
Abstract
Lysine crotonylation (Kcr) is an important post-translational modification, which is present in both histone and non-histone proteins, and plays a key role in a variety of biological processes such as metabolism and cell differentiation. Therefore, rapid and accurate identification of this modification has become a key task to study its biological effects. In the past few years, some calculation methods have been developed, but there is room for improvement in prediction performance. In this paper, we propose an effective model named DeepMM-Kcr, which is based on multiple features and an innovative deep learning framework. Multiple features are extracted from natural language processing features and hand-crafted features, where natural language processing features include token embedding and positional embedding encoded by transformer, and hand-crafted features include one-hot, amino acid index and position-weighted amino acid composition, and encoded by bidirectional long short-term memory network. Then natural language processing features and hand-crafted features are fusing by multi-head self-attention mechanism. Finally, a deep learning framework is constructed based on convolutional neural network, bidirectional gated recurrent unit and multilayer perceptron for robust prediction of Kcr sites. On the independent test set, the accuracy of DeepMM-Kcr is highest among the existing models. The experimental results show that our model has very good performance in predicting Kcr sites. The source datasets and codes (in Python) are publicly available at https://github.com/yunyunliang88/DeepMM-Kcr.
Similar content being viewed by others

Introduction
Protein post-translational modifications (PTMs) alter the properties of proteins by proteolytic cleavage and the addition of modification groups. Any protein in the proteome can be modified during or after translations1,2. PTMs play an important role in many biological processes such as DNA replication, cell differentiation, embryonic development, gene activation and gene regulation3,4,5. These PTMs can regulate protein stability and biological activity by affecting protein structure. With the upgrading and development of mass spectrometry detection technology, more and more PTMs have been identified, including a series of short-chain lysine acylation modifications, such as butyrylation, propionylation, succinylation, malonylation, crotonylation and benzoylation6.
Lysine crotonylation (Kcr) is a crucial post-translational modification (PTM), where “K” represents lysine, “cr” represents crotonylation, that is, Kcr represents lysine crotonylation. In 2011, Tan et al. defined Kcr using mass spectro proteomics in human cells and mouse germ cells7. This modification is widespread in both histone and non-histone proteins, particularly in chromatin regions associated with active transcription. Kcr plays significant roles in regulating gene expression, maintaining chromatin structure, and controlling reproductive processes1. Additionally, research has shown that Kcr is involved in various physiological and pathological processes, such as tissue injury, inflammatory responses, and cancer development. It also plays important roles in neuropsychiatric disorders, telomere maintenance, and HIV latency8,9,10,11,12. With the advancement of proteomics technologies, including high-performance liquid chromatography-tandem mass spectrometry, stable isotope labeling, and specific antibody techniques, scientists have been able to comprehensively detect Kcr sites13. Traditional high-throughput experimental methods are time consuming and expensive, so computational predictors based on machine learning algorithms are becoming mainstream solutions. Moreover, the development of machine learning, especially deep learning techniques, has allowed for accurate prediction of Kcr sites on large-scale datasets. These technological advancements not only enhance our understanding of Kcr, but also provide powerful tools for studying the dynamic regulation of proteins in various cellular contexts. Research into Kcr has revealed its multifaceted functions in gene transcription, cell cycle regulation, tissue repair, and more, offering new possibilities for the diagnosis and treatment of related diseases.
In recent years, several traditional machine learning-based tools have been developed for predicting Kcr sites. The earliest is CroPred, which is designed by using a discrete hidden Markov model (DHMM) based on amino acid sequence data to predict Kcr sites in histones14. Subsequently, the models of Qiu et al.15, CKSAAP-CrotSite16, iKcr-PseEns17, iCrotoK-PseAAC18, LightGBM-CroSite19, Wang et al.20 and predML-Site21 are successively proposed.
With the widespread application of deep learning in bioinformatics, some prediction tools based on deep learning have been proposed one after another. In 2020, lv et al. developed the prediction tool named Deep-Kcr based on convolutional neural network (CNN)22. In 2021, Chen et al. developed a novel deep learning-based computational framework termed as CNNrgb for Kcr sites prediction on nonhistone proteins by integrating different types of features, and implemented an online server called nhKcr23. In 2022, Qiao et al. proposed a computational method called BERT-Kcr, which extracted features from the BERT model and placed them into the bidirectional long short-term memory network (BiLSTM)-based classifier for Kcr sites prediction24. Later, Khanal et al. proposed a model named DeepCap-Kcr by using a capsule network (CapsNet) based on CNN and LSTM25. Also in 2022, Li et al. proposed a new deep learning model called Adapt-Kcr, which utilized adaptive embedding based on CNN as well as BiLSTM and attention mechanism26. Meanwhile, in 2022, Dou et al. developed a CNN-based model framework called iKcr_CNN, which applied the focal loss function instead of the standard cross-entropy as the indicator to overcome the imbalance issue27. In 2023, Khanal et al. developed a new model based on capsule network CapsNh-KCR, particularly focusing on non-histone proteins, which more effectively improves the experimental efficiency28.
