
Abstract
The link between gut microbiota and COVID-19 has been previously established, but the role of the oral fungal microbiota in this context remains underexplored. This study aimed to characterize the oral mycobiome of COVID-19 convalescents. Saliva samples were collected from three groups: COVID-19 patients treated with antibiotics (group I), COVID-19 patients without antimicrobial treatment (group II), and healthy volunteers (group III) from the University Hospital and from the University Dental Clinic in Kraków. The samples were analyzed using next-generation sequencing (NGS) targeting the ITS-1 region. Statistically significant differences in dental indices (Plaque Index—PI, Bleeding on Probing—BOP, Winkel Tongue Coating Index—WTCI) were observed between the convalescent groups (I and II) and the control group (III). At the phylum level, significant alpha diversity differences were noted across all groups. At the genus level, alpha diversity was significant for all tested indices. Beta diversity analysis revealed no significant differences between groups I and II at either the phylum or genus levels (p > 0.05). The most abundant genera were Candida and Malassezia, with Candida being more prevalent in group I (88.11%) compared to group II (78.20%) and group III (45.81%). Linear discriminant analysis (LDA) indicated Candida as overrepresented in group I, and Malassezia as a characteristic marker in group II. Additionally, we observed the higher evenness of mold species like Aspergillus and Penicillium in the control group compared to the COVID-19 convalescents. COVID-19 convalescents, particularly those treated with antibiotics, exhibited worse oral condition compared to healthy controls, with Candida overgrowth strongly associated with antibiotic use.
Similar content being viewed by others

Introduction
The oral cavity is an entry route for many pathogens into the human body, and its health condition influences the frequency and occurrence of various diseases, both within the oral cavity itself and systemic diseases such as blood diseases (leukemia, anemia)1 atherosclerosis, pulmonary infections, diabetes mellitus, osteoporosis, and kidney diseases2. Recurrent gingival inflammation, periodontal disease, and other dental ailments activate the host inflammatory response and tend to the chronic inflammatory condition in the oral cavity and the whole body3,4. Chronic inflammation can lead to further consequences, including qualitative and quantitative changes in the oral microbiome, which is the second largest microbial community in the human body, following the gut in terms of diversity and abundance5.
The human oral microbiome, a dynamic and complex of bacteria, fungi, viruses, archea and protozoa coexists in a tightly regulated community. The interactions between the host and microbiota are multifaceted and reciprocal, influencing numerous biological functions crucial for health and immune homeostasis. Digestive beneficial microbiota are considered key drivers in shaping protective mucosal immunity4. However, the diverse microbial groups can exhibit both synergistic and antagonistic interactions, where the suppression of one group can impact the growth of another. The disruption of microbial balance, including oral bacterial and fungal microbiota, can contribute to the onset or progression of various diseases4,6. Previous studies have reported that the oral-gut axis affects human health and disease states such as influenza, respiratory syncytial virus, human immunodeficiency virus, HBV, asthma6,7, and bacterial, parasitic, and fungal infections.
Early in the COVID-19 pandemic, it became clear that the oral cavity was a critical site of infection with the SARS-CoV-2 virus, and saliva was identified as a potential vector for virus transmission8. In this context, many researchers have evaluated the link between the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection and the microbiota since the COVID-19 pandemic until now9,10. Likewise, our team also reported the changes in the bacterial community composition of patients recovered from COVID-19 disease11,12.
Conversely, a viral infection can disrupt the indigenous microbiome, resulting in altered susceptibility and disease severity through changes in community structure and function. Evidence suggests that SARS-CoV-2 infection may predispose patients to bacterial co-infections and superinfections, leading to increased disease severity and mortality13. Furthermore, dysbiosis in individuals with COVID-19 tends to progress toward microbiota homeostasis as viral clearance and recovery occur, indicating that the health status of microbial community is a reliable marker of disease recovery6,14.
Moreover, during the pandemic COVID-19 commonly requires treatment using antibiotics, which usage is frequently associated with the onset of microbiome disturbation15. The rate of antimicrobial prescriptions for COVID-19 patients was significantly higher (94–100%) than necessary, given that secondary infections occur in only 10–15% of these patients. Furthermore, the prevalence of antibiotic use among COVID-19 patients was markedly higher in low- and middle-income countries compared to high-income countries (89% vs. 58%)16.
However, limited focus has been placed on the human oral mycobiome in relations to COVID-19 progression and outcomes. It may be attributed to the relatively low proportion of fungi within the human microbiome6,17. Additionally, many oral fungi are not cultivable with traditional microbiological techniques. Nonetheless, metagenomic studies have revealed that the commensal oral microbiome harbors more fungi than previously recognized4,6. Furthermore, evidence has suggested that fungi play significant roles in the development of host immune responses, thereby modulating the extent of the inflammatory response and impacting human health and disease4,6,15,18.
