% with PRS alone.
PRS-based disease prediction models vs. Rare variants
Evidence from cumulative incidence curves, including the UKB (Figure S8) and additional cohorts like eMERGE and PAGE MEC (Figure S9), supports the notion that higher PRSs correlates with a lifelong increased probability of developing health conditions. However, current health systems prioritize rare mutations that confer significant risks for disease development, while neglecting PRSs that can aggregate comparable risks through the accumulation of hundreds to thousands of common variants with minor effects. Indeed, for numerous prevalent diseases, genes have been discovered where rare mutations significantly increase the risk, often several-fold, for carriers who are heterozygous86 In line with Thompson et al. (2024)8, we analyzed and contrasted the risk profiles of rare mutations and PRSs, using cardiovascular disease, breast cancer and bowel cancer as illustrative case studies.
We identified carriers of high-risk mutations in key functional genes in UKB participants’ whole exome sequencing data and aimed to match their risk profiles by selecting individuals within the top percentiles of higher risk based on their PRS profiles. For example, the prevalence of coronary artery disease among carriers of pathogenic mutations in genes associated with familial hypercholesterolemia (APOB, APOE, LDLR and PCSK9) was approximately 12% by age 70 in our testing cohort. A similar lifetime risk was noted among participants in the top 20% of the PRS distribution outlined by Thompson et al. (2024)8 for this trait, consistent with what they reported. Our PRS ensemble model identified even more individuals – up to 22% of those in the top distribution – as having an equivalently high risk (Fig. 6A). Remarkably, the high risk PRS group identified 55 to 80 times more true coronary artery disease events than those detected among high-risk variant carriers, depending on the age group analyzed (Fig. 6B). This incidence surpassed the one reported by Thompson et al., which identified between 50 and 73 times the cases found in carriers. We found that rare pathogenic mutations contributed more significantly to early-onset disease, evidenced by a lower ratio of high PRS individuals to rare mutation carriers among those diagnosed before age 50. We also explored the interplay between monogenic risk variants and PRS profiles, categorizing individuals into low, intermediate, or high PRS risk groups. The risk among mutation carriers varied from a 1.22-fold odds ratio (OR) in the lowest PRS risk group to 7.05 OR in the highest PRS risk group, compared to non-carriers with an intermediate PRS (Fig. 6C). This illustrates how PRS can modulate the effects of high-risk variants typically evaluated in clinical settings. Parallel insights emerged when examining breast cancer in relation to mutations in BRCA1, BRCA2, ATM, PALB2, and CHECK2, and bowel cancer in relation to rare variants in MSH2, MSH6, MLH1, and PMS2 (Figure S10).
Towards the clinical implementation of PRS-based disease prediction models
Many of the PRS-based disease prediction models we have developed demonstrated sufficient overall accuracy to potentially merit being used in a clinical setting. To further elucidate the effectiveness of these models as diagnostic tests, we calculated the diagnostic odds ratio (DOR)85 for each model. As can be seen in Fig. 6D and E, DORs ranged from a low of 3.04 for obesity in Africans to a high of 411.67 for type 1 diabetes in non-Europeans (Table S12). However, when we restricted our observations to condition-ancestry pairs that had 100 or more cases, the highest DOR of 66.2 was observed for Alzheimer’s disease in Europeans. The DOR across all ancestry groups exceeded five for 25 out of 30 traits, and 10 for 19 out of 30 traits, suggesting that the majority of the tests were highly predictive.

Towards the clinical implementation of PRS-based disease prediction models. Comparison between PRS risk profiles and functional variant carriers in UKB Europeans. (A) Cumulative incidence of coronary artery disease among carriers with pathogenic or likely-pathogenic mutations in familial hypercholesterolemia genes vs. individuals in the top 22% of our PRS distribution from the ensemble method, where a percentile was chosen to match the risk up to age 70 with that of mutation carriers. The median 25%-75% of the PRS distribution serves as the population reference. (B) Percentage of coronary artery disease (CAD) cases diagnosed in individuals aged under 50, under 65, and across all age groups, occurring either in mutation carriers or in those at the top of the PRS distribution. Plots display the ratio of high risk PRS cases to mutation carrier cases in each age category. (C) Forest plot displays odds ratios for the observed risk in coronary artery disease. Horizontal lines represent 95% confidence intervals. Individuals were categorized into three groups based on their polygenic scores – low, intermediate, and high, corresponding to the lowest 25%, middle 25–75%, and highest 25% of the distribution, respectively. The intermediate PRS group served as the reference category for calculating odds ratios. (D) Diagnostic test effectiveness of PRS-based disease prediction models. Odds ratios are reported per ancestry group, defined as the ratio of the odds that an individual with a clinical condition will score above a specified risk threshold, to the odds of scoring below this threshold without the condition. Error bars represent 95% confidence intervals. (E) ‘High risk threshold’ illustrates the percentile cutoff for a specific condition, above which results are considered high-risk. Odds ratios were optimized by looking for the largest high risk group for which the DOR exceeded the pre-set threshold of 10 using cutoffs at the top 1%, 2%, 3%, 5%, 10%, and 20% percentiles.