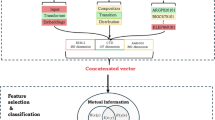
Although many models have been used to identify Kcr sites, the prediction accuracy of existing methods still needs to be improved, and better integration of hand-crafted features and natural language processing (NLP) features is needed. In this paper, a novel model named DeepMM-Kcr for Kcr sites prediction by fusing multi-features based on multi-head self-attention mechanism. The multi-head self-attention maps NLP deep features and hand-crafted features to the same space through independent attention heads, avoiding conflicts caused by differences in feature types, each head can adaptively learn the importance of different features. First, multi-features are extracted based on NLP technology using word embedding combined with transformer encoder. Hand-crafted features are extracted using one-hot, amino acid index (AAindex) and position-weighted amino acid composition (PWAA), which are concatenated with BiLSTM for encoding. Then, NLP features are point-wise added with hand-crafted features, and multi-head self-attention mechanism is adopted for features fusing. Finally, prediction is performed by integrating CNN, bidirectional gated recurrent unit (BiGRU) and multilayer perceptron (MLP) based on K-fold cross-validation and independent test. The prediction accuracy has reached 85.86% for independent test set, and improved 0.4% than that of best model called Adapt-Kcr26.
Materials and methods
Datasets
Datasets play an important role in establishment of the model. The datasets used in this paper are the same as those used by lv et al.22, which are originally constructed by Yu et al. in 202013 from HeLa cell data, comprising 14,311 Kcr sites across 3734 proteins. After removing redundant sequences using CD-HIT29 with 30% identity, 9964 non-redundant Kcr sites are retained as positive samples. To balance the dataset, 9964 non-redundant normal lysine sites are randomly selected as negative samples also from HeLa cells. Each sequence is truncated into 31 amino acids with lysine centrally located. If the center K of the segment with 31 amino acids is crotonylation, it is defined as a positive sample (Kcr), otherwise it is defined as a negative sample (non-Kcr). Then, the non-redundant dataset is divided into the benchmark set and the test set with a ratio of 7:3. The benchmark set includes 6975 positive samples and 6975 negative samples for cross-validation, and the test set includes 2989 positive samples and 2989 negative samples for independent test. More details of the datasets are shown in Table 1.
Transformer-based natural Language processing
Word embedding
Word embedding (WE) is a key technique in natural language processing, which transforms words in natural language into dense vectors, where similar words are represented by similar vectors. This transformation facilitates the exploration of features between words and sentences in text30,31.
The Token embedding is a technique that converts text into a series of numbers, Token can be understood as the smallest unit used to segment and encode input text, which can be word, subword or character. Token embedding includes two parts: segmentation and encoding. Segmentation divides text into individual words or characters, while encoding assigns each word or character a unique numerical identifier. In this paper, each character represents an amino acid residue in an amino acid sequence, called a “token”. This technique helps computer models capture the relationship and similarity between different unit in a sequence. In token embedding, we obtain the representation vector for each amino acid through a table lookup. Words need to be encoded according to different word segmentation methods and the words must exist in the vocabulary.
Positional embedding is a technique that helps a model understand the relative positions of elements in a sequence. Its core purpose is to solve the problem that the model cannot directly perceive the order of elements in the analysis of biological sequences such as DNA, RNA or proteins. It was originally derived from the work of Vaswani et al.32. They tested two approaches, one adaptive position embedding that can be learned and the other fixed position encoding. We choose to use fixed position encoding, using the sine and cosine functions to compute fixed representation vectors. The sequence length is \(N\), and the positional embedding vector (PE) formula is shown as following:
where \(k\) represents the position of each residue in the sequence (\(0 \leqslant k \leqslant N - 1\)), where \(N\) is the length of the sequence. \(d\) represents the dimension of positional embedding and must match the dimension of the token embedding. 2i represents even dimensions, 2i + 1 represents odd dimensions, \(0 \leqslant i<d/2\), meaning only half of the embedding dimensions are used for the sine and cosine calculations. The constants \(b\)and \(d\) are predefined, \(d\) must match the dimension of the token embeddings. In this study, the values of \(b\) and \(d\) are set to 1000 and 128, respectively.
The combination of token embedding and positional embedding can improve the performance of the model more effectively26. Hence, token embedding and positional embedding are point-wise addition (Add) as word embedding, which is expressed as follows:
Encoder with transformer
The transformer, introduced by Vaswani et al.32, has gained widespread attention and popularity in the field of natural language processing (NLP)33. It is structured around an encoder-decoder framework. In this paper, we use the transformer’s encoder block to process the input word embedding. The encoder block structure of transformer is composed of multi-head self-attention mechanism (MHA), feed forward neural network (FFN), residual connection (Add) and layer normalization (Norm).
