
Abstract
On September 1, 2019, China implemented the “Blood Station Technical Operation Procedures (2019 Edition),” specifying Enzyme-Linked Immunosorbent Assay (ELISA) and Chemiluminescent Immunoassay (CLIA) for serological testing. However, no CLIA HIV reagent has been approved for blood donor screening in China. This study evaluates the WT CMIA reagents for HIV screening in blood stations, providing evidence for their approval as a national screening tool. A controlled trial compared WT CMIA and ELISA reagents, as well as CLIA reagents from other manufacturers, through clinical experiments and reference serum panel testing. Evaluation metrics included precision, sensitivity, specificity, detection limit, method comparison, and reference range. Clinical Experiments: Among 10,470 blood donor samples, WT CMIA detected 100% of confirmed HIV-infected donors (42/42), compared to 97.62% for ELISA (41/42). CMIA and Wantai ELISA both had a specificity of 99.96%, Lizhu ELISA 99.98%. Kappa values between CMIA and ELISA methods were 0.90 and 0.92. Reference Serum Testing: Both CMIA and ELISA demonstrated 100% sensitivity (148/148). CMIA had a CV under 5%, while ELISA methods exceeded 5%. WT CMIA detected 97.03% of early HIV infections, compared to 93.07% for Roche and Abbott CLIA. Detection titers for HIV subtypes were not lower than 1:40, with a detection limit of 1.25 IU/mL for the P24 antigen, meeting WHO standards. WT CMIA shows high consistency with ELISA and superior sensitivity for early HIV detection, meeting WHO standards for blood donor screening in China.
Similar content being viewed by others
Introduction
In 2022, there were 39 million people globally infected with HIV, including 1.3 million new infections and 630,000 AIDS-related deaths. Notably, 86% of HIV-positive individuals were aware of their status, though approximately 5.5 million remained unaware of their infection. Among those aware of their HIV status, 86% [73–> 98%] knew their condition, and 89% [75–> 98%] received treatment. Of those receiving treatment, 93% [79–> 98%] achieved viral suppression. A total of 29.8 million people received antiretroviral therapy1.
Before 1995, China did not implement universal HIV testing for all blood donors; testing was limited to specific regions. For instance, the Chengdu Blood Center in Sichuan Province began testing in March 19882, and the Kunming Blood Center in Yunnan Province started in July 19933. HIV transmission occurred not only among blood recipients but also among donors. In February 1995, an HIV-1 infection case was discovered among a paid blood donor at a hospital in Tianjin, Hebei Province. Subsequently, a screening of former paid donors in Hebei from February to April 1995 revealed an infection rate of 1.5% (146/9811). From 1995 to 2013, Hebei Province reported 326 cases of HIV/AIDS transmission through plasma donation4.
Since the implementation of the Blood Donation Law in 1998, comprehensive HIV screening has been conducted on blood donors by blood collection and supply organizations and community health institutions, utilizing third-generation and/or fourth-generation ELISA testing methods5. In 2010, the Ministry of Health in China initiated nucleic acid testing trials at several blood centers6, and by 2015, nucleic acid testing was implemented for all blood donors. CLIA technology, valued for its convenience, high throughput, sensitivity, stability, and reproducibility, has been widely adopted in major hospitals across China7, and has replaced ELISA for blood screening in several developed countries8,9. The “Blood Station Technical Operating Procedures (2019 Edition),” effective September 1, 2019, allows the use of CLIA methods for testing transfusion-related infectious disease markers10. This study aims to evaluate the diagnostic performance of WanTai CMIA reagents on the Wan200 + analyzer for HIV detection, compare it with ELISA and other CLIA methods, and verify its suitability for HIV screening in Chinese blood donors, providing data for its approval as a “pharmaceutical” blood screening reagent.
Materials and methods
Specimens
Clinical experiment samples
Blood samples were collected from routine blood donors in Shandong Province, China, and stored at 2–8 °C. On the day of collection, samples were centrifuged and tested the following day using ELISA, nucleic acid testing (NAT), and chemiluminescent immunoassay (CMIA). Samples with discordant results were re-tested on the third day. The study was conducted in two phases: Phase 1 included 5130 donor samples collected between August 19 and August 31, 2021, along with 21 archived HIV-positive samples retained by the Shandong Blood Center since 2015. Phase 2 included 5,298 donor samples collected between May 8 and May 20, 2023, as well as a separate set of 21 archived HIV-positive samples. All archived HIV-positive samples were confirmed by the Jinan Center for Disease Control and Prevention using Western blot (WB) and the Wantai NAT platform. In total, 10,470 samples were analyzed, including 42 confirmed HIV-positive archived samples.