Discussion
Polygenic risk scores (PRSs) are at the frontier of personalized medicine, with a potential to predict an individual’s susceptibility to disease, enable population-level risk stratification and provide targeted interventions for those at high risk1,2,3,6,7,9. This could lead to refined screening protocols, more precise preventive measures and tailored therapeutic strategies, ultimately improving clinical outcomes. However, the utility of PRS is currently hampered by several limitations. Chief among these is the reduced predictive power in diverse populations due to the majority of GWAS being conducted in individuals of European ancestry1,8. Additionally, the integration of PRS into clinical practice faces challenges, including the need for validated models that have the same level of accuracy and utility of traditional diagnostic tests used in the clinic9,10.
With those issues in mind, we first generated novel genome-wide summary statistics for 30 medically-related traits, leveraging trans-ancestry GWAS meta-analyses of diverse cohort data, including datasets such as the UKB, the FinnGen project, BioBank Japan, and the Global Lipids Genetics Consortium. Meta-analyses often yielded notable improvements in PRS accuracy. We believe there are two main reasons for that: (1) the meta-analyses had increased power due to a higher number of cases compared to UKB, on average 6.4 times greater for binary traits and 7 times greater for quantitative traits; and (2) the meta-analyses increased the representation of non-European ancestries where such data was available. Prior research shows that including diverse ancestries often enhances trait-association. This may be attributed to better tagging of true casual variants, due to more heterogeneous allele frequencies and linkage disequilibrium patterns in the data87. We should note, however, that performing meta-analyses is not without hurdles. Data harmonization was often challenging, with datasets in different formats, some of those with incomplete or missing information. It is our hope that, as more GWAS summary statistics become available, better standardization and integration practices will follow.
Next, we benchmarked six PRS algorithms and found that SBayesRC had the overall best performance. Leveraging functional genomic annotations in SBayesRC may play a critical role in distinguishing causal from non-causal variants, boosting PRS accuracy77. SCT performed well in cardiovascular traits, while LDpred2 and lassosum2 showed stronger performance in neuropsychiatric and gastrointestinal conditions. Notably, the simplified maxCT method, an optimized version of classical clumping and thresholding, delivered competitive results, making it an attractive choice for resource-constrained settings. A key advantage of SBayesRC may be that it exclusively utilizes summary statistics from GWAS without depending on a training cohort with individual-level data for tuning hyper-parameters. In contrast, many other PRS algorithms still require tuning parameters via grid searches on external datasets, which can compromise statistical power by requiring split testing samples. Methods like LDpred2 and PRS-CS also offer auto-tuned options that remove the need for a dedicated training cohort; however, their authors still recommend using a small subset of the target population to improve accuracy. We did not explore these auto-tuned setups here, but future work could assess whether doing so enhances model generalizability across diverse cohorts. SCT is also known to gain predictive power with larger training cohorts22, suggesting that future work could explore how performance scales with sample size – particularly in scenarios where other methods may plateau. SCT can also be extended to account for other parameters, such as allele frequency, potentially rivaling SBayesRC, though at a higher computational cost. Including quantitative traits would further broaden comparative insights, however, such analyses were beyond the scope of the present study. It is important to note that SBayesRC and PRS-CS use UKB as an LD reference panel. It is possible that this may inadvertently inflate the results in UKB, and may be partially responsible for the drop in accuracy when applied to eMERGE and PAGE MEC datasets. However, a meta-analysis incorporating multiethnic datasets would likely alter the LD configuration compared to UKB alone. Furthermore, despite PRS-CS also using UKB LD as a reference, it still did not achieve the same accuracy levels as SBayesRC.
To assist researchers in method selection based on available resources and study context, we have summarized each method’s tuning strategy, computational demand, strengths and limitations in Table S4. For groups with limited computational capacity, SBayesRC (using the 1 M HapMap SNP set) remains a strong standalone option, and combinations like SBayesRC + SCT (even under strict P value filtering to control resource usage) may offer an efficient and complementary ensemble strategy, as supported by low correlation patterns (Fig. 1C).
