
Abstract
Exosomes are crucial in the development of non-small cell lung cancer (NSCLC), yet exosome-associated genes in NSCLC remain insufficiently explored. The present study identified 59 exosome-associated differentially expressed genes (EA-DEGs) from the Gene Expression Omnibus (GEO) and GeneCards databases. Functional analysis indicated the involvement of the EA-DEGs in NSCLC-related pathways, including the cell cycle, DNA replication, and the immune response. Logistic regression, Least Absolute Shrinkage and Selection Operator (LASSO), support vector machine-recursive feature elimination (SVM-RFE), and random forest (RF) models were used to identify four key biomarkers, namely, PAICS, SLC2A1, A2M, and GPM6A, with diagnostic potential. Gene expression, pathological staging, and prognosis were analyzed in the lung adenocarcinoma (LUAD) subtype. Potential drugs targeting these biomarkers were identified, and an RNA-binding protein (RBP) and transcription factor (TF) regulatory network was constructed. Single-sample Gene Set Enrichment Analysis (ssGSEA) analysis highlighted the involvement of changes in the immune microenvironment. A diagnostic model providing new insight into the molecular mechanisms underlying NSCLC is proposed. However, further experimental verification is required to assess its practical value for NSCLC and other lung cancer subtypes before clinical application.
Similar content being viewed by others
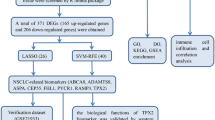


Introduction
Lung cancer remains the most prevalent malignancy worldwide, including in China, and is the leading cause of cancer-related mortality1. The annual death toll from lung cancer exceeds that of colorectal, breast, and prostate cancers combined. Despite an overall decline in cancer incidence since 2006, lung cancer continues to account for a substantial proportion of cancer-related deaths, largely due to late-stage diagnosis and limited treatment options2. Non-small cell lung cancer (NSCLC) constitutes approximately 85% of all lung cancer cases, with lung adenocarcinoma (LUAD) being the most common histological subtype. Evidence shows the presence of significant transcriptomic alterations and disruptions in cellular regulatory networks in NSCLC compared with normal lung tissues, underscoring the importance of identifying key genetic drivers and prognostic biomarkers for improved clinical outcomes3.
The early detection of NSCLC remains a major clinical challenge. Conventional diagnostic methods, such as X-rays, computed tomography (CT), positron emission tomography (PET), and tissue biopsies, often fail to detect lung cancer at an early stage, primarily due to inadequacies of the screening programs and the asymptomatic nature of early-stage disease4. As a result, many patients are diagnosed when the disease is already advanced when treatment options are limited and prognosis is poor. This highlights the urgent need for the discovery of novel biomarkers that could facilitate early detection, particularly in high-risk populations, such as smokers and individuals exposed to occupational carcinogens5. The identification of reliable biomarkers would not only improve early diagnosis but also aid personalized treatment approaches, thereby enhancing patient survival and quality of life.
The potential of using exosomes in cancer diagnosis and therapy has been recognized in recent years. Exosomes are small membrane-bound vesicles, typically between 30 and 150 nm in diameter, that are released into the extracellular environment by various cell types, including stem cells, immune cells, and cancer cells6,7. These vesicles play a crucial role in intercellular communication through their transfer of biologically active molecules, such as proteins, lipids, and nucleic acids, between cells. Notably, exosomes contribute to cancer progression by promoting angiogenesis, immune evasion, and metastasis, indicating their relevance for both tumor biology and potential clinical applications8. Their ability to shuttle molecular cargo between cells enables their modulation of the tumor microenvironment, influencing key processes such as immune response suppression and drug resistance.
Beyond oncology, exosomes have been implicated in various physiological and pathological conditions, including chronic obstructive pulmonary disease (COPD), neurodegenerative disorders, and regenerative medicine. Studies have demonstrated a variety of potential therapeutic applications, such as delivering bioactive molecules to damaged tissues, promoting wound healing, and even supporting hair regeneration in alopecia treatment9,10,11. Given their normal presence in body fluids, exosomes offer a minimally invasive approach to disease monitoring and biomarker discovery. In the context of cancer, tumor-derived exosomes carry molecular signatures reflective of the parental tumor, making them valuable candidates for liquid biopsy-based diagnostics12.
Recent research on the tumor microenvironment and immune modulation has further underscored the significance of exosomes in cancer research. Tumor-derived exosomes have been shown to alter the immune landscape by their transportation of immunosuppressive molecules, facilitating tumor progression and metastasis13. Consequently, exosome-based immunotherapies are being explored as potential strategies to counteract these tumor-supportive mechanisms. Moreover, as exosomes can carry tumor-specific biomarkers that can be detected in liquid biopsies, they hold great promise for early cancer detection and real-time monitoring of disease progression14.
Despite the recognized involvement of exosomes in lung cancer progression, research on their specific molecular contributions remains limited. The present study aimed to identify key genes associated with both exosomes and NSCLC, to elucidate the molecular mechanisms through which exosomes influence lung cancer development. It is hoped that the clarification of these interactions will provide novel insights that may aid in the development of targeted therapeutic interventions and improve prognosis prediction in NSCLC.