Reference serum samples
A total of 328 reference serum samples were provided by The National Center for Clinical Laboratories (NCCL), comprising 148 confirmed HIV-positive samples from 14 blood centers, 78 confirmed HIV-negative samples, 31 medium-control samples, 31 low-control samples, 12 gradient-diluted virus samples from three different cultures, 10 HIV antibody strong positive gradient-diluted samples, 5 samples with unclear status, and 13 negative controls. Additionally, 101 commercially obtained early HIV infection serum samples (HIV RNA-positive) were included. Detailed information on the serum panel samples is provided in Supplementary Table S1.
Investigation of the sensitivity of anti-HIV antibody and HIV-1 p24 antigen detection
The sensitivity of antibody detection were evaluated using the WHO 1 st International Reference Panel for Anti-HIV covering HIV-1 subtypes A, B, C, CRF01_AE, and Group O and HIV-2-positive clinical specimen (NIBSC, Code 02/210). was used to investigate the lower limit of antigen detection. A 5-step, 2-fold serial dilution series of these samples was prepared and measured in 3 different lots. (NIBSC, Code 90/636). Detailed sample information is provided in the WHO product inserts (Codes 02/210 and 90/636; see Supplementary Documents 1 and 2).
Assays and instruments
ELISA detection system
The ELISA detection system consisted of an automated sample processor (STAR, Hamilton, Switzerland) and an automated ELISA analyzer (FAME, Hamilton, Switzerland) for sample preparation and analysis. HIV antigen and antibody detection was performed using the Wantai HIV Ab-Ag ELISA kit and the Livzon HIV Ab-Ag ELISA kit.
Chemiluminescence detection system
The chemiluminescence detection system utilized the Wan200 + analyzer (Beijing Wantai Biological Pharmacy Enterprise Co., Ltd., China) for HIV screening. The detection was conducted using the Wantai HIV Ag/Ab CMIA kit, which operates on the electrochemiluminescence principle. Results were automatically generated, and sample reactivity was determined based on the Cutoff Index (COI).
Nucleic acid testing (NAT) system
Nucleic acid testing (NAT) was performed to detect HIV RNA using the Roche Cobas 201 system (Roche, Switzerland) and the Huayi Mei nucleic acid testing system (STAR, Slan). NAT was primarily employed to confirm samples with discordant ELISA or CMIA results.
Western blot (WB) confirmation system
Western Blot (MP HIV Blot 2.2) confirmation was conducted at the National Center for Clinical Laboratories (NCCL, China) using Western Blot assays for HIV antibody confirmation.
Evaluation method
Sensitivity and specificity evaluation
Sensitivity was calculated as (number of positive test results/number of confirmed positive cases) × 100%, while specificity was calculated as (number of negative test results/number of confirmed negative cases) × 100%.
Consistency evaluation
The test results of each ELISA assay were compared with those of the CMIA assay using the Kappa test. A Kappa value > 0.75 indicated excellent agreement.
Precision evaluation
The S/CO values of the quality control samples C1 and C2 were calculated, and the coefficient of variation (CV) value was analyzed.
Sensitivity and specificity evaluation
Sensitivity was calculated as (number of positive test results/number of confirmed positive cases) × 100%, while specificity was calculated as (number of negative test results/number of confirmed negative cases) × 100%.
Consistency evaluation
The agreement between ELISA and CMIA assays was assessed using the Kappa test. A Kappa value > 0.75 indicated excellent agreement between methods.
Precision evaluation
The S/CO values of the quality control samples C1 (moderate positive) and C2 (low positive) were calculated, and the coefficient of variation (CV) value was analyzed. A lower CV indicated better precision.
Dilution sensitivity evaluation
To assess dilution sensitivity, a strongly positive HIV antibody sample was serially diluted. The detection limit was determined by the highest dilution at which the assay remained reactive.