To harness the strengths of each individual method and enhance overall predictive accuracy, we next trained an ensemble PRS model using logistic regression. The main drawbacks of ensemble models are reduced interpretability and the assumptions underlying weight optimization. Unlike individual PRS algorithms, where the contribution of each variant is explicit, the ensemble model integrates multiple PRS algorithms, making it more challenging to isolate the precise influence of each component. Additionally, the logistic regression framework assumes linear contributions from each PRS method, which may not fully capture complex interactions between models. In fact, while cross-validation helps mitigate overfitting, using five correlated PRS profiles as model inputs could introduce unintended biases. However, if designed carefully, we demonstrate that the ensemble approach effectively balances these trade-offs, leveraging the strengths of multiple PRS methods to achieve notable improvements in predictive performance. Our ensemble PRSs performed better than the SBayesRC algorithm for 77% of the traits in our UKB testing cohort. Moreover, our results were superior in AUC and OR per SD to those reported by Thompson et al. (2024)8 for 87% of the traits, achieving a median AUC improvement of 5.07%. Using eMERGE, our ensemble PRSs significantly outperformed those by Lennon et al. (2024)14 for six out of eight traits when using AUC as a metric, for seven out of eight traits when it comes to OR per SD, and for all eight traits when we measured OR at the top 20%. We used the models developed by Thompson et al. as examples of the most recent, highly optimized PRSs that outperformed over 70 other published PRS models8 Similarly, we compared our models to Lennon et al., which are cutting edge models developed for clinical implementation in diverse populations14.
Improvements over Thompson et al. (2024) and Lennon et al. (2024) can likely be partially attributed to larger sample sizes and more diverse datasets included in the meta-analyses, with further gains achieved by our ensemble PRS model. When it comes to sample size, specifically, our study achieved a 1.25-fold increase in controls and a 1.22-fold increase in cases compared to Thompson et al. Twenty-two out of the 30 clinical conditions had a higher number of cases, notwithstanding some overlap in the meta-analyzed cohorts. Only three traits, epithelial ovarian cancer, Alzheimer’s disease and breast cancer, had fewer cases and controls simultaneously compared to those reported by Thompson et al. The results were particularly poor for epithelial ovarian cancer within our UKB validation cohort. For Alzheimer’s disease and breast cancer, although our initial outcomes were weaker, the application of our ensemble method yielded improvements that rivaled Thompson et al.‘s results. Our ensemble approach proved efficient by training PRS algorithms on a computationally manageable cohort and then combining them within a larger training dataset, optimizing the computational workflow while achieving greater accuracy, an approach made feasible by the simplicity and scalability of logistic regression.
It is important to note that methodological differences between our study and those cited, such as variations in phenotype definitions and the cohorts used for evaluation, may contribute to discrepancies in published accuracy metrics. For instance, the results by Lennon et al.14 were derived from a smaller eMERGE cohort consisting of 2,500 individuals, whereas our evaluation used a substantially larger sample from the same project. In this regard, AUC metric tends to be more robust with larger sample sizes, which is essential particularly in contexts of conditions with low prevalence. Although this likely contributed to the minor discrepancies in accuracy metrics reported across the studies, we believe the significance of our findings remains robust.
When applying our ensemble PRS models, developed on UKB data, to eMERGE or PAGE MEC cohorts, the drop in performance did not exceed a 4% median reduction. This suggests that our ensemble PRS model was well calibrated. As previously noted88, we observed a decline in PRS accuracy for non-European ancestries. The current overrepresentation of European-ancestry cohorts inevitably leads to more robust estimates and greater PRS predictive performance in that group. In our data, 78.7% of UK Biobank participants were of European ancestry, even after maximizing non-European representation in PRS training and testing. Similarly, 82.3% of eMERGE participants were European, dominating the overall signal in global evaluations. Notably, not all non-European groups were equally affected: while individuals of African ancestry, genetically more divergent from Europeans, exhibited more pronounced accuracy losses, Asians and Latinos showed only modest reductions in PRS performance. This was likely bolstered by GWAS sources like the Japan BioBank and FinnGen. Finns, known to have a distinctive Siberian heritage, may have contributed to refining effect-size estimates by leveraging finer population structure differentiation, improving trans-ancestry PRS performance. Ultimately, these observations reinforce the need for larger and more diverse GWAS to broaden PRS applicability across all global ancestries. Future research might involve re-weighting underrepresented groups in PRS training – though at the risk of sacrificing the statistical power afforded by larger European samples and potentially overcompensating smaller cohorts, introducing new biases. Recent methods like PRS-CSx89 offer promising alternatives through cross-ancestry PRS tuning, and we plan to explore such methods going forward.
Next, we incorporated ancestry information and other easily obtained clinical characteristics: sex, age and known risk factors, to make a final logistic regression model that would serve as a prospective diagnostic test. This resulted in an improvement in a large subset of the traits tested, with 12 models surpassing the 80% accuracy range with the AUC metric. Age and sex improved performance for all but one trait, with a median AUC improvement of 8.4%. Addition of risk factors, where available, added an extra 1.3% on average, while ancestry information improved accuracy by a modest 0.4% on average. Although the addition of ancestry information did not substantially enhance performance over PRS alone, when we look at the contributions of individual features to the full risk model, up to 22% of the model’s weight was still attributable to ancestry, suggesting that our PRS ensemble model might have implicitly captured the effects of ancestry. This is presumably because each algorithm in the ensemble optimizes differently across ancestry groups, collectively yielding a slight accuracy boost through the integration of various PRS models tailored to different ancestries. Notably, local ancestry inference had a more pronounced contribution than traditional PCs, suggesting that detailed ancestry insights might lead to greater predictive accuracy.