The self-attention mechanism, enables transformer to capture remote dependencies between sequence elements, and the core is to update the representation of each element in the input sequence by calculating its relationship to the other elements in the sequence. To achieve this, the self-attention mechanism introduces three matrixes: Query (Q), Key (K) and Value (V). These three vectors are generated from the representation of the input matrix X by linear transformation:
\({W^Q}\), \({W^K}\) and \({W^V}\) are the weight matrix. We can calculate the output of self-attention by the following formula:
where the inner product of each row vector of the matrix Q and K is calculated by \(Q{K^T}\), which represents correlation score for each input word. To improve the stability of training and prevent gradient explosion, correlation score is normalized by dividing by the square root of \({d_k}\), \({d_k}\) is the dimension of K. The score vector between each word is converted into a probability distribution between [0,1] using the softmax function. The softmax matrix can be multiplied by V to get the final output Z. The multi-head self-attention is formed by concatenating the input for multiple self-attention.
The encoder block structure of transformer is expressed as follows:
where \({\text{FFN(}}{X_{MHA}}{\text{)}}+{X_{MHA}}\) and \({\text{FFN(}}{X_{MHA}}{\text{)}}+{X_{MHA}}\) stands for residual connection commonly used to solve the problem of multi-layer network training, allowing the network to focus only on the part that is currently different. Norm refers to layer normalization and is commonly used in RNN structure, where layer normalization converts the inputs to each layer of neurons to have the same mean and variance, which speeds up convergence. The feed forward neural network is a two-layer fully connected layer, the first layer uses the Relu activation function, the second layer does not use the activation function, the formula is as follows:
where \({W_1}\) and \({b_1}\) are the weight matrixes and bias vectors of the first layer in the FFN. \({W_2}\) and \({b_2}\) are the weight matrixes and bias vectors of the second layer in the FNN.
Hand-crafted features
Hand-crafted (HF) features refer to the extraction of representative features from the original data, which is generally used in data analysis, modeling and prediction. Feature extraction is a very important step in machine learning. In this paper, we use one-hot, AAindex and PWAA for extracting hard-crafted features.
One-hot
One-hot (OH) coding34,35, also known as one-bit effective encoding, mainly uses N-bit status registers to encode N states, each state is composed of its own independent register bits, and only one bit is valid at any time. One-hot encoding is a representation of categorical variables as binary vectors. This first requires mapping class values to integer values. Each integer value is then represented as a binary vector that is zero except for the index of the integer, which is labeled 1. The one-hot coding solves the problem that the classifier is not good at processing attribute data, and plays a role in expanding the feature to a certain extent. Its values are only 0 and 1, and the different types are stored in vertical space. Finally, the dimension of the OH-based feature vector is 620.
AAindex
The amino acid index (AAindex) is a widely used database that contains indices based on multiple physicochemical and biochemical properties of amino acids36. The AAindex database is used to extract major physicochemical properties based on the amino acid index to characterize protein fragments (31 residue long fragments in this study). For such fragments, the amino acids at each location are represented as 12 values based on their physicochemical and biochemical properties. These 12 values represent the following properties: net charge; normalized frequency of alpha-helix; alpha helix propensity of position 44 in T4 lysozyme; composition of amino acids (AA) in intracellular proteins; AA composition of membrane of multi-spanning proteins; the volume of crystallographic water; information value for accessibility; transfer energy, organic solvent/water; AA composition of membrane proteins; entropy of formation; conformational preference for all beta-strands and optimized relative partition energies22. Thus, for each fragment of 31 residues, the amino acids at each position are described by these 12 properties, resulting in a 372-dimensional feature vector. Such feature vector can comprehensively characterize various physicochemical and biochemical properties of protein fragments, and provide rich information for further bioinformatics analysis.
Position-weighted amino acid composition
Position-weighted amino acid composition (PWAA) is a method to encode sequence information based on the weight of the position of amino acids in a protein sequence37. It takes into account the relative positions of amino acid residues in the sequence and reflects the position information by calculating weights. It takes the center of the sequence as the observation point and calculates the position weight of each amino acid residue relative to the center point. The size of the weight depends on the distance between the residue and the center point, usually the closer the distance, the greater the weight.
The specific steps are as follows: (1) Define a sequence fragment \(P\) of length 2L + 1 (L is the number of upstream or downstream residues). (2) For each amino acid residue, and calculate its position weight \({C_i}\) \({\text{(}}i=1{\text{,}}\,2{\text{,}}\, \ldots {\text{,}}\,20{\text{)}}\) in sequence fragment \(P\). (3) The calculation of position weights is usually based on a predefined function or rule that considers the position of the residue relative to the center point. (4) Finally, the dimension of the feature vector based on PWAA is 20 (corresponding to 20 amino acids), and the value of each dimension represents the sum of the position weights of the corresponding amino acids in the whole sequence22. The formula is as follows:
where L indicates the number of upstream residues or downstream residues from the central site in the protein sequence fragment \(P\), if the jth positional residue is \({a_i}\) in protein sequence fragment \(P\), \({x_{i,j}}=1\), otherwise \({x_{i,j}}=0\). Finally, the dimension of the PWAA-based feature vector is 20.