Detection of HIV subtypes
Detection capability for different HIV subtypes was evaluated using WHO standard samples, including HIV-1 subtypes A, B, C, E, and O, as well as HIV-2. The detection threshold was defined as the lowest dilution at which samples remained positive.
Limit of detection (LOD) for HIV p24 antigen
The LOD for HIV p24 antigen was determined using WHO standard samples at varying concentrations. The lowest concentration at which all replicates remained positive was recorded as the assay’s LOD.
Comparison with reference methods
The performance of the CMIA assay was compared with ELISA, CLIA, and NAT using clinical and reference serum samples. The overall agreement rate, positive agreement rate, and negative agreement rate were calculated.
Early HIV Infection DetectionTo evaluate early infection detection capability, HIV RNA-positive samples were tested with CMIA and compared to Roche and Abbott assaysThe percentage of RNA-positive samples correctly identified was recorded.
Criteria for determining reference serum panel results
The National Center for Clinical Laboratories (NCCL) confirmed HIV status in the serum panel using eight HIV ELISA assays and seven chemiluminescence assays. Samples reactive in all assays were classified as true positive, while those non-reactive in all assays were classified as true negative. If ELISA and chemiluminescence assay results were inconsistent, samples were classified as true negative if both nucleic acid testing (NAT) and Western blot (WB) were negative, true positive if NAT was positive, true positive if NAT was negative but WB was positive, and indeterminate if NAT was negative and WB was indeterminate. WB interpretation followed specific criteria: negative if no virus-specific bands were detected or only the p17 antibody was present; HIV-1 positive if at least two ENV bands (gp160, gp41, or gp120) were detected along with either one GAG band (p17, p24, or p55) or one POL band (p31, p51, or p66); and indeterminate if specific bands were present but did not meet the criteria for positivity.
Sample requirements
Clinical trial samples were obtained from post-routine testing plasma, with quality ensured through specific criteria. Samples with turbidity or precipitates were centrifuged or filtered to obtain a clear supernatant, while those containing fibrin strands or clots were excluded from testing. All samples were required to be free from microbial contamination. Regarding storage conditions, samples tested within 7 days were stored at 2–8 °C, whereas those intended for long-term storage were kept at ≤ −20 °C.
Inclusion and exclusion criteria
Samples were included in the study if they met all of the following criteria: they were derived from routine clinical tests or retained samples matching trial grouping requirements, had complete and traceable clinical trial data, were free from fibrinogen, aggregates, or severe hemolysis, had a remaining volume of approximately 900 µL, and complied with preservation conditions and the required number of freeze-thaw cycles.
Exclusion criteria
Samples were excluded if they did not strictly adhere to the clinical protocol or test kit instructions, had insufficient volume to complete the trial, or did not meet clinical trial requirements for other reasons.
Statistical analysis
Statistical analysis was performed using Stata 22.0 with a significance level of P < 0.05. Calculated metrics include concordance rates, detection rate, sensitivity, specificity, precision, and minimum detection limit.
Results
Clinical performance evaluation
Clinical experiment process
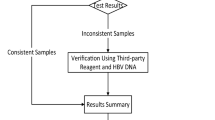
The flowchart depicting the clinical experiment process is shown in Fig. 1.

Clinical experiment flowchart.
Formulas used in the clinical experiment
The data codes used in the clinical experiment are presented in Table 1, and the agreement rates are calculated using the given formulas.
The calculation formula for the 95% confidence interval (95% CI) is as follows:
Positive agreement rate 95% confidence interval:
Negative agreement rate 95% confidence interval:
Overall agreement rate 95% confidence interval:
Clinical experiment results
The results and analysis from the trials conducted in 2021 and 2023 are detailed in Tables 2 and 3. A total of 10,470 tests were conducted, including 42 true positives and 10,428 negatives.
Reference serum sample testing results
Sensitivity and specificity
Among the 226 samples on the reference serum panel, both CMIA and ELISA detected all 148 positive samples, yielding 100% sensitivity. For the 78 negative samples, CMIA had 5 false positives (specificity 93.59%), Wantai ELISA had 3 false positives (specificity 96.15%), and LivZon ELISA had 1 false positive (specificity 98.72%). The negative samples included in the reference panel were collected from blood donors who showed reactivity with ELISA assays from a single manufacturer or with assays from two different manufacturers, but were confirmed negative by supplemental testing. As a result, the panel likely contains a relatively high proportion of cross-reactive samples, and therefore, the specificity results derived from this panel should be interpreted for reference only. Detailed information is provided in Table 4.