It is important to highlight the variability in the contributions of individual features to the model that was evident across traits. We observed higher contribution of genetics (PRS) to models such as melanoma, age to models such as Alzheimer’s disease, gender to models such as systemic lupus erythematosus, ancestry to models such as Crohn’s disease and known risk factors to models such as coronary artery disease. We conclude that depending on the condition, incorporating easily measured clinical factors alongside genetic data into predictive models, can be an easy way to increase model accuracy, even without considering environmental variables such as smoking status or lifestyle.
Finally, we compared the predictive accuracy of our PRS models to that obtained by looking at rare pathogenic variants, for three well studied conditions: coronary artery disease, breast cancer and bowel cancer. For coronary artery disease, our PRS model was able to identify between 55 and 80 times more true coronary artery disease events than models using rare pathogenic variants. Rare pathogenic mutations seemed to contribute more to early-onset disease, while more common genetic variants with modest effects, as captured by PRSs, seemed to contribute relatively more to late-onset disease cases, in line with Thompson et al.8. When we look at the interplay between rare pathogenic risk variants and PRS profiles, the risk among rare pathogenic mutation carriers varied from a 1.22-fold odds ratio in the lowest PRS risk group to 7.05 in the highest PRS risk group. Similar patterns were observed for breast and bowel cancer. This underscores the critical need to consider all genetic components and further suggests that the polygenic component can modulate the effects of high-risk variants typically evaluated in clinical settings. Moreover, after applying our PRS-based disease prediction models as diagnostic tests, we found that their effectiveness, measured by diagnostic odds ratios, exceeded five for 25 out of 30 traits, and 10 for 19 out of 30 traits across all ancestry groups, suggesting that the majority of the tests were highly predictive.
With PRS models on the cusp of entering the clinic, some of the remaining barriers to wide-scale clinical implementation include a need for (1) clinical guidelines for the interpretation of PRS-based models, (2) guidelines for actionable and effective disease prevention strategies for those with high risk scores, and (3) robust regulatory frameworks to ensure that PRS-based risk assessments are used safely and effectively. Incorporating the PRSs into disease prediction models with clear binary outcomes can hopefully help simplify the interpretation and understanding of these genetic models and ease their more wide-scale adoption. With groups already working on pipelines for clinical implementations and frameworks for regulatory compliance14, we are a step closer to using PRS in a clinical setting across diverse populations.
In this paper, we have optimized PRS models both by increasing GWAS power through meta-analysis and by using ensemble models that leverage the best features of individual PRS models. In addition, we showed the importance of integrating a variety of data types to systematically improve diagnostic accuracy. We demonstrated that many of our PRS-based disease prediction models have sufficient accuracy to warrant consideration of being used in a clinical setting. An important next step would be to fully validate the entire process from instrumental genotype measurements to final classification in individuals that do not belong to cohorts used for training and validation in this paper. Further, we are working on models that would provide a unified measure of genetic risk, combining information from both PRSs and rare high impact variants. Finally, applying these models in clinical trials is needed to demonstrate real-world benefits. With ever increasing GWAS sample sizes, refinements, and improvements in PRS algorithms, we expect that PRS-based disease prediction models will further improve with time. Here, we have shown that PRS-based disease prediction models already have sufficient accuracy and portability to warrant consideration of being used in the clinic.
Data availability
This study used the openly available GWAS catalog, FinnGen and deCODE genetics datasets and the UK Biobank, dbGaP and The Million Veterans Program datasets that are available to researchers upon application. UK Biobank research was conducted under application number #84038. dbGaP access was obtained for phs001868.v1.p1 (Landi et al., 2020), phs001584.v2.p2 (eMERGE cohort), phs000220.v2.p2 (PAGE MEC cohort) and phs001672.v11.p1 (The Million Veterans Program). All data produced in the present study are available upon request to the corresponding author.
References
Lewis, C. M. & Vassos, E. Polygenic risk scores: from research tools to clinical instruments. Genome Med. 12, 44. https://doi.org/10.1186/s13073-020-00742-5 (2020).
Fahed, A. C. et al. Polygenic background modifies penetrance of Monogenic variants for tier 1 genomic conditions. Nat. Commun. 11, 3635. https://doi.org/10.1038/s41467-020-17374-3 (2020).
Khera, A. V. et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to Monogenic mutations. Nat. Genet. 50, 1219–1224. https://doi.org/10.1038/s41588-018-0183-z (2018).