Materials and methods
Data sources
Gene expression profiles of NSCLC and normal tissues were retrieved from the Gene Expression Omnibus (GEO) database15 (https://www.ncbi.nlm.nih.gov/geo/). The datasetutilized were generated using different microarray platforms, ensuring a comprehensive analysis of gene expression profiles obtained via various analytical procedures. Specifically, the GSE116959 dataset was based on the Agilent-039494 SurePrint G3 Human GE v2 8 × 60 K Microarray 039381 (GPL17077) and included 68 samples, comprising 57 NSCLC samples and 11 peritumoral samples of normal tissue (Table S1). The GSE136043 dataset, generated using the Agilent-026652 Whole Human Genome Microarray 4 × 44 K v2 (GPL13497), consisted of five paired samples of fresh lung cancer and adjacent normal tissues (Table S2). GSE18842, processed on the Affymetrix Human Genome U133 Plus 2.0 Array (GPL570), contained 91 samples, including 46 tumor samples and 45 normal controls, the majority of which were paired except for three unpaired samples (Table S3). Meanwhile, GSE7670, based on the Affymetrix Human Genome U133A Array (GPL96), comprised 33 matched pairs of NSCLC and normal tissue samples (Table S4), along with corresponding clinical data, including age, sex, disease stage, subtype, and survival outcomes. Additional data on clinical factors and gene expression from patients with lung adenocarcinoma were obtained from The Cancer Genome Atlas database16 (Table S5), which included a total of 557 patients. Genes related to exosomes were sourced from the GeneCards database17 (https://www.genecards.org/) and literature searches; specific details are provided in Table S6.
Identification of EA-DEGs
Initially, the raw GEO microarray data (not log2-transformed) were normalized using the limma package (version 3.62.1) in R (version 4.4.2)18,19. The four GEO datasets were then merged, averaging the values of genes with multiple entries and retaining only a single representative row, and removing data with missing values. Batch effects were then eliminated using the empirical Bayesian method, which adjusts gene expression data by computing the batch-specific mean and variance for each gene, ensuring a similar distribution across different batches. This adjustment was performed using the Combat function from the R sva package (version 3.54.0)20. Visualization of the processed data was performed with the ggplot2 package (version 3.5.1)21. Principal component analysis (PCA) was conducted to verify the integrity of the resulting expression matrix; detailed information is provided in Table S7. Following this, differential expression was examined in the consolidated gene set, using the threshold criteria of logFC = 1 (equivalent to a 2-fold change) and adj.P.Val = 0.05 to identify differentially expressed genes (DEGs) between NSCLC and normal tissues. The results were displayed using a volcano plot, and a heatmap of the top 50 DEGs was created using the pheatmap package (version 1.0.12) in R22. Finally, the DEGs were cross-referenced with exosome-associated genes previously compiled, using the ggvenn package (version 0.1.10) to identify exosome-associated differentially expressed genes (EA-DEGs).
Functional enrichment analyses
The functions of the EA-DEGs were investigated using Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Gene Set Enrichment Analysis (GSEA). These analyses were performed using the clusterProfiler package (version 4.14.3) in R23, with a significance threshold set at p < 0.05. The enriched pathways and biological processes were visualized using the ggplot2 package in R.
Identification of potential diagnostic biomarkers for NSCLC
To develop the most effective diagnostic model, logistic regression was used to assess the EA-DEGs using the R glmnet package (version 4.1.8)24 to identify NSCLC-associated gene sets. Candidate genes were then identified using Least Absolute Shrinkage and Selection Operator (LASSO) regression. Subsequently, the e1071 package (version 1.7.16)25 was utilized to build a Support Vector Machine Recursive Feature Elimination (SVM-RFE) model. Additionally, the randomForest package (version 4.7.1.2) in R26 was employed to produce random forest (RF) curves and identify NSCLC-specific diagnostic genes. Genes that were identified consistently using all three methods were selected as the most promising candidate biomarkers.
To verify the performance of the candidate biomarkers, receiver operating characteristic (ROC) curves were plotted using the pROC package (version 1.18.5) in R27, and the diagnostic accuracy of the genes was assessed quantitatively by measuring the areas under the curves (AUCs). The rms package (version 6.8.2)28 was then used to develop nomograms providing a risk score assessment for predicting disease based on the identified diagnostic biomarkers.
Biological functions of model genes in NSCLC
Firstly, differences in the expression of four model genes were visualized between NSCLC and adjacent normal tissues using the ggplot2 package, offering a clear depiction of which genes were upregulated and which were downregulated. Variations in the expression levels of these genes were further analyzed in 483 LUAD and 347 normal tissue samples utilizing the GEPIA2 database29 (http://gepia2.cancer-pku.cn), applying a significance threshold of log2FC > 1 and a p-value < 0.01 to establish statistical significance. Subsequently, the expression patterns of these model genes in relation to the four principal pathological stages of the tumor were examined. Next, using the median expression levels as a cutoff, patients were categorized into groups with high and low expression as follows: PAICS high expression (239 patients), low expression (238 patients); GPM6A high expression (239 patients), low expression (237 patients); SLC2A1 and A2M each had 239 patients in both high- and low-expression groups. Kaplan-Meier curves were used to assess the associations of the genes with survival in LUAD patients, shedding light on their potential prognostic value.
Analysis of differential expression and survival in the TCGA database
Transcriptomic and clinical data from TCGA were used to investigate the differential expression and prognostic significance of the four candidate genes. Differential expression was analyzed using the “limma” package in R to compare expression levels in tumor tissues with those in neighboring normal tissues. The significance of differences was determined using the Wilcoxon rank sum test, and error discovery rate (FDR) adjustments were made for multiple comparisons. After the adjustment, genes fulfilling the criteria of p < 0.05, | log 2x change | > 1 were considered differentially expressed. For survival analysis, Kaplan-Meier curves and log-rank tests were used to assess the relationship between gene expression levels and overall survival (OS). The optimal cut-off value was determined using the survminer (version 0.5.0) and survival packages (version 3.7.0) in R based on the expression of each gene, and the patients were divided into high-expression and low-expression groups.
Analysis of immune infiltration
Single-sample Gene Set Enrichment Analysis (ssGSEA) was conducted using the GSEAbase package (version 1.68.0) to assess the associations of the EA-DEGs with immune function and to determine the infiltration scores of various immune cell types30, with detailed information provided in Table S8. The scores were visualized using the ggplot2 package. Lastly, to explore the correlations among immune checkpoint-related genes, the reshape2 (version 1.4.4), ggpubr (version 0.6.0), corrplot (version 0.95), and tidyverse packages (version 2.0.0) in R were employed for data manipulation and visualization31,32.