Dilution sensitivity evaluation
In the detection of the gradient dilution series of HIV antibody strongly positive samples, CMIA was able to detect samples diluted up to 1:400 (Table 5).
Evaluation of HIV virus detection capability
In Table 6, B90-1, B24, and P94 are codes for three different HIV virus samples.
Within-batch precision
A total of 30 weakly positive quality control samples were tested, with 1 sample excluded due to insufficient serum. For moderately positive samples, 31 were tested, and the highest value was excluded (Table 7).
Early HIV infection patient detection results
When testing early HIV RNA-positive samples, CMIA detected 98 cases, while Roche and Abbott detected only 94 cases. CMIA’s detection rate is higher than that of Roche and Abbott (Table 8).
WHO International Standard HIV antibody, HIV-1 P24 Antigen) Test Results (Limit of Detection and Ability to Detect HIV Subtypes)
Using three batches of CMIA reagents, we tested five different titers of the WHO 1 st International Reference Panel for Anti-HIV (NIBSC) for four HIV-1 subtypes (A, B, C, CRF01_AE) and Group O, as well as HIV-2. Each sample was tested three times per batch, and the average value was calculated. All samples were positive at dilutions of 1:40 or lower, indicating a detection titer of no less than 1:40 (Table 9).
Using three batches of CMIA reagents, we also tested five different titers of the HIV-1 p24 antigen samples from the WHO 1 st International Reference Panel for Anti-HIV (NIBSC). Each sample was tested three times per batch, and the average value was calculated. All samples were positive at a concentration of 1.25 IU/mL (Table 10).
WHO standard samples test results (Limit of detection and ability to detect HIV subtypes)
Using three batches of CMIA reagents, we tested five different titers of WHO standard samples for five subtypes of HIV-1 and HIV-2. Each sample was tested three times for each batch, and the average value was calculated respectively. All samples were positive at dilutions of 1:40 or lower, indicating detection titers of no less than 1:40 (Table 9).
Using three batches of CMIA reagents, we tested five different titers of WHO standard samples for the P24 antigen. Each sample was tested three times for each batch, and the average value was calculated respectively. All samples were positive at 1.25 IU/mL (Table 10).
Discussion
The sensitivity of HIV screening methods is critical for ensuring the safety of blood transfusions. Clinical HIV tests primarily include nucleic acid testing, antigen/antibody testing, CD4 + T cell counts, and HIV genotyping. In China, blood centers currently use fourth-generation HIV ELISA reagents from two manufacturers, along with nucleic acid testing for donor screening. According to the “Diagnosis of AIDS and HIV Infection” (WS-293–2019) standard, effective since July 1, 2019, the window periods for detecting HIV nucleic acids, antigens, and antibodies are roughly 1, 2, and 3 weeks post-infection, respectively. In many developed countries, CLIA has replaced ELISA as the routine method for blood donor screening, including in Australia11, Italy12, South Africa13, Japan14, and others. Research has demonstrated that CLIA meets regulatory standards in terms of stability, sensitivity, and specificity for screening HIV infection15. According to the “Technical Operating Procedures for Blood Stations” (2019 Edition)10 in China, reagents must be selected from in vitro diagnostic reagents approved by the National Medical Products Administration for blood source screening or approved in vitro diagnostic reagents. In other words, blood screening reagents must comply not only with the relevant provisions of the “Regulations for the Registration and Management of Medical Devices"16 but also with the “Drug Registration Management Measures.“17In recent years, the NCCL has organized various blood centers to evaluate CLIA for donor screening and conducted extensive comparisons with ELISA18,19,20.
The clinical experiment included 10,470 samples, encompassing routine, confirmed HIV-positive, ELISA false-positive, and HBV, HCV, TP positive samples, as well as those with severe hyperlipidemia and hemolysis. This diverse sample set represented nearly all donor scenarios, ensuring unbiased test accuracy. Across two trials, CMIA demonstrated 100% sensitivity, surpassing that of the two ELISA reagents currently used. CMIA also shortened the HIV detection window compared to international CLIA methods. Notably, Roche’s chemiluminescence methods, like Elecsys HIV combi PT, have shown 100% sensitivity in multiple studies, including those by Uettwiller-Geiger Denise L15.