Encoder with BiLSTM
LSTM is an improved RNN model38. Traditional RNN is prone to gradient disappearance or gradient explosion when dealing with long sequences, while LSTM solves these problems by introducing gating mechanisms (input gate, forget gate, output gate) to control the flow of information, thus better capturing long-term dependencies. The calculation formula is as follows:
where \({f_t}\), \({x_t}\), \({i_t}\), \({o_t}\), \({h_t}\), \({C_t}\) and \({\tilde {C}_t}\) represent forget gate, input data, update gate, output gate, cell state, memory cell and candidate state, respectively. \(W\) and \(b\) represent weight matrix and bias, respectively. \(\odot\) represents point-wise multiplication.
A one-way LSTM processes sequence data in only a single direction, usually from left to right of the sequence, and cannot combine the dependencies of sequence information. Therefore, it is a better way to choose BiLSTM for feature extraction. BiLSTM is essentially two LSTMs, one to process the sequence forward and one to process the sequence backward, and after processing, the output of the two LSTMs is concatenated. BiLSTM captures both forward and backward information of the sequence, which can greatly improve the effect of the model. The forward LSTM processes the sequence from beginning to end, generating a feature vector represented as \({h_L}=[{h_{L,0}},{h_{L,1}}, \ldots ,{h_{L,T}}]\). Meanwhile, the backward LSTM analyzes the sequence in reverse, producing a feature vector \({h_R}=[{h_{R,0}},{h_{R,1}}, \ldots ,{h_{R,T}}]\). To form the final output, the corresponding elements from these two vectors are combined, resulting in \(h=[{h_{L,0}},{h_{R,T}},{h_{L,1}},{h_{R,T - 1}} \ldots ,{h_{L,T}},{h_{R,0}}].\)This approach ensures that each feature at position incorporates both past and future context. Generally, BiLSTM outperforms standard LSTM when handling context-sensitive data. One-hot, AAindex and PWAA are concatenated (Concat) and encoded with BiLSTM, which can integrate the temporal context information of various features, enrich the feature representation, and further optimize the feature extraction effect.
Deep learning framework
Attention fusion layer
The multi-head self-attention is formed by the combination of multiple self-attention39. In this paper, word embedding with transformer coding (NLP features) is combined with hand-crafted features with BiLSTM coding by point-wise addition as follows:
where X represents multiple features, which are fused by multi-head self-attention mechanism.
The multi-head self-attention enables effective feature fusion by: (1) Capturing heterogeneous feature interactions via independently learned attention patterns across heads; (2) Implementing dynamic feature re-weighting to enhance discriminative elements; (3) Enriching representation spaces through decomposed attention subspaces without significant parameter overhead.
CNN layer
CNN refers to convolutional neural networks, a deep learning model that is widely used in computer vision fields such as image classification, object detection, and image segmentation40. The main feature of CNN is the use of convolutional layers to automatically extract image features, and filters (or convolution cores) slide over the image to capture local patterns, which are then used to identify image content. In this paper, one-dimensional convolution (Conv1d) is used, which efficiently extracts local features of input data through local perception and parameter sharing mechanisms. The batch normalization layer, used to normalize input features to zero mean and unit variance, helps speed up the training process and improve model performance. Dropout layer can prevent overfitting, which randomly drops a certain percentage of the neuronal output in the network during training. ReLU activation function is used to increase the nonlinearity of the network. It sets all negative values to zero and keeps all positive values. The one-dimensional maximum pooling layer (MaxPool1d) is used to reduce the dimension (height) of the feature map.
BiGRU layer
The bidirectional gated recurrent unit (BiGRU) was proposed in 2014 by Cho et al.41. It is a recurrent neural network (RNN)42,43 that consists of two independent GRU units, one processing data forward in time series and the other processing data backward in time series. Similar to BiLSTM, it combines forward and backward hidden states to capture contextual dependencies in the sequence. Because GRU is simpler than LSTM, the computational efficiency of BiGRU is higher than that of BiLSTM.
The CNN operates as a local feature extractor for short-range spatial patterns, whereas the BiGRU captures long-range temporal dependencies across the entire sequence. This integrated framework by combining CNN with BiGRU enables synergistic spatial-temporal representation learning.
Output layer
MLP is a feedforward neural network that can model complex nonlinear relationships through multilayer linear transformations and nonlinear activation functions. MLP is widely used in a variety of tasks, such as classification, regression, and sequence prediction. Through the steps of forward propagation, loss calculation and backpropagation, MLP can gradually adjust the parameters and optimize the model performance. In this paper, we use the batch normalization layers and dropout layers, which can improve the robustness of the model.
Detailed structure and parameter settings
In order to present the model structure clearly, more detailed introduction and parameter settings are shown in Table 2.