Prediction of targeted drugs
The Drug-Gene Interaction Database (DGIdb)33 (https://dsigdb.tanlab.org/DSigDBv1.0/) was utilized to identify potential drug targets related to the candidate biomarkers. Target drug enrichment analysis was conducted using the colorspace (version 2.1.1) and stringi packages (version 1.8.4)34, and the findings were visualized using Cytoscape35 (version 3.10.0). Additionally, the 3D structures of the target drugs and proteins encoded by the candidate biomarker genes were retrieved from the PubChem database36 (https://pubchem.ncbi.nlm.nih.gov/) and the Protein Data Bank (PDB)37 (https://www.rcsb.org/), respectively. Molecular docking simulations were undertaken to evaluate the interactions between drugs and their target proteins using the online platform CB-Dock238 (https://cadd.labshare.cn/cb-dock2/index.php).
Chromosomal localization and construction of RBP and TF networks
To further investigate the four genes, their chromosomal locations were evaluated using the circlize package (version 0.4.16)39. Information on RNA-binding proteins (RBPs) influencing the expression of the genes was sourced from the ENCORI database40 (https://rnasysu.com/encori/). Furthermore, transcription factors (TFs) that regulate the expression of the candidate biomarker genes were identified using the TRRUST database41 (https://www.grnpedia.org/trrust/). Relationship networks depicting the interactions between RBPs, TFs, and the diagnostic genes were developed and visualized using Cytoscape. This provided a detailed perspective on the regulatory mechanisms affecting these genes. Separate networks for RBPs and TFs were constructed to assess their specific contributions to gene regulation.
Quantitative PCR analysis in BEAS-2B and A549 cell lines
To verify the expression levels of the four model genes in vitro, quantitative reverse-transcription polymerase chain reaction (qRT-PCR) analysis was performed on BEAS-2B (normal bronchial epithelial) and A549 (lung adenocarcinoma) cell lines. Total RNA was extracted from cultured cells using the Animal RNA Isolation Kit with Spin Column (Beyotime, China) following the manufacturer’s protocol. The purity and concentration of the RNA were assessed using a NanoDrop 2000 spectrophotometer (Thermo Fisher, USA). Reverse transcription was carried out using the PrimeScript™ RT Master Mix (Takara, Japan), as directed. qRT-PCR was conducted using the TB Green Premix Ex Taq 2 (Takara) on a real-time PCR system (Bio-Rad, USA). The thermal cycling conditions were as follows: initial denaturation at 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s and 60 °C for 30 s. Gene-specific primers were designed using Primer-BLAST (NCBI) and were synthesized by Wanze Bio (Shenyang, China) (Table S9). GAPDH was used as the internal control, and relative gene expression levels were calculated using the 2−ΔΔCt method. Each reaction was performed in triplicate to ensure reproducibility. Statistical analysis was performed using GraphPad Prism42 (version 8.0.2). Expression differences between BEAS-2B and A549 cells were compared using independent t-tests, and a p-value < 0.05 was considered statistically significant.
Results
The identification of exosome-associated differentially expressed genes (EA-DEGs)
The GSE116959, GSE136043, GSE18842, and GSE7670 datasets were obtained from the GEO database. After normalization and correction for batch effects (illustrated in Fig. 1A and B), a consolidated gene expression profile of 276 samples was established, including 133 from normal and 143 from NSCLC tissues. The presence of batch effects was further confirmed by PCA analysis (Figure S1). Analysis of differentially expressed genes in the amalgamated dataset identified 917 genes that differed significantly in expression between NSCLC and normal tissues (Fig. 1C), as shown in Table S10. Figure 1D shows the top 50 most upregulated and 50 most downregulated genes. Additionally, 878 exosome-related genes sourced from the GeneCards database and related studies were included, resulting in the identification of 59 EA-DEGs (Fig. 1E).

Acquisition of EAD-EGs. (A,B) Comparison of the merged dataset before (A) and after (B) batch effect correction. (C) Differential expression analysis performed on the newly constructed dataset (logFC = 0.585, adj.P.Val = 0.05). (D) Identification of the top 100 differentially expressed genes(upregulates and downregulated genes). (E) Intersection of the identified genes with exosome-related genes.
Identification of molecular pathways and biological functions
GO, KEGG, and GSEA analyses were undertaken to. The results of the GO analysis are shown in Fig. 2A. In terms of the GO biological process (BP) category, EA-DEGs were found to be involved in the negative regulation of catalytic activity, cell adhesion, and the humoral immune response. In the cellular component (CC) category, associations with apical and basolateral plasma membranes, collagen-containing extracellular matrix, and tertiary granules were found, while in the molecular function (MF) category, the genes were enriched in enzyme activity inhibition, calcium-dependent protein binding, and molecular sequestration (Fig. 2B). KEGG pathway analysis indicated associations between EA-DEGs and various biological themes, including malaria, Salmonella infection, and processes in the hematopoietic system. Notably, EA-DEGs were also significantly enriched in resistance to antifolate drugs, pointing to their potential role in enhancing pathogen or cancer cell resistance to treatments such as methotrexate and sulfonamides (Fig. 2C). The GSEA results revealed the top five most significantly enriched signaling pathways (Fig. 2D), showing that EA-DEGs were associated with the cell cycle, DNA replication, amino acid biosynthesis, and base excision repair processes in NSCLC. Moreover, these genes were involved in interactions involving cytokines and their receptors, the p53 signaling pathway, JAK-STAT signaling, and chemokine signaling transduction pathways (Figure S2). These results underscore the potentially pivotal role of the EA-DEGs in NSCLC, particularly in nucleic acid metabolism, immune regulation, and the modulation of enzyme activity.

Exploration of the biological functions of EAD-EGs. (A) Results of GO enrichment analysis. (B) Visualization of GO enrichment analysis categorized into BP (Biological Process), CC (Cellular Component), and MF (Molecular Function). (C) Results of KEGG enrichment analysis, including pathways such as malaria, Salmonella infection, and antifolate resistance. (D) Results of GSEA enrichment analysis, highlighting pathways such as cell cycle and DNA replication.