The specificity of Wantai HIV CMIA when testing samples from routine blood donors was 99.96% and 99.98%. Uettwiller-Geiger Denise L reported a specificity of 99.94% 15. A joint study from six centers in Asia revealed specificities of 99.86% for Elecsys® combi PT, 99.78% for ARCHITECT® HIV, 99.52% for ADVIA Centaur® HIV, and 99.85% for Elecsys® HIV21. Research in Korea indicated specificities of 99.6% for AxSYM, 99.6% for ARCHITECT, 99.0% for Elecsys 2010 HIV Combi, and 99.5% for Elecsys HIV Combi PT22. A study conducted across 12 centers in Europe, Australia, and Thailand for Elecsys® HIV combi PT showed an overall specificity of 99.88%23. Beijing Hospital in China reported a specificity of 99.93% for Abbott CMIA (i2000SR)7.
This study used an NCCL-provided serum panel to evaluate CMIA. The WT CMIA detected all 148 HIV-positive samples with 100% sensitivity, matching ELISA. Among 78 HIV-negative samples, CMIA had 5 false positives, versus 3 and 1 for ELISA. These findings suggest CMIA may have a slightly higher false-positive rate. In previous testing of the same 78 HIV-negative samples, Roche CLIA had 10 false positives, while Bio-Rad ELISA had 6, indicating CLIA’s slightly lower specificity24..
In detecting HIV viral culture samples, CMIA detected all samples with a viral load of 1 IU/mL. The second ELISA had one false negative, and CMIA showed slightly better sensitivity based on S/CO values. At equivalent concentrations, CMIA’s S/CO values were higher than Roche CLIA’s. For WHO HIV P24 antigen, CMIA detected all samples at 1.25 IU/mL. A 2013 study indicated that Elecsys® HIV combi PT had an analytical sensitivity of 0.90 IU/mL for HIV-1 p24 antigen21, which was even superior to the 1.05 IU/mL reported in 201214, indirectly confirming the superior sensitivity of the WT CMIA in detecting HIV antigens. A multicenter study in South Africa found that the fourth-generation Elecsys HIV combi reduced the diagnostic window by 1.56 to 5.32 days compared to third-generation assays25.
Based on these findings, the Wantai CMIA meets WHO standards for HIV blood screening. Its performance metrics are comparable to or exceed those of the two ELISA assays commonly used in Chinese blood centers, confirming CMIA’s suitability for blood donor screening.
Data availability
All data generated or analysed during this study are included in this published article.
Abbreviations
- ELISA:
-
Enzyme-linked immunosorbent assay
- CMIA:
-
Chemiluminescence microparticle immunoassay
- CLIA:
-
Chemiluminescence immunoassay
- S/CO:
-
Signal to cut-off
- WB:
-
Western blot
- NCCL:
-
National Center for Clinical Laboratories
- RNA:
-
Ribonucleic acid
- NAT:
-
Nucleic acid testing
- WHO:
-
World Health Organization
- PCR:
-
Polymerase chain reaction
- CV:
-
Coefficient of variation
- PPV:
-
Positive predictive value
- NPV:
-
Negative Predictive Value
- WanTai/WT:
-
WanTai BioPharm
- LivZon/LZ:
-
Zhuhai Livzon Diagnostics Inc
- InTec/XC:
-
InTec PRODUCTS, INC
- Pos:
-
Positive
- Neg:
-
Negative
- NR:
-
Non-reactive
- R:
-
Reactive
- Val:
-
Value
- LOD:
-
limit of detection
References
Sheet, G. H. A. s. F. Global HIV & AIDS statistics — Fact sheet. (2023).
Yang, J. et al. HIV antibody testing report for blood donors and blood products in ten cities and counties in Sichuan Province. Chin. J. Blood Transf. 8, 28–29. https://doi.org/10.13303/j.cjbt.issn.1004-549x.1990.01.013 (1990).
Xiang, Z. et al. Analysis of the Test Situation of the Blood Donors HIV Antibody from1993to 1995in Kunming Area. Chin. J. AIDS STD 8, 7–8 (1997).