Model evaluation
In this paper, five-fold cross-validation is applied to the model on the benchmark set, and independent test is used to evaluate the model on the test set44,45,46. The five-fold cross-validation is a way to divide the data set into 5 subsets of equal size, usually randomly, and use 4 subsets as the training set in turn, and the remaining subset as the test set for model training and evaluation. The experiment is run five times, ensuring that each subset is used as the test set once. Finally, the results of 5 tests are averaged to get the final performance evaluation of the model. The independent test utilizes a test set, entirely independent from the training set, to evaluate the generalization ability of the trained model. We use five popular statistical measures to assess the model performance, including accuracy (ACC), sensitivity (Sn), specificity (Sp), Matthew’s correlation coefficient (MCC) and area under the ROC curve (AUC or auROC)47,48,49,50,51. The formulas are as follows:
where TP, TN, FP and FN denote the numbers of true positive, true negative, false positive and false negative, respectively. In addition, we adopted the receiver operating characteristic (ROC) curve and the precision-recall (PR) curve. The ROC curve is a graphical way to evaluate the performance of a classification model. The closer the AUC (auROC) value is to 1, the better the prediction performance of the model. The PR curve shows the relationship between precision and recall at different thresholds. The area under PR curve (auPRC) can also be used to quantify the performance of a model, but usually more attention is paid to the shape and position of the PRC curve itself.
Results and discussion
Experimental settings and runtime
In this paper, we train the DeepMM-Kcr model under the Python PyTorch framework (https://pytorch.org/). The hardware environment is 11th Gen Intel(R) Core(TM) i5 1135G7 CPU @ 2.40 GHz 2.42 GHz with 16.0 GB of RAM. The cross-entropy is used as the loss function, and the weight parameters are optimized by the Adam algorithm. The batch size is 64, the learning rate is 0.0001 for our training model, and the iteration epoch number is 50. The average training time and independent test time for each epoch are 115 s and 10 s, respectively.
Prediction performance of our model
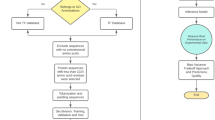
In this paper, a new model named DeepMM-Kcr is proposed, the workflow of the model is shown in Fig. 1. The mean values and standard deviation of five-fold and values of each fold for ACC, Sn, Sp, MCC and AUC are calculated by the five-fold cross-validation on the benchmark set, as well as the values of ACC, Sn, Sp, MCC and AUC are calculated by the independent test on the test set, and the results are shown in Table 3. As can be seen from Table 3, the ACC of the benchmark set and the independent test set reaches 82.03% and 85.56%, respectively. Sn, Sp, MCC and AUC also obtained satisfactory results. The results of five-fold cross-validation show that our model has strong stability, and independent test results show that our model has good generalization ability.

The flowchart of the DeepMM-Kcr model.
Ablation experiments
Feature ablation
Features extraction plays an important role in the prediction performance of the model. To validate the impact of feature representation methods on model performance, ablation experiments are performed. We construct the different feature groups, including One-hot + BiLSTM, AAindex + BiLSTM, PWAA + BiLSTM, HF + BiLSTM, WE + transformer and Add, which are all without multi-head self-attention. One-hot + BiLSTM refers to One-hot features encoded by BiLSTM, AAindex + BiLSTM and PWAA + BiLSTM are similar to One-hot + BiLSTM. HF + BiLSTM refers to hand-crafted features derived from the concatenation of features One-hot, AAindex and PWAA encoded by BiLSTM. WE + transformer refers to word embedding encoded by transformer. Add refers to the features that combine the features of WE + transformer and HF + BiLSTM by point-wise addition. The ablation experimental results on the benchmark set and independent test set are shown in Table 4.
From Table 4, for the three types of hand-crafted features, the order of feature importance from high to low is One-hot, AAindex and PWAA. Meanwhile, we can also see that WE + transformer has better identification ability than HF + BiLSTM, because it can provide richer and more delicate feature representations. Combining the features of WE + transformer and HF + BiLSTM by point-wise addition, as a whole, better effect is to be generated than that using single type features. In conclusion, WE + transformer contribute most significantly to model performance.
Attention mechanism ablation
Multi-head self-attention is composed of the concatenation of multiple self- attention results. The number of heads is selected by conducting attention mechanism ablation. Self-attention (1-head), 4-head self-attention and 8-head self-attention are listed for ablation experiments. The ablation experimental results on the benchmark set and independent test set are shown in Table 5; Fig. 2. As is shown in Tables 5 and 8-head self-attention obtain the best prediction performance. ROC and PR curves, as well as auROC and auPRC in Fig. 2 intuitively shows that 8-head attention is the best method.

ROC and PR curves for ablation experiments on attention methods.