Identification of EA-DEGs associated with NSCLC diagnosis
To explore the differences between NSCLC patients and healthy individuals, univariate logistic regression analysis was used to identify 59 EA-DEGs associated with NSCLC (Table S11). Three machine learning techniques, namely, LASSO, SVM-RFE, and RF, were used to identify critical EA-DEGs that could differentiate NSCLC from normal samples. Ten-fold cross-validation in LASSO regression resulted in the identification of 18 potential genes (Fig. 3A and B). The SVM-RFE algorithm identified 29 candidate genes (Fig. 3C and D), while RF analysis revealed 11 candidate genes (Fig. 3E and F). Insection of the LASSO, SVM-RFE, and RF findings led to the identification of four genes, namely, PAICS, SLC2A1, A2M, and GPM6A, as the most promising candidate biomarkers (Fig. 3G). To evaluate the diagnostic capability of these EA-DEGs, a logistic regression model using these four genes was developed in R to assess both the individual genes and the overall model. The ROC curves showed high diagnostic accuracy for each gene: PAICS (AUC = 0.944), SLC2A1 (AUC = 0.938), A2M (AUC = 0.927), and GPM6A (AUC = 0.992) (Fig. 4A). The constructed EAD-EG model exhibited an even higher AUC of 0.996 (Fig. 4B), surpassing the performance of the individual genes in distinguishing between NSCLC and healthy samples. Additionally, a nomogram was constructed for risk assessment of NSCLC (Fig. 4C), and the calibration curve exhibited high accuracy in predicting NSCLC risk based on the nomogram. Decision curve analysis confirmed that the EA-DEG model was effective in differentiating between NSCLC patients and healthy controls (Fig. 4D and E).

Identification of diagnostic genes for NSCLC. (A,B): Selection of 18 candidate genes from EAD-EGs using lasso logistic regression with 10-fold cross-validation. (C,D) Selection of 29 candidate genes using the SVM-RFE algorithm. (E,F) Analysis of the RF model yielding 11 candidate genes. (G) Integration of the results from the three machine learning models to identify the 4 most significant diagnostic genes.

This study validates the model and genes. (A) AUC curves of the 4 genes included in the model (PAICS = 0.944, SLC2A1 = 0.938, A2M = 0.927, GPM6A = 0.992). (B) AUC curve of the overall model (AUC = 0.996). (C) Construction of a nomogram for scoring the model’s genes. (D,E) Prediction of disease risk using the nomogram (D) and evaluation of the accuracy in distinguishing NSCLC from normal tissues (E).
Four candidate diagnostic biomarker genes exhibit significant differences in expression in NSCLC and influence patient prognosis
To further evaluate the biological attributes of the candidate biomarker EA-DEGs in NSCLC, the limma package in R was used to analyze differences in the expression of the four genes between normal and tumor tissues (Fig. 5A). The analysis revealed that PAICS (logFC = 1, adj.P.Val < 0.01) and SLC2A1 (logFC = 1, adj.P.Val < 0.01) were significantly upregulated in tumors, whereas A2M (logFC = 1, adj.P.Val < 0.01) and GPM6A (logFC = 1, adj.P.Val < 0.01) were significantly downregulated. Further validation using the GEPIA2 database demonstrated that the expression patterns of these genes in the LUAD subtype were similar to those observed in the GEO database (Fig. 5B). It was also found that the expression levels of PAICS (p < 0.01) and SLC2A1 (p < 0.01) were increased at more advanced tumor stages, whereas A2M expression (p < 0.05) decreased as tumor staging progressed; however, GPM6A expression showed no significant correlation with cancer staging (Fig. 5C). A correlation analysis of these genes indicated that PAICS was positively correlated with SLC2A1 but negatively correlated with A2M and GPM6A, while A2M was positively correlated with GPM6A (Fig. 5D). In a further assessment of the clinical significance of the genes, analysis using the GEPIA2 database indicated that PAICS (p < 0.01) and SLC2A1 (p < 0.01) overexpression was associated with worse clinical prognosis (Fig. 5E and F), whereas A2M and GPM6A did not significantly affect patient prognosis (Figure S3). In summary, PAICS, SLC2A1, and A2M show promise as biomarkers for assessing clinical staging in NSCLC, with PAICS and SLC2A1 also potentially serving as prognostic markers for these patients.

This study primarily observes the clinical data of the four genes in NSCLC. (A) Differential expression of the 4 genes between NSCLC and normal tissues determined by the LIMMA package. (B) Expression differences of the four genes between LUAD and normal tissues (log2FC > 1, p-value < 0.01). (C) Expression levels of the genes in LUAD at different stages (p < 0.05). (D) Correlation analysis between the feature genes. (E,F) Survival curves for the feature genes PAICS (p < 0.01) and SLC2A1 (p < 0.01).
Differential expression and survival analysis of the four model genes in TCGA data
To further validate the findings derived from the GEO dataset, the expression patterns and prognostic significance of the four model genes, namely, PAICS, SLC2A1, A2M, and GPM6A, were evaluated using transcriptomic and clinical data from the TCGA LUAD cohort. Differential expression was analyzed using the limma package in R, with statistical significance assessed via the Wilcoxon rank-sum test and FDR correction. Consistent with the GEO results, PAICS and SLC2A1 were found to be significantly upregulated in LUAD tumor tissues compared to adjacent normal tissues (adjusted p < 0.001, |log₂FC| > 1), while A2M and GPM6A were significantly downregulated (adjusted p < 0.01, |log₂FC| > 1) (Fig. 6A–D). To assess the prognostic relevance of the genes, Kaplan-Meier survival analysis was conducted to assess the OS of patients in the high- and low-expression subgroups using optimal cut-off values calculated via the survminer and survival R packages. The results showed that elevated expression of PAICS and SLC2A1 was significantly associated with worse OS (log-rank p < 0.001), while high expression of A2M and GPM6A was correlated with better OS (log-rank p < 0.05) (Fig. 6E–H). These TCGA-based results were highly consistent with those from the GEO database, providing robust validation of the differential expression and prognostic significance of the four model genes in LUAD. Together, they highlight the potential of these genes as clinically relevant biomarkers.