Chen, S. et al. Eighteen-year follow-up report of the surveillance and prevention of an HIV/AIDS outbreak amongst plasma donors in Hebei Province, China. BMC Infect. Dis. 15, 316. https://doi.org/10.1186/s12879-015-1073-y (2015).
Wenqian, S., Li, Z., Yong, G., Xiaohua, L. & Wanxin, A. Survey of HIV detection rates in blood donors tested by 357 provincial and municipal blood collection agencies nationwide. Chin. J. Transf. Med. 25, 1244–1246. https://doi.org/10.13303/j.cjbt.issn.1004-549x.2012.12.032 (2012).
Yongbao, Z. et al. Analysis of nucleic acid test results in unpaid blood donors in the Jinan Region from 2010 to 2012. Chin. J. Transf. Med. 26, 722–724. https://doi.org/10.13303/j.cjbt.issn.1004-549x.2013.08.044 (2013).
Cui, C. et al. Evaluation of the clinical effectiveness of HIV antigen/antibody screening using a chemiluminescence microparticle immunoassay. J. Virol. Methods 214, 33–36. https://doi.org/10.1016/j.jviromet.2014.07.026 (2015).
Arcot, P. et al. Comparative evaluation of ADVIA Centaur® XP chemiluminescence system for screening of HBV, HCV, HIV and syphilis in Indian blood donors. Transf. Apheresis Sci. 61, 103318, doi: 10.1016/j.transci.2021.103318 (2022).
Schmidt, M. et al. Head-to-head comparison between two screening systems for HBsAG, anti-HBc, anti-HCV and HIV combination immunoassays in an international, multicentre evaluation study. Vox sanguinis 109, 114–121. doi: 10.1111/vox.12259 (2015).
China, N. H. C. o. (April 2019).
Kiely, P. & Wilson, D. Results of HCV screening of volunteer blood donors with a chemiluminescent immunoassay and a second- or third-generation EIA: overlap of false-positive reactivity and its impact on donor management. Transfusion 40, 580–584. https://doi.org/10.1046/j.1537-2995.2000.40050580.x (2000).
Sommese, L. et al. Screening tests for hepatitis B virus, hepatitis C virus, and human immunodeficiency virus in blood donors: evaluation of two chemiluminescent immunoassay systems. Scand. J. Infect. Dis. 46, 660–664. https://doi.org/10.3109/00365548.2014.926564 (2014).
Vermeulen, M., Swanevelder, R., Van Zyl, G., Lelie, N. & Murphy, E. An assessment of hepatitis B virus prevalence in South African young blood donors born after the implementation of the infant hepatitis B virus immunization program: Implications for transfusion safety. Transfusion 61, 2688–2700. https://doi.org/10.1111/trf.16559 (2021).
Taira, R. et al. Residual risk of transfusion-transmitted hepatitis B virus (HBV) infection caused by blood components derived from donors with occult HBV infection in Japan. Transfusion 53, 1393–1404. https://doi.org/10.1111/j.1537-2995.2012.03909.x (2013).
Uettwiller-Geiger, D. et al. Analytical and clinical performance evaluation of the elecsys HIV combi PT assay on the cobas e 602 analyzer for the diagnosis of human immunodeficiency virus. Am. J. Clin. Pathol. 151, 377–385. https://doi.org/10.1093/ajcp/aqy153 (2019).
Administration, N. M. P. Notice from the National Medical Products Administration regarding the release of the Technical Guidelines for clinical experiments of In Vitro Diagnostic Reagents (Announcement No. 72, 2021). (September 16, 2021).
Regulation, S. A. f. M. Drug Registration Management Measures. (January 22, 2020).
Chang, L. et al. Comparative Evaluation and measure of accuracy of ELISAs, CLIAs, and ECLIAs for the detection of HIV infection among blood donors in China. Can. J. Infect. Dis. Med. Microbiol. 2020, 2164685, doi: 10.1155/2020/2164685 (2020).
Ji, H. et al. Evaluation of ELISA and CLIA for Treponema pallidum specific antibody detection in China: A multicenter study. J. Microbiol. Methods 166, 377 (2019).
Chen, J. et al. Performance evaluation of the CLIA and ECLIA for anti-tp screening in blood donors. Zhongguo shi yan xue ye xue za zhi 27, 260–265. https://doi.org/10.7534/j.issn.1009-2137.2019.01.042 (2019).