Model structure ablation
In order to fully evaluate and prove the superiority and effectiveness of the constructed deep learning framework, ablation experiments on model structure are performed. We list six model structures, including No-attention-CNN, Attention-CNN, No-attention-BiGRU, Attention-BiGRU, No-attention-CNN-BiGRU and Attention-CNN-BiGRU, these results for ablation experiments on the benchmark set and independent test set are shown in Table 6.
As is shown in Table 6, attention represents multi-head self-attention. CNN, BiGRU and CNN-BiGRU with attention are better than CNN, BiGRU and CNN-BiGRU without attention, respectively. Prediction accuracies of all substructures including No-attention-CNN, Attention-CNN, No-attention-BiGRU, Attention-BiGRU, No-attention-CNN-BiGRU are lower than that of our constructed deep learning framework (Attention-CNN-BiGRU). The results for ablation experiments demonstrate the effectiveness of multi-head self-attention, the structure Attention-CNN-BiGRU is the most outstanding. Attention, CNN and BiGRU all make positive contributions for our model.
Visualization of learned features
To reflect the performance of learning features, we visualize the significant relevant model outputs by employing t-distributed stochastic neighbor embedding (t-SNE) to reduce dimensions and visualize the distribution of positive and negative samples. We extract and visualize fused features by multi-head self-attention from natural language processing features and hand-crafted features, and outputs of the CNN and BiGRU in Fig. 3. It clearly demonstrated that the fused features are disorderly, after CNN and BiGRU, the distribution of learned features tends to be separated as Kcr and non-Kcr. The visualization results using t-SNE tool further illustrate that the DeepMM-Kcr model can effectively capture subtle differences between Kcr and non-Kcr.

Visualization of samples on the benchmark set using t-SNE.
Performance comparison with different existing models
To demonstrate the validity and superiority of our model DeepMM-Kcr, we search for existing models for discriminating Kcr sites, including Position_weight15, CKSAAP_CrotSite16, Lightbm-crotsite19, Deep-Kcr22, BERT-Kcr24, Adapt-Kcr26. The measurements of ACC, Sn, Sp, MCC and AUC of the seven models are shown in Table 7.
As can be seen from Table 7, the ACC, Sn, Sp, MCC and AUC of our model DeepMM-Kcr on the independent test set are 0.856, 0.876, 0.835, 0.712 and 0.931, respectively. It is obvious that the performance of our model has improved significantly. The ACC of our model is improved by 0.256 compared with the Position_weight of the earlier model and by 0.004 compared with the better model Adapt-Kcr. We can see that although the Sp of our model is 0.835, which is 0.036 lower than that of the Deep-Kcr model, Sp and Sn have better balance in our model, as well as ACC, Sn, MCC and AUC all achieve the best results. The experimental results show that our model DeepMM-Kcr is superior to the existing model and is a very good calculation tool.
Discussion
The DeepMM-Kcr model use word embedding with transformer encoder, hand-crafted features with BiLSTM encoder, multi-head self-attention, CNN and BiGRU. The transformer encoding based on word embedding directly captures the global context by calculating the correlation weights of all position pairs. Hand-crafted features, which reflect statistical regularities and exhibit robustness to distributional changes. BiLSTM coding with hand-crafted features can better capture bidirectional information in the sequence, better establish long-term dependence relationship. Hand-crafted features can serve as complementary inputs in NLP. The multi-head self-attention fuses multiple features by learning multiple sets of attention weights in parallel. The attention weights of different heads can explain the focus of the model’s attention and help understand the decision-making process of the model. The combination of CNN and BiGRU can achieve excellent performance in complex tasks that require both detailed features and contextual understanding through collaborative design of local perception and global modeling.
Although our model has achieved good prediction performance, there are still some limitations, such as potential overfitting and limited data sources. In future work, for potential overfitting, we will use regularization to reduce model complexity and data augmentation methods to further address overfitting issue. Our model is only appropriate for dataset from HeLa cells. For restricted data sources, we will strengthen academic exchanges with peers to obtain richer data.
Kcr plays an important role in gene expression regulation, metabolism, and disease occurrence, and is a key node in the regulation of “metabolism epigenetics”. Kcr sites prediction using DeepMM-Kcr model provides a new target for cancer treatment, and in the future, high-resolution detection technology and site-specific editing tools need to be developed to deepen research.
Conclusions
In this paper, we combine the hand-crafted features and natural language processing features, and then use multi-head self-attention mechanism to fuse these features. A deep learning framework is constructed based on CNN, BiGRU and MLP for classification. Our model named DeepMM-Kcr, has better predictive accuracy and more balanced performance than previous tools. Therefore, our model is valid and greatly improved. The accurate identification of Kcr sites will not only contribute to the in-depth understanding of biological mechanisms, promote disease diagnosis and treatment, promote the development of biotechnology and agricultural applications, but also promote scientific research progress and interdisciplinary cooperation.
Data availability
Data is provided within the manuscript.
References
Soffer, R. L. Post-translational modification of proteins catalyzed by aminoacyl-tRNA-protein transferases. Mol. Cell. Biochem. 2 (1), 3–14 (1973).