Differential expression and survival analysis of four genes. (A–D) Significant differences in the expression levels of PAICS, SLC2A1, A2M, and GPM6A were observed between tumor tissues and adjacent non-tumorous tissues (p < 0.01 for all genes). (E–H) High expression of PAICS (p < 0.001) and SLC2A1 (p < 0.001) was associated with poorer survival, whereas low expression of A2M (p = 0.0018) and GPM6A (p = 0.032) also correlated with reduced overall survival.
Immune infiltration scores
Exosomes play a significant role in the tumor immune microenvironment (TME), which is critical for the initiation and progression of NSCLC. To explore this further, ssGSEA was used to examine differences in the infiltration of immune cells between NSCLC and normal tissues (Fig. 7A). The analysis revealed that in tumor tissues, the levels of memory B cells (p < 0.05) and activated CD4 + T cells (p < 0.05) were elevated, whereas those of mast cells (p < 0.05) and eosinophils (p < 0.05) were significantly reduced compared to normal tissues. The correlations between immune cell infiltration and the four candidate NSCLC diagnostic genes were then assessed. The findings indicated that PAICS and SLC2A1 showed significant positive correlations with memory B cells (p < 0.05) and activated CD4 + T cells (p < 0.05), and negative correlations with mast cells (p < 0.05) and eosinophils (p < 0.05) in tumor tissues (Fig. 7B). These results suggest that the candidate diagnostic genes are intricately linked with the tumor immune microenvironment and may influence tumor progression by modulating the activity of immune cells.

Aim to analyze the immune infiltration levels of the diagnostic genes. (A) ssGSEA enrichment analysis to evaluate the expression differences of immune cells between NSCLC and normal tissues. (B) Correlation of the four feature genes with immune cells in NSCLC.
Screening of drugs affecting the candidate diagnostic genes
The results above indicate that the maintenance of normal levels of the four EA-DEGs is crucial for patient prognosis. As a further investigation, chemical compounds potentially influencing these genes were explored using the DSigDB database43 (https://dsigdb.tanlab.org/) for retrieving data on interactions. A drug enrichment analysis was then performed to identify chemical compounds significantly associated with the target genes (p < 0.05), with the findings presented using Cytoscape (Fig. 8A). The analysis showed that quinoline could significantly influence PAICS, while 7-hydroxyflavone and 16 other compounds influenced SLC2A1. Additionally, 6-aminohexanoic acid and 11 other compounds were significantly linked to A2M. A notable finding was the impact of dibenz[a, h]anthracene, a polycyclic aromatic hydrocarbon (PAH) (Fig. 8B and C), known for its potential carcinogenicity to humans, which is typically produced by the incomplete combustion of organic materials44. The results indicated that dibenz[a, h]anthracene could directly alter the expression levels of two target genes, SLC2A1 and A2M. Consistently, molecular docking analysis revealed strong binding affinities between the compound and both targets, with binding energies of -11.0 kcal/mol for SLC2A1 (Fig. 8D) and − 8.7 kcal/mol for A2M (Fig. 8E), respectively, underscoring its potential role in regulating the levels of these diagnostic genes.

Prediction of targeted drugs for diagnostic genes. (A) Network of targeted drugs related to PAICS, SLC2A1, and A2M. (B,C) Drug enrichment analysis of the intersected feature genes (p-value < 0.05, adj. p-value < 0.05), where SLC2A1 and A2M are significantly enriched for Dibenz[a, h]anthracene (B) and its 3D structure (C). (D) Visualization of molecular docking between SLC2A1 and targeted drugs and the binding energy (-11.0) (D). (E) Molecular docking of A2M with targeted drugs and the minimum binding energy (-8.7) (E).
Identification of the chromosomal localizations of the diagnostic genes and regulatory networks of RNA-Binding proteins and transcription factors
To gain a deeper understanding of the four EA-DEGs, the chromosomal locations of genes were mapped (Fig. 9A). The ENCORI database was used to identify RBP targets for the genes, which were then visualized using Cytoscape. It was observed that each gene interacted with 20 RBPs, of which CSTF2T and CPSF1 could interact with all four diagnostic genes (Fig. 9B). Additionally, the TRRUST database was used to identify to identify TFs that regulate the diagnostic genes (Fig. 9C). Four TFs, namely, HDAC5, ATM, TP53, and HIF1A, were identified that could effectively regulate SLC2A1 expression, whereas NFKB1, STAT3, and TFCP2 were found to modulate the expression of A2M.

Regulatory network of feature genes. (A) Chromosomal localization of the feature genes. (B) RNA-binding protein (RBP) regulatory network of the four genes. (C) Transcription factor network regulating SLC2A1 and A2M.
qRT-PCR analysis of the four model genes in BEAS-2B and A549 cells
The results of the qRT-PCR analysis revealed significant differential expression of the four model genes between the BEAS-2B (normal bronchial epithelium) and A549 (lung adenocarcinoma) cell lines. The relative expression levels of each gene were normalized to GAPDH and analyzed using the 2−ΔΔCt method. Compared to BEAS-2B cells, PAICS (P = 0.0061) and SLC2A1 (P = 0.0001) were significantly upregulated in A549 cells, suggesting a potential oncogenic role. Conversely, A2M (P = 0.01) and GPM6A (P = 0.0002) exhibited significant downregulation in A549 cells, implying a possible tumor-suppressive function. The fold-change values for each gene are presented in Fig. 10. Statistical analysis using independent t-tests confirmed that the expression differences between BEAS-2B and A549 cells were statistically significant for all four genes (p < 0.05). These findings indicate that these model genes may play distinct roles in the progression of lung adenocarcinoma and could serve as potential biomarkers for further investigation.