Wray, N. R. et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668–681. https://doi.org/10.1038/s41588-018-0090-3 (2018).
Mars, N. et al. The role of polygenic risk and susceptibility genes in breast cancer over the course of life. Nat. Commun. 11, 6383. https://doi.org/10.1038/s41467-020-19966-5 (2020).
Mavaddat, N. et al. Polygenic risk scores for prediction of breast Cancer and breast Cancer subtypes. Am. J. Hum. Genet. 104, 21–34. https://doi.org/10.1016/j.ajhg.2018.11.002 (2019).
Riveros-Mckay, F. et al. Integrated Polygenic Tool Substantially Enhances Coronary Artery Disease Prediction. Circ. Genom Precis Med. 14, e003304. https://doi.org/10.1161/CIRCGEN.120.003304. (2021).
Thompson, D. J. et al. A systematic evaluation of the performance and properties of the UK biobank polygenic risk score (PRS) release. PLoS One. 19, e0307270. https://doi.org/10.1371/journal.pone.0307270 (2024).
Torkamani, A., Wineinger, N. E. & Topol, E. J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 19, 581–590. https://doi.org/10.1038/s41576-018-0018-x (2018).
Sud, A. et al. Realistic expectations are key to realising the benefits of polygenic scores. BMJ 380, e073149. https://doi.org/10.1136/bmj-2022-073149 (2023).
Hingorani, A. D. et al. Performance of polygenic risk scores in screening, prediction, and risk stratification: secondary analysis of data in the polygenic score catalog. BMJ Med. 2, e000554. https://doi.org/10.1136/bmjmed-2023-000554 (2023).
Albiñana, C. et al. Multi-PGS enhances polygenic prediction by combining 937 polygenic scores. Nat. Commun. 14, 4702. https://doi.org/10.1038/s41467-023-40330-w (2023).
Wang, Y., Tsuo, K., Kanai, M., Neale, B. M. & Martin, A. R. Challenges and opportunities for developing more generalizable polygenic risk scores. Annu. Rev. Biomed. Data Sci. 5, 293–320. https://doi.org/10.1146/annurev-biodatasci-111721-074830 (2022).
Lennon, N. J. et al. Selection, optimization, and validation of ten chronic disease polygenic risk scores for clinical implementation in diverse populations. Nat. Med. 30, 480–487. https://doi.org/10.1038/s41591-024-02796-z (2024).
Bycroft, C. et al. The UK biobank resource with deep phenotyping and genomic data. Nature 562, 203–209. https://doi.org/10.1038/s41586-018-0579-z (2018).
McCarty, C. A. et al. The eMERGE network: a consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med. Genomics. 4, 13. https://doi.org/10.1186/1755-8794-4-13 (2011).
Hofmeister, R. J., Ribeiro, D. M., Rubinacci, S. & Delaneau, O. Accurate rare variant phasing of whole-genome and whole-exome sequencing data in the UK Biobank. Nat. Genet. 55, 1243–1249. https://doi.org/10.1038/s41588-023-01415-w. (2023).
Picard v.2.26.7 v.3.0.0 (The Broad Institute Picard, & 2021). (2020). https://broadinstitute.github.io/picard/
Danecek, P. et al. Twelve years of SAMtools and BCFtools, GigaScience 10, giab008. (2021). https://doi.org/10.1093/gigascience/giab008
Browning, B. L., Zhou, Y. & Browning, S. R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 103, 338–348. https://doi.org/10.1016/j.ajhg.2018.07.015. (2018).
Kolonel, L. N. et al. A multiethnic cohort in Hawaii and Los Angeles: baseline characteristics. Am. J. Epidemiol. 151, 346–357. https://doi.org/10.1093/oxfordjournals.aje.a010213 (2000).
Privé, F., Vilhjálmsson, B. J., Aschard, H. & Blum, M. G. B. Making the most of clumping and thresholding for polygenic scores. Am. J. Hum. Genet. 105, 1213–1221. https://doi.org/10.1016/j.ajhg.2019.11.001 (2019).
Kurki, M. I. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613, 508–518. https://doi.org/10.1038/s41586-022-05473-8 (2023).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 53, 1415–1424. https://doi.org/10.1038/s41588-021-00931-x (2021).
Kanai, M. et al. Insights from complex trait fine-mapping across diverse populations. medRxiv (2021). https://doi.org/10.1101/2021.09.03.21262975
Wojcik, G. L. et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature 570, 514–518. https://doi.org/10.1038/s41586-019-1310-4 (2019).
Guindo-Martínez, M. et al. The impact of non-additive genetic associations on age-related complex diseases. Nat. Commun. 12, 2436. https://doi.org/10.1038/s41467-021-21952-4 (2021).