Wold, F. In vivo chemical modification of proteins (post-translational modification). Annu. Rev. Biochem. 50 (1), 783–814 (1981).
Fu, J. Q., Wu, M. & Liu, X. Y. Proteomic approaches beyond expression profiling and PTM analysis. Anal. Bioanal. Chem. 410 (17), 4051–4060 (2018).
Huang, H. et al. SnapShot: Histone modifications. Cell 159(2), 458e1 (2014).
Wang, Y. et al. Identification of the YEATS domain of GAS41 as a pH-dependent reader of histone succinylation. Proc. Natl. Acad. Sci. U.S.A. 115 (10), 2365–2370 (2018).
Ramazi, S., Allahverdi, A. & Zahiri, J. Evaluation of post-translational modifications in histone proteins: A review on histone modification defects in developmental and neurological disorders. J. Biosci. (Bangalore). 45 (1), 135 (2020).
Tan, M. J. et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146 (6), 1016–1028 (2011).
Jiang, G. Y. et al. Protein lysine crotonylation: Past, present, perspective. Cell Death Dis. 12 (7), 1–11 (2021).
Wei, W. et al. Class I histone deacetylases are major histone decrotonylases: Evidence for critical and broad function of histone crotonylation in transcription. Cell Res. 27 (7), 898–915 (2017).
Li, K. & Wang, Z. Q. Histone crotonylation-centric gene regulation. Epigenetics Chromatin. 14 (1), 1–6 (2021).
Liu, S. M. et al. Chromodomain protein CDYL acts as a crotonyl-coA hydratase to regulate histone crotonylation and spermatogenesis. Mol. Cell. 67 (5), 853–866 (2017).
Jiang, G. C. et al. HIV latency is reversed by ACSS2-driven histone crotonylation. J. Clin. Invest. 128 (3), 1190–1198 (2018).
Yu, H. J. et al. Global Crotonylome reveals CDYL-regulated RPA1 crotonylation in homologous recombination-mediated DNA repair. Sci. Adv. 6 (11), eaay4697 (2020).
Huang, G. & Zeng, W. A discrete hidden Markov model for detecting histone Crotonyllysine sites. MATCH-Commun. Math. Comput. Chem. 75 (3), 717–730 (2016).
Qiu, W. R. et al. Identify and analysis crotonylation sites in histone by using support vector machines. Artif. Intell. Med. 83, 75–81 (2017).
Ju, Z. & He, J. J. Prediction of lysine crotonylation sites by incorporating the composition of k-spaced amino acid pairs into Chou’s general PseAAC. J. Mol. Graph. Model. 77 (0), 200–204 (2017).
Qiu, W. R. et al. iKcr-PseEns: Identify lysine crotonylation sites in histone proteins with pseudo components and ensemble classifier. Genomics 110 (5), 239–246 (2018).
Malebary, S. J., Rehman, M. S. U. & Khan, Y. D. iCrotok-PseAAC: Identify lysine crotonylation sites by blending position relative statistical features according to the Chou’s 5-step rule. PLoS ONE. 14 (11), e0223993 (2019).
Liu, Y. N. et al. Prediction of protein crotonylation sites through LightGBM classifier based on SMOTE and elastic net. Anal. Biochem. 609 (0), 113903 (2020).
Wang, R. L. et al. Characterization and identification of lysine crotonylation sites based on machine learning method on both plant and mammalian. Sci. Rep. 10 (1), 1–12 (2020).
Ahmed, S. et al. predML-Site: Predicting multiple lysine PTM sites with optimal feature representation and data imbalance minimization. IEEE/ACM Trans. Comput. Biol. Bioinf. 19 (6), 3624–3634 (2022).
Hao, L. et al. Deep-Kcr: Accurate detection of lysine crotonylation sites using deep learning method. Brief. Bioinform. 22 (4), 1–10 (2021).
Chen, Y. Z. et al. NhKcr: A new bioinformatics tool for predicting crotonylation sites on human nonhistone proteins based on deep learning. Brief. Bioinform. 22 (6), bbab146 (2021).
Qiao, Y., Zhu, X. & Gong, H. BERT-Kcr: Prediction of lysine crotonylation sites by a transfer learning method with pre-trained BERT models. Bioinformatics 38 (3), 648–654 (2022).
Khanal, J. et al. DeepCap-Kcr: Accurate identification and investigation of protein lysine crotonylation sites based on capsule network. Brief. Bioinform. 23 (1), bbab492 (2022).
Li, Z. T. et al. Adapt-Kcr: A novel deep learning framework for accurate prediction of lysine crotonylation sites based on learning embedding features and attention architecture. Brief. Bioinform. 23 (2), bbac037 (2022).
Dou, L. J. et al. iKcr_CNN: A novel computational tool for imbalance classification of human nonhistone crotonylation sites based on convolutional neural networks with focal loss. Comput. Struct. Biotechnol. J. 20, 3268–3279 (2022).