Expression levels of the four genes in BEAS-2B and A549 cells. (A,B) PAICS (p = 0.0061) and SLC2A1 (p = 0.0001) were significantly upregulated in A549 cells relative to BEAS-2B cells. (C,D) The expression levels of A2M (p = 0.01) and GPM6A (p = 0.0002) were significantly lower in A549 cells compared to BEAS-2B cells.
Discussion
NSCLC is the most prevalent form of lung cancer, with distinct manifestations and outcomes compared to other lung cancer subtypes. Despite advancements in diagnostic and therapeutic strategies, lung cancer remains a leading cause of cancer-related deaths globally, primarily due to challenges in early detection, diminishing returns from adjuvant and neoadjuvant treatments, and advanced stage at diagnosis. Lung cancer frequently lacks distinctive symptoms, complicating the identification of early-stage disease and delaying the onset of intervention. Prior studies have shown that low-dose computed tomography (LDCT) screening can reduce lung cancer mortality by up to 20%; however, its widespread implementation is restricted by costs, radiation risks, and a high rate of false positives45. Although traditional diagnostic approaches such as LDCT and liquid biopsy have improved clinical screening and monitoring, they are associated with notable limitations. LDCT, while effective for imaging-based early detection, is associated with high false-positive rates and limited molecular insight. Liquid biopsy methods, including analysis of circulating tumor DNA (ctDNA) and circulating tumor cells (CTCs), offer minimally invasive alternatives but often lack sensitivity in early-stage disease and provide insufficient information about the TME. In contrast, molecular biomarkers, particularly those derived from exosomes, offer a promising combination of early detectability, biological specificity, and functional relevance. These vesicles not only reflect the molecular status of early-stage tumors but also play active roles in cancer progression and immune modulation. As such, the integration of exosome-based biomarkers into current diagnostic workflows may improve the precision of early detection and support more individualized therapeutic strategies.
Moreover, NSCLC cells have evolved mechanisms to escape immune detection, contributing to poor responses to treatment. Specifically, patients exhibiting a clinical pattern of minimal acquired resistance Oligo (OligoAR < 4) show a unique pattern of acquired resistance to PD-(L)1 therapy46. Although treatment with PD-1/PD-L1 checkpoint inhibitors has marked a significant advance in lung cancer therapy, their efficacy is limited to a subset of patients. Furthermore, resistance, both primary and acquired, ultimately leads to disease progression in these patients. A deeper investigation into this resistance has highlighted issues such as T-cell dysfunction, impaired antigen presentation, and alterations within the TME47.
Exosomes, as vesicles secreted by cells, play a pivotal role in tumor progression by their transport of bioactive molecules. These exosomes contribute to the growth and spread of lung cancer, as well as influencing the immune environment of the tumor. For instance, CircSATB2 is found in high levels within the serum exosomes of lung cancer patients and is associated with promoting the growth, migration, and invasion of NSCLC cells via exosomal transfer. This molecule also has the potential to trigger abnormal proliferation in normal human lung epithelial cells48. Furthermore, exosomes carrying CircVMP1 promote NSCLC progression and resistance to the chemotherapy drug cisplatin by affecting the miR-524-5p-METTL3/SOX2 signaling pathway49. Additionally, cancer-derived CircUSP7 exosomes impact the miR-934/SHP2 axis in NSCLC, leading toCD8 + T cell dysfunction, facilitating immune evasion through PD-1 resistance, and thus expediting the progression of the disease50. The four candidate genes identified in this study may contribute to immune evasion in NSCLC through distinct but complementary mechanisms. PAICS and SLC2A1 promote metabolic reprogramming and a suppressive TME, limiting immune cell infiltration and enhancing PD-L1 expression51,52. A2M modulates cytokine signaling and antigen presentation, potentially facilitating immune tolerance53. Although the immunoregulatory role of GPM6A remains less defined, emerging evidence suggests its involvement in vesicle trafficking and exosomal immune modulation54. Together, these genes may act synergistically to facilitate tumor immune escape and progression.
Exosomes can promote tumor progression in various cancers by influencing the TME. However, information on the role of exosomes in NSCLC is limited. Therefore, the present research focused on identifying exosome-associated genes to develop a diagnostic model based on multiple methodologies and analytical layers, to improve the detection of early-stage NSCLC and suggest novel clinical treatment strategies. The enrichment analyses indicated significant associations between exosomal activity in the TME and critical cellular processes such as cell cycle progression and DNA replication in tumor cells. The constructed diagnostic model included four disease-specific genes, and their relevance and diagnostic accuracy were confirmed using comprehensive correlation analysis. Further investigations demonstrated that the expression levels of these key genes in LUAD were significantly associated with the pathological stage of the tumor, underscoring their potential utility for diagnosing early-stage NSCLC in clinical settings. Moreover, it was also found that these disease-specific genes could interact directly with polycyclic aromatic hydrocarbons (PAHs), compounds identified as Group 2 A carcinogens and produced predominantly by the incomplete combustion of organic materials. PAHs are frequently encountered in daily life, emanating from sources such as cigarette smoke, barbecued foods, and emissions from vehicles or industrial activities55,56.
This study presents an exosome-based gene signature with potential application in NSCLC diagnosis, prognosis, and mechanistic understanding. the findings provide a foundation for future translational research. Targeting exosome release, uptake, or cargo content may open new avenues for early detection and therapeutic intervention in lung cancer. However, this study is not without limitations. First, most of the findings were based on bioinformatics analyses and in vitro experiments. Second, despite the identification of key exosomal components and associated pathways, the precise molecular mechanisms linking these elements to immune modulation and metastasis require further experimental elucidation. Third, the reliance on public datasets such as TCGA and the GEO database could have introduced potential biases, and several of the external validation cohorts had relatively small sample sizes, which may limit generalizability. Future studies incorporating large-scale clinical samples, animal models, and functional assays will be essential to confirm and extend these findings toward clinical translation.