Moreno-Grau, S. et al. Genome-wide association analysis of dementia and its clinical endophenotypes reveal novel loci associated with Alzheimer’s disease and three causality networks: the GR@ACE project. Alzheimers Dement. 15, 1333–1347. https://doi.org/10.1016/j.jalz.2019.06.4950 (2019).
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. https://doi.org/10.1038/ng.2802 (2013).
Low, S. K. et al. Identification of six new genetic loci associated with atrial fibrillation in the Japanese population. Nat. Genet. 49, 953–958. https://doi.org/10.1038/ng.3842 (2017).
Christophersen, I. E. et al. Large-scale analyses of common and rare variants identify 12 new loci associated with atrial fibrillation. Nat. Genet. 49, 946–952. https://doi.org/10.1038/ng.3843 (2017).
Choi, S. H. et al. Association between Titin Loss-of-Function variants and Early-Onset atrial fibrillation. JAMA 320, 2354–2364. https://doi.org/10.1001/jama.2018.18179 (2018).
Demenais, F. et al. Multiancestry association study identifies new asthma risk loci that colocalize with immune-cell enhancer marks. Nat. Genet. 50, 42–53. https://doi.org/10.1038/s41588-017-0014-7
Chang, X. et al. Genetic architecture of asthma in African American patients. J. Allergy Clin. Immunol. 151, 1132–1136. https://doi.org/10.1016/j.jaci.2022.09.001 (2022).
Michailidou, K. et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat. Genet. 47, 373–380. https://doi.org/10.1038/ng.3242 (2015).
Mullins, N. et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat. Genet. 53, 817–829. https://doi.org/10.1038/s41588-021-00857-4 (2021).
Nikpay, M. et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 47, 1121–1130. https://doi.org/10.1038/ng.3396 (2015).
de Lange, K. M. et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 49, 256–261. https://doi.org/10.1038/ng.3760 (2017).
Garcia-Etxebarria, K. et al. Local genetic variation of inflammatory bowel disease in Basque population and its effect in risk prediction. Sci. Rep. 12, 3386. https://doi.org/10.1038/s41598-022-07401-2 (2022).
Garcia-Etxebarria, K. et al. Performance of the use of genetic information to assess the risk of colorectal Cancer in the Basque population. Cancers (Basel). 14, 4193. https://doi.org/10.3390/cancers14174193 (2022).
Tanikawa, C. et al. GWAS identifies two novel colorectal cancer loci at 16q24.1 and 20q13.12. Carcinogenesis 39, 652–660. https://doi.org/10.1093/carcin/bgy026 (2018).
Fernandez-Rozadilla, C. et al. Deciphering colorectal cancer genetics through multi-omic analysis of 100,204 cases and 154,587 controls of European and East Asian ancestries. Nat. Genet. 55, 89–99. https://doi.org/10.1038/s41588-022-01222-9 (2023).
Phelan, C. M. et al. Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat. Genet. 49, 680–691. https://doi.org/10.1038/ng.3826 (2017).
Lawrenson, K. et al. Genome-wide association studies identify susceptibility loci for epithelial ovarian cancer in East Asian women. Gynecol. Oncol. 153, 343–355. https://doi.org/10.1016/j.ygyno.2019.02.023 (2019).
Oh, S. W. et al. Genome-wide association study of metabolic syndrome in Korean populations. PLoS One. 15, e0227357. https://doi.org/10.1371/journal.pone.0227357 (2020).
Malik, R. et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat. Genet. 50, 524–537. https://doi.org/10.1038/s41588-018-0058-3 (2018).
Mishra, A. et al. Stroke genetics informs drug discovery and risk prediction across ancestries. Nature 611, 115–123. https://doi.org/10.1038/s41586-022-05165-3 (2022).
Landi, M. T. et al. Genome-wide association meta-analyses combining multiple risk phenotypes provide insights into the genetic architecture of cutaneous melanoma susceptibility. Nat. Genet. 52, 494–504. https://doi.org/10.1038/s41588-020-0611-8 (2020).
Schumacher, F. R. et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat. Genet. 50, 928–936. https://doi.org/10.1038/s41588-018-0142-8 (2018).
Ito, S. et al. Androgen receptor binding sites enabling genetic prediction of mortality due to prostate cancer in cancer-free subjects. Nat. Commun. 14, 4863. https://doi.org/10.1038/s41467-023-39858-8 (2023).
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102. https://doi.org/10.1016/S1474-4422(19)30320-5 (2019).
Rodrigo, L. M. & Nyholt, D. R. Imputation and Reanalysis of ExomeChip Data Identifies Novel, Conditional and Joint Genetic Effects on Parkinson’s Disease Risk. Genes (Basel). 12, 689. (2021). https://doi.org/10.3390/genes12050689
Le Guen, Y. et al. Common X-Chromosome variants are associated with Parkinson disease risk. Ann. Neurol. 90, 22–34. https://doi.org/10.1002/ana.26051 (2021).