Khanal, J., Kandel, J. & Tayara, H. CapsNh-Kcr: Capsule network-based prediction of lysine crotonylation sites in human non-histone proteins. Comput. Struct. Biotechnol. J. 21, 120–127 (2022).
Gao, Y. et al. CD-HIT suite: A web server for clustering and comparing biological sequences. Bioinformatics 26 (5), 680–682 (2010).
Gwak, H. J. & Rho, M. ViBE: A hierarchical BERT model to identify eukaryotic viruses using metagenome sequencing data. Brief. Bioinform. 23 (4), bbac204 (2022).
Zhang, Y. et al. A novel antibacterial peptide recognition algorithm based on BERT. Brief. Bioinform. 22 (6), bbab200 (2021).
Vaswani, A. et al. Attention is all you need. Learning 5, 1-15 (2017).
Jumper, J. et al. Highly accurate protein structure prediction with alphafold. Nature 596 (7873), 583–589 (2021).
Shen, C. C., Wang, Q. Z. & Priebe, C. E. One-hot graph encoder embedding. IEEE Trans. Pattern Anal. Mach. Intell. 45 (6), 7933–7938 (2023).
Hasan, M. M. et al. Deepm5C: A deep-learning-based hybrid framework for identifying human RNA N5-methylcytosine sites using a stacking strategy. Mol. Ther. 30 (8), 2856–2867 (2022).
Kawashima, S., Pokarowski, P. & Pokarowska, M. AAindex: Amino acid index database, progress report 2008. Nucleic Acids Res. 36, D202–D205 (2008).
Hao, L. et al. iRice-MS: An integrated XGBoost model for detecting multitype post-translational modification sites in rice. Brief. Bioinform. 23 (1), bbab486 (2022).
Wang, L. Y. et al. NcRFP: A novel end-to-end method for non-coding RNAs family prediction based on deep learning. IEEE/ACM Trans. Comput. Biol. Bioinf. 18 (2), 784–789 (2021).
Chen, S. Z. et al. NeuroPred-CLQ: Incorporating deep Temporal convolutional networks and multi-head attention mechanism to predict neuropeptides. Brief. Bioinform. 23 (5), bbac319 (2022).
Long, H. X., Wang, M. & Fu, H. Y. Deep convolutional neural networks for predicting hydroxyproline in proteins. Curr. Bioinform. 12 (3), 233–238 (2017).
Cho, K. et al. Learning phrase representations using RNN encoder-decoder for statistical machine translation. Comput. Sci., 2014, 1–14, (2014).
Elman, J. L. Finding structure in time. Cogn. Sci. 14 (2), 179–211 (1990).
Jordan, M. I. Attractor dynamics and parallelism in a connectionist sequential machine. Cogn. Sci., 10(4), 531–546 (1986).
Li, J. W. et al. DeepAVP: A dual-channel deep neural network for identifying variable-length antiviral peptides. IEEE J. Biomedical Health Inf. 24 (10), 3012–3019 (2020).
Shoombuatong, W. et al. THRONE: A new approach for accurate prediction of human RNA N7-methylguanosine sites. J. Mol. Biol. 434 (11), 167549 (2022).
Zou, X. D. et al. Accurately identifying hemagglutinin using sequence information and machine learning methods. Front. Med. 10, 1281880 (2023).
Guo, X. Y., Jiang, Y. Z. & Zou, Q. Structured sparse regularized TSK fuzzy system for predicting therapeutic peptides. Brief. Bioinform. 23 (3), 1–11 (2022).
Fang, Y. T. et al. AFP-MFL: Accurate identification of antifungal peptides using multi-view feature learning. Brief. Bioinform. 24 (1), 1–12 (2023).
Zhang, S. L. & Jing, Y. Y. PreVFs-RG: A deep hybrid model for identifying virulence factors based on residual block and gated recurrent unit. IEEE/ACM Trans. Comput. Biol. Bioinf. 20 (3), 1926–1934 (2023).
Li, X. J., Zhang, S. L. & Shi, H. Y. An improved residual network using deep fusion for identifying RNA 5-Methylcytosine sites. Bioinformatics 38 (18), btac532 (2022).
Shi, H. Y., Zhang, S. L. & Li, X. J. R5hmCFDV: Computational identification of RNA 5-hydroxymethylcytosine based on deep feature fusion and deep voting. Brief. Bioinform. 23 (5), bbac341 (2022).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 12101480), and the Fundamental Research Funds for the Central Universities (No. QTZX23002).
Author information
Authors and Affiliations
Contributions
Yunyun Liang and Minwei Li wrote the main manuscript text and E.F. prepared Figs. 1, 2 and 3. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liang, Y., Li, M. A deep learning model for prediction of lysine crotonylation sites by fusing multi-features based on multi-head self-attention mechanism. Sci Rep 15, 18940 (2025). https://doi.org/10.1038/s41598-025-04058-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-04058-5