Conclusion
This study used the GeneCards database along with published literature to pinpoint genes associated with exosomes, which were then integrated with an analysis of differentially expressed genes between NSCLC and normal tissue samples to construct an EA-DEGs dataset. The findings indicated that these genes are biologically active in tumors, notably in promoting tumor cell cycle progression and DNA replication. Three machine learning approaches were applied, leading to the identification of four candidate diagnostic genes, namely, PAICS, SLC2A1, A2M, and GPM6A, for the construction of a disease diagnostic model. The performance of the model was found to be excellent, as shown by the AUC values of ROC curves and nomograms. Extensive analysis of clinical data for each of the genes revealed their specific roles in the development and progression of NSCLC. Immune infiltration analysis was also conducted to assess the effectiveness of the model in terms of the tumor immune microenvironment. Moreover, drugs that interacted with the proteins encoded by the genes were identified, including a PAH compound targeting SLC2A1 and A2M directly. Additionally, the chromosomal locations of the model genes were evaluated, as were regulatory networks involving RBPs and TFs that influence the expression of the genes. Overall, the study offers novel approaches and insights for the early detection and treatment of NSCLC, with profound clinical implications.
Data availability
Data is provided within the manuscript or supplementary information files.
Abbreviations
- NSCLC:
-
Non-small cell lung cancer
- EA-DEGs:
-
Exosome-associated differentially expressed genes
- GEO:
-
Gene Expression Omnibus
- LASSO:
-
Least absolute shrinkage and selection operator
- SVM-RFE:
-
Support vector machine recursive feature elimination
- RF:
-
Random Forest
- LUAD:
-
Lung adenocarcinoma
- RBPs:
-
RNA-binding proteins
- TFs:
-
Transcription factors
- ssGSEA:
-
Single-sample gene set enrichment analysis
- TME:
-
Tumor immune microenvironment
- COPD:
-
Chronic obstructive pulmonary disease
- PCA:
-
Principal Component Analysis
- DEGs:
-
Differentially expressed genes
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- GSEA:
-
Gene Set Enrichment Analysis
- AUC:
-
Area under the curve
- DGIDB:
-
Drug-gene interaction database
- PDB:
-
Protein Data Bank
- qRT-PCR:
-
Quantitative real-time polymerase chain reaction
- BP:
-
Biological process
- CC:
-
Cellular component
- MF:
-
Molecular function
- PAH:
-
Polycyclic aromatic hydrocarbon
- LD-CT:
-
Low-dose computed tomography
References
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 74(3), 229–263 (2024).
Siegel, R. L., Giaquinto, A. N. & Jemal, A. Cancer statistics. CA Cancer J. Clin. 74(1), 12–49 (2024).
Relli, V. et al. Abandoning the notion of non-small cell lung cancer. Trends Mol. Med. 25(7), 585–594 (2019).
Gridelli, C. et al. Non-small-cell lung cancer. Nat. Rev. Dis. Primers. 1, 15009 (2015).
Nooreldeen, R. & Bach, H. Current and future development in lung cancer diagnosis. Int. J. Mol. Sci. 22(16), 8661 (2021).
Lee, K. W. A. et al. Clinical applications of exosomes: a critical review. Int. J. Mol. Sci. 25(14) (2024).
He, C. et al. Exosome theranostics: biology and translational medicine. Theranostics 8(1), 237–255 (2018).
Zhang, J. et al. Exosome and exosomal microrna: trafficking, sorting, and function. Genom. Proteom. Bioinform. 13(1), 17–24 (2015).
Popowski, K. et al. Exosome therapeutics for lung regenerative medicine. J. Extracell. Vesicles. 9(1), 1785161 (2020).
Kost, Y. et al. Exosome therapy in hair regeneration: A literature review of the evidence, challenges, and future opportunities. J. Cosmet. Dermatol. 21(8), 3226–3231 (2022).
Salarpour, S. et al. The application of exosomes and exosome-nanoparticle in treating brain disorders. J. Mol. Liq. 350, 118549 (2022).
Sun, Y. & Liu, J. Potential of cancer cell-derived exosomes in clinical application: a review of recent research advances. Clin. Ther. 36(6), 863–872 (2014).
Xu, Z. et al. Exosome-based immunotherapy: a promising approach for cancer treatment. Mol. Cancer. 19(1), 160 (2020).
Yu, W. et al. Exosome-based liquid biopsies in cancer: opportunities and challenges. Ann. Oncol. 32(4), 466–477 (2021).
Edgar, R., Domrachev, M. & Lash, A. E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30(1), 207–210 (2002).
Tomczak, K., Czerwińska, P. & Wiznerowicz, M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp. Oncol. (Poznan Poland). 19(1a), A68–77 (2015).
Stelzer, G. et al. The GeneCards Suite: from gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 54, 1.30.1–1.3 (2016).
R Core Team. R: A language and environment for statistical computing. https://www.r-project.org/ (R Foundation for Statistical Computing, 2023).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43(7), e47 (2015).
Leek, J. T. et al. The Sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28(6), 882–883 (2012).
Gustavsson, E. K. et al. Ggtranscript: an R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinformatics 38(15), 3844–3846 (2022).
Wang, Y. et al. Unveiling the key genes, environmental toxins, and drug exposures in modulating the severity of ulcerative colitis: a comprehensive analysis. Front. Immunol. 14, 1162458 (2023).
Wu, T. et al. ClusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2(3), 100141 (2021).
Li, Y., Lu, F. & Yin, Y. Applying logistic LASSO regression for the diagnosis of atypical Crohn’s disease. Sci. Rep. 12(1), 11340 (2022).
Shi, H. et al. Identifying and validating GSTM5 as an Immunogenic gene in diabetic foot ulcer using bioinformatics and machine learning. J. Inflamm. Res. 16, 6241–6256 (2023).