Gharahkhani, P. et al. Genome-wide meta-analysis identifies 127 open-angle glaucoma loci with consistent effect across ancestries. Nat. Commun. 12, 1258. https://doi.org/10.1038/s41467-020-20851-4 (2021).
Stuart, P. E. et al. Transethnic analysis of psoriasis susceptibility in South Asians and Europeans enhances fine-mapping in the MHC and genomewide. HGG Adv. 3, 100069. https://doi.org/10.1016/j.xhgg.2021.100069 (2022).
Ishigaki, K. et al. Multi-ancestry genome-wide association analyses identify novel genetic mechanisms in rheumatoid arthritis. Nat. Genet. 54, 1640–1651. https://doi.org/10.1038/s41588-022-01213-w (2022).
Langefeld, C. D. et al. Transancestral mapping and genetic load in systemic lupus erythematosus. Nat. Commun. 17, 16021. https://doi.org/10.1038/ncomms16021 (2017).
Wang, Y. F. et al. Identification of 38 novel loci for systemic lupus erythematosus and genetic heterogeneity between ancestral groups. Nat. Commun. 12, 772. https://doi.org/10.1038/s41467-021-21049-y (2021).
Song, Q. et al. Genome-wide association study on Northern Chinese identifies KLF2, DOT1L and STAB2 associated with systemic lupus erythematosus. Rheumatol. (Oxford). 60, 4407–4417. https://doi.org/10.1093/rheumatology/keab016 (2021).
Trubetskoy, V. et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604, 502–508. https://doi.org/10.1038/s41586-022-04434-5 (2022).
Inshaw, J. R. J. et al. Analysis of overlapping genetic association in type 1 and type 2 diabetes. Diabetologia 64, 1342–1347. https://doi.org/10.1007/s00125-021-05428-0 (2021).
Robertson, C. C. et al. Fine-mapping, trans-ancestral and genomic analyses identify causal variants, cells, genes and drug targets for type 1 diabetes. Nat. Genet. 53, 962–971. https://doi.org/10.1038/s41588-021-00880-5 (2021).
Chen, J. et al. Genome-wide association study of type 2 diabetes in Africa. Diabetologia 62, 1204–1211. https://doi.org/10.1007/s00125-019-4880-7 (2019).
Mansour Aly, D. et al. Genome-wide association analyses highlight etiological differences underlying newly defined subtypes of diabetes. Nat. Genet. 53, 1534–1542. https://doi.org/10.1038/s41588-021-00948-2 (2021).
Spracklen, C. N. et al. Identification of type 2 diabetes loci in 433,540 East Asian individuals. Nature 582, 240–245. https://doi.org/10.1038/s41586-020-2263-3 (2020).
Gaziano, J. M. et al. Million Veteran Program: A mega-biobank to study genetic influences on health and disease. J. Clin. Epidemiol. 70, 214 – 23. (2016). https://doi.org/10.1016/j.jclinepi.2015.09.016
Cai, L. et al. Genome-wide association analysis of type 2 diabetes in the EPIC-InterAct study. Sci. Data. 7, 393. https://doi.org/10.1038/s41597-020-00716-7 (2020).
Loh, M. et al. Identification of genetic effects underlying type 2 diabetes in South Asian and European populations. Commun. Biol. 5, 329. https://doi.org/10.1038/s42003-022-03248-5 (2022).
Scott, R. A. et al. An expanded Genome-Wide association study of type 2 diabetes in Europeans. Diabetes 66, 2888–2902. https://doi.org/10.2337/db16-1253 (2017).
Graham, S. E. et al. The power of genetic diversity in genome-wide association studies of lipids. Nature 600, 675–679. https://doi.org/10.1038/s41586-021-04064-3 (2021).
Liu, J. Z. et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986. https://doi.org/10.1038/ng.3359 (2015).
Ghouse, J. et al. Genome-wide meta-analysis identifies 93 risk loci and enables risk prediction equivalent to Monogenic forms of venous thromboembolism. Nat. Genet. 55, 399–409. https://doi.org/10.1038/s41588-022-01286-7 (2023).
Fritsche, L. G. et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 48, 34–43. https://doi.org/10.1038/ng.3448 (2015).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191. https://doi.org/10.1093/bioinformatics/btq340 (2010).
Sollis, E. et al. The NHGRI-EBI GWAS catalog: knowledgebase and deposition resource. Nucleic Acids Res. 51, D977–D985. https://doi.org/10.1093/nar/gkac1010 (2023).
Ge, T., Chen, C. Y., Ni, Y., Feng, Y. A. & Smoller, J. W. Polygenic prediction via bayesian regression and continuous shrinkage priors. Nat. Commun. 10, 1776. https://doi.org/10.1038/s41467-019-09718-5 (2019).