Tao, C. et al. Validation of the Elecsys® HIV combi PT assay for screening and reliable early detection of HIV-1 infection in Asia. J. Clin. Virol. 58, 221–226, doi:10.1016/j.jcv.2013.05.012 (2013).
Song, E. et al. Performances of four fourth-generation human immunodeficiency virus-1 screening assays. J. Med. Virol. 84, 1884–1888. https://doi.org/10.1002/jmv.23423 (2012).
Mühlbacher, A. et al. Performance evaluation of a new fourth-generation HIV combination antigen-antibody assay. Med. Microbiol. Immunol. 202, 77–86. https://doi.org/10.1007/s00430-012-0250-5 (2013).
Jianfeng, C. et al. Evaluation of two CLIA reagents for blood screening HIV. Chin. J. Transf. Med. 31, 846–850. https://doi.org/10.13303/j.cjbt.issn.1004-549x.2018.08.009 (2018).
Weber, B. et al. Multicenter evaluation of a new 4th generation HIV screening assay Elecsys HIV combi. Clin. Lab. 52, 463–473 (2006).
Acknowledgements
We extend our sincere gratitude to Dr. Wang Lunan, who served as the Director and Researcher at the Blood Safety and Immunohematology Laboratory, National Clinical Laboratory Center of the National Health Commission. Her invaluable guidance and assistance were instrumental in the success of this study. We are also deeply appreciative of Dr. Chang Le for her invaluable technical support during the analysis of experimental data. Finally, we express our heartfelt thanks to Xiamen Innodx for their unwavering support in supplying scientific reagents and providing technical assistance. Their collaboration and assistance were genuinely invaluable and greatly appreciated.
Funding
This research was financially supported by the Shandong Blood Center through the Shandong Provincial Medical and Health Science and Technology Project(No. 202011000934) and generously supported by the Shandong Province Humanities and Social Science Project (No. 2021-ZXJK-24). We also acknowledge the valuable research reagent support provided by Xiamen Innodx Biotechnology Co., Ltd.
Author information
Authors and Affiliations
Contributions
J.C.and F.W. contributed to the experimental design, conducted laboratory testing, performed data analysis, drafted the initial manuscript, and participated in its review and revision. L.J., R.J., and Y.L. assisted with data analysis, data curation, and drafting sections of the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
NCCL
The National Center for Clinical Laboratories (NCCL), established in 1982 under the National Health Commission of China, oversees clinical laboratory quality management nationwide. It conducts external quality assessment (EQA) programs, develops reference measurement procedures, and produces standard materials to ensure test accuracy and traceability. NCCL supports over 140 EQA programs with participation from more than 9,000 institutions and adheres to international standards such as ISO/IEC 17043 and ISO 15195. Currently, it has developed more than 100 national primary or secondary reference materials for routine laboratory tests.
Ethical approval
This study was conducted in full accordance with the Declaration of Helsinki and was approved by the Institutional Review Board of the Shandong Blood Center prior to the enrollment of any participants (IRB approval number: “LXB Ethics [2020] No. 4”). The study was subsequently registered and approved in the National Health Insurance Platform’s Medical Research Registration and Record Information System (registration number: MR-37-23-026261). Written informed consent was obtained from all participants involved in the study, and their consent forms are archived in accordance with institutional requirements.
Generative AI and AI-assisted technologies in the writing process
Generative AI-assisted technologies were not used in the writing process of this manuscript.
Declaration of competing interest
None are declared.
Author contribution statement
J.C. contributed to the experimental design, conducted laboratory testing, performed data analysis, drafted the initial manuscript, and participated in its review and revision. F.W. oversaw the experiments, supervised laboratory testing, validated the data, and contributed to the manuscript’s review and revision. L.J., R.J., and Y.L. assisted with data analysis, data curation, and drafting sections of the manuscript. All authors critically reviewed and revised the manuscript, approved the final version, and take full responsibility for the integrity of the data and the accuracy of the data analysis.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, J., Jiang, L., Ju, R. et al. Analytical and clinical performance evaluation of the Wantai chemiluminescence microparticle immunoassay on the Wan200 + analyzer for diagnosing HIV. Sci Rep 15, 17821 (2025). https://doi.org/10.1038/s41598-025-03077-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-03077-6