Alderden, J. et al. Predicting pressure injury in critical care patients: a machine-learning model. Am. J. Crit. Care. 27(6), 461–468 (2018).
Robin, X. et al. pROC: an open-source package for R and S + to analyze and compare ROC curves. BMC Bioinform. 12(77) (2011).
Li, M. et al. Recognition of refractory Mycoplasma pneumoniae pneumonia among Myocoplasma pneumoniae pneumonia in hospitalized children: development and validation of a predictive nomogram model. BMC Pulm. Med. 23(1), 383 (2023).
Tang, Z. et al. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 47(W1), W556–w60 (2019).
Xiong, X. et al. Identification of a novel defined inflammation-related long noncoding RNA signature contributes to predicting prognosis and distinction between the cold and hot tumors in bladder cancer. Front. Oncol. 13, 972558 (2023).
Bian, J. et al. Identification and prognostic biomarkers among ZDHHC4/12/18/24, and APT2 in lung adenocarcinoma. Sci. Rep. 14(1), 522 (2024).
Li, C., Deans, N. C. & Buell, C. R. Simple tidy genecoex: A gene co-expression analysis workflow powered by tidyverse and graph-based clustering. R Plant. Genome. 16(2), e20323 (2023).
Freshour, S. L. et al. Integration of the Drug-Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 49(D1), D1144–d51 (2021).
Shi, H. et al. Mechanism of action of tripterygium wilfordii for treatment of idiopathic membranous nephropathy based on network pharmacology. Ren. Fail. 44(1), 116–125 (2022).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13(11), 2498–2504 (2003).
Kim, S. et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 47(D1), D1102–d9 (2019).
Berman, H. M. et al. Protein Data Bank. Nucleic Acids Res., 28(1): 235–242. (2000).
Liu, Y. et al. CB-Dock2: improved protein-ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 50(W1), W159–w64 (2022).
Gu, Z. et al. Circlize implements and enhances circular visualization in R. Bioinformatics 30(19): 2811–2812. (2014).
Li, J. H. et al. StarBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 42(Database issue), D92–D97 (2014).
Han, H. et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 46(D1), D380–d6 (2018).
GraphPad, S. GraphPad Prism version 9.5.0 for Windows. https://www.graphpad.com/ (GraphPad Software, 2024).
Vasudevan, V., Gayathri, K. V. & Krishnan, M. E. G. Bioremediation of a pentacyclic PAH, Dibenz(a,h)Anthracene- A long road to trip with bacteria, fungi, autotrophic eukaryotes and surprises. Chemosphere 202, 387–383 (2018).
Yoo, M. et al. DSigDB: drug signatures database for gene set analysis. Bioinformatics 31(18), 3069–3071 (2015).
Gasparri, R., Sabalic, A. & Spaggiari, L. The early diagnosis of lung cancer: critical gaps in the discovery of biomarkers. J. Clin. Med. 12(23) (2023).
Schoenfeld, A. J. et al. Systemic and oligo-acquired resistance to PD-(L)1 blockade in lung cancer. Clin. cancer Research: Official J. Am. Association Cancer Res. 28(17), 3797–3803 (2022).
Sun, J. Y. et al. Resistance to PD-1/PD-L1 blockade cancer immunotherapy: mechanisms, predictive factors, and future perspectives. Biomark. Res. 8, 35 (2020).
Zhang, N. et al. Circular RNA circSATB2 promotes progression of non-small cell lung cancer cells. Mol. Cancer. 19(1), 101 (2020).
Xie, H. et al. Exosome-transmitted circVMP1 facilitates the progression and cisplatin resistance of non-small cell lung cancer by targeting miR-524-5p-METTL3/SOX2 axis. Drug Deliv. 29(1), 1257–1271 (2022).
Chen, S. W. et al. Cancer cell-derived exosomal circUSP7 induces CD8(+) T cell dysfunction and anti-PD1 resistance by regulating the miR-934/SHP2 axis in NSCLC. Mol. Cancer. 20(1), 144 (2021).
Liu, X. S. et al. NPM1 is a prognostic biomarker involved in immune infiltration of lung adenocarcinoma and associated with m6A modification and glycolysis. Front. Immunol. 12, 724741 (2021).
Hao, B. et al. Identification of SLC2A1 as a predictive biomarker for survival and response to immunotherapy in lung squamous cell carcinoma. Comput. Biol. Med. 171, 108183 (2024).
Lee, H. C. et al. Downregulated ADAMTS1 incorporating A2M contributes to tumorigenesis and alters tumor immune microenvironment in lung adenocarcinoma. Biology. 11(5) (2022).
Wang, Z. et al. Induction of m(6)A methylation in adipocyte exosomal LncRNAs mediates myeloma drug resistance. J. Exp. Clin. Cancer Res. 41(1), 4 (2022).
Zelinkova, Z. & Wenzl, T. The occurrence of 16 EPA PAHs in food - a review. Polycycl. Aromat. Compd. 35(2-4), 248–284 (2015).
Corona, L. et al. Removal of two high molecular weight PAHs from soils with different water content. Bull. Environ Contam. Toxicol. 99(5), 619–624 (2017).
Acknowledgements
The authors would like to thank the researchers who provided open access to the raw data.
Author information
Authors and Affiliations
Contributions
ZS: Conceptualization, Data curation, Methodology, Software, Validation, Visualization, Writing – original draft TD: Conceptualization, Data curation, Methodology, Software, Writing – original draft GY: Conceptualization, Data curation, Writing – original draft YS: Conceptualization, Data curation XX: Conceptualization, Methodology, Supervision, Writing – review & editing.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Ethics Committee approved the protocol of this study at First Affiliated Hospital of Jinzhou Medical University (Approval No: KYLL2024316).
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, Z., Du, T., Yang, G. et al. Identification of exosome-related genes in NSCLC via integrated bioinformatics and machine learning analysis. Sci Rep 15, 22962 (2025). https://doi.org/10.1038/s41598-025-04485-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-04485-4