Zheng, Z. et al. Leveraging functional genomic annotations and genome coverage to improve polygenic prediction of complex traits within and between ancestries. Nat. Genet. 56, 767–777. https://doi.org/10.1038/s41588-024-01704-y (2024).
Weissbrod, O. et al. Leveraging fine-mapping and multipopulation training data to improve cross-population polygenic risk scores. Nat. Genet. 54, 450–458. https://doi.org/10.1038/s41588-022-01036-9 (2022).
Chun, S. et al. Non-parametric polygenic risk prediction via partitioned GWAS summary statistics. Am. J. Hum. Genet. 107, 46–59. https://doi.org/10.1016/j.ajhg.2020.05.004 (2020).
Privé, F., Arbel, J., Aschard, H. & Vilhjálmsson, B. J. Identifying and correcting for misspecifications in GWAS summary statistics and polygenic scores. HGG Adv. 3, 100136. https://doi.org/10.1016/j.xhgg.2022.100136 (2022).
Privé, F., Arbel, J. & Vilhjálmsson, B. J. LDpred2: better, faster, stronger. Bioinformatics 36, 5424–5431. https://doi.org/10.1093/bioinformatics/btaa1029 (2021).
Lerga-Jaso, J. et al. Tracing human genetic histories and natural selection with precise local ancestry inference. Nat. Commun. 16, 4576. https://doi.org/10.1038/s41467-025-59936-3 (2025)
Chora, J. R., Medeiros, A. M., Alves, A. C. & Bourbon, M. Analysis of publicly available LDLR, APOB, and PCSK9 variants associated with Familial hypercholesterolemia: application of ACMG guidelines and implications for Familial hypercholesterolemia diagnosis. Genet. Med. 20, 591–598. https://doi.org/10.1038/gim.2017.151 (2018).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. https://doi.org/10.1038/gim.2015.30 (2015).
Šimundić, A. M. Measures of diagnostic accuracy: basic definitions. EJIFCC 19, 203–211 (2009).
Patel, A. P. et al. Association of rare pathogenic DNA variants for Familial hypercholesterolemia, hereditary breast and ovarian Cancer syndrome, and Lynch syndrome with disease risk in adults according to family history. JAMA Netw. Open. 3, e203959. https://doi.org/10.1001/jamanetworkopen.2020.3959 (2020).
Kullo, I. J. et al. Polygenic scores in biomedical research. Nat. Rev. Genet. 23, 524–532. https://doi.org/10.1038/s41576-022-00470-z (2022).
Ding, Y. et al. Polygenic scoring accuracy varies across the genetic ancestry continuum. Nature 618, 774–781. https://doi.org/10.1038/s41586-023-06079-4 (2023).
Ruan, Y. et al. Stanley Global Asia Initiatives;, Improving polygenic prediction in ancestrally diverse populations. Nat. Genet. 54, 573–580. (2022). https://doi.org/10.1038/s41588-022-01054-7
Web resources
R Project: https://www.R-project.org/
GWAS catalog: https://www.ebi.ac.uk/gwas/
FinnGen: https://www.finngen.fi/en
deCODE genetics: https://www.decode.com/
UK Biobank: https://www.ukbiobank.ac.uk/
dbGaP (PAGE MEC): https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000220.v2.p2
dbGAP (eMERGE): https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001584.v2.p2
dbGAP (The Million Veterans Program): https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001672.v11.p1
Author information
Authors and Affiliations
Contributions
J.L.J. and P.G.Y. designed this study. J.L.J, S.B. and A.D.M. processed genomic data for training and testing. J.L.J, A.T. and B.N. gathered and processed summary statistics for the meta-analyses. J.L.J., A.T. and M.K. performed the meta-analyses. J.L.J., A.T., A.O., and C.M. developed the integrated PRS disease prediction models. J.L.J., B.N. and P.G.Y. wrote the main manuscript text. J.L.J. prepared the figures. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
J.L.J., A.T., B.N., A.O., C.M., S.B., A.D.M. and P.G.Y. are either employed by and/or hold stock or stock options in Omics Edge, a subsidiary of Genius Labs. In addition, P.G.Y. has equity in Systomic Health LLC and Ethobiotics LLC. This does not alter our adherence to journal policies on sharing data and materials. M.K. has no conflicts of interest to declare. There are no other relevant activities or financial relationships which have influenced this work. All work was funded by a commercial source, Omics Edge, a subsidiary of Genius Labs Company. Omics Edge provided only funding for the study, but had no additional role in study design, data collection and analysis, decision to publish or preparation of the manuscript beyond the funding of the contributors salaries.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lerga-Jaso, J., Terpolovsky, A., Novković, B. et al. Optimization of multi-ancestry polygenic risk score disease prediction models. Sci Rep 15, 17495 (2025). https://doi.org/10.1038/s41598-025-02903-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-02903-1