In connection with the above, in the present study, we analyzed the profile of salivary mycobiome using MiSeq (Illumina) ITS-1 sequencing in response to convalescent COVID-19 received antibiotics during hospitalization and without antimicrobials. Additionally, we evaluated oral indices to assess the oral hygiene status of the studied populations. Furthermore, we assessed the relationships between salivary fungal and bacterial communities in the patients and volunteers participating in the study.
Materials and methods
Study population, dental examination and sample collection
Initially, 60 participants were enrolled in the study. Of these, 60 saliva samples were sequenced, but 15 were excluded from the analysis due to chimeric reads. Consequently, 45 samples underwent bioinformatic analysis and were included for further examination. Participants were divided into three groups:
-
group I—convalescents who suffered from COVID-19 and received antibiotics during hospitalisation (n = 16),
-
group II—COVID-19 convalescents who did not receive antibiotics during hospitalisation (n = 17)
-
group III—healthy volunteers (control group) who have not been infected with the SARS-CoV-2 virus and not treated with antibiotics (n = 12).
The study was conducted between June 2021 and February 2022. The inclusion criteria for the study groups were the absence of systemic diseases, with exception of hypertension. Patients from groups I and II were previously treated in hospital conditions due to COVID-19 disease at the Temporary Hospital for COVID-19 patients at the Trauma Center of Emergency Medicine and Disasters of the University Hospital in Krakow. Volunteers for the third (control) group were recruited from the University Dental Clinic in Kraków. Treatment in group I was administered according to the guidelines of the Polish Ministry of Health, based on the recommendations of the World Health Organization19. Patients were mainly treated with ceftriaxone (i.v. 2 g/12 h). Four patients from group I were treated in different schemes: two of them with levofloxacin alone (i.v. 500 mg/12 h), third with ceftriaxone (i.v. 2 g/12 h) and ciprofloxacin (i.v. 400 mg/12 h), fourth with ceftriaxone (i.v. 2 g/12 h) and levofloxacin (i.v. 500 mg/12 h). The treatment lasted 6–7 days. Antifungals were not prescribed either to COVID-19 patients or to control volunteers. Additionally, an acrylic partial denture was present in one patient from group I (no. 2812) and one patient from group II (no. 2793).
The dental inclusion criteria were having at least six teeth, refraining from oral hygiene activities, and not eating for 12 h before sampling. For the control group (group III), as for group II, an additional inclusion criterion was not taking antibiotics for at least 3 months before the study. In addition, the inclusion criterium was not taking probiotics at least 3 months before the study. The lack of any of the criteria resulted in exclusion from the study. The selected indices such as Decayed, Missing and Filled Teeth index (DMFT) and Plaque Index simplified (PI) and selected plaque-related indices such us Bleeding on Probing (BOP) were measured. Additionally, tongue type and Winkel Tongue Coating Index (WTCI) were determined during a dental examination as we described before12. Dental examination, biological materials collection, protection, and transport were performed by a dentist as we also described before12.
The biological material tested in this study was unstimulated saliva, collected when the patients were considered convalescents (negative result of the PCR test for SARS-CoV-2) on the day of discharge as described before12. For patients from groups I and II, a dental examination was performed on the day of discharge from the hospital. The dental examination procedure was the same for patients in all study groups.
ITS sequencing preparation and next generation sequencing
The collected samples were used for DNA isolation beginning with the placement of the clinical material in a plastic tube with glass beads (OMNI International a PerkinElmer Company, United States). Next, 200 µl of 75 mM NaOH (POCH, Poland) was added and the mixture was shaken in the ball homogenizer Bead Ruptor Elite (Bead Mill Homogenizer, OMNI International, United States) Then, it was incubated for 10 min at 95 °C in thermoblock (Eppendorf, Hamburg, Germany). Following incubation, 500 µl of buffer with β-mercaptoethanol was added (POCH, Poland), along with the enzyme: 5.0 µl of lyticase (4000 U; A&A Biotechnology, Poland). Samples were incubated at 37° C for 30 min. The remaining steps of the isolation were performed in an automated nucleic acid extraction instrument CroBEE (GeneProof, Brno, Czech Republic) as described previously11. After extraction, the isolates of DNA were amplified by PCR (T100 Thermal Cycler, BioRad, California, United States), with primers targeting the ITS-1 regions of the rDNA gene (5′→3′; F: GTAAAAGTCGTAACAAGGTTTC 2 R: GTTCAAAGAyTCGATGATTCAC with attached overhang adapter sequences (F:TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG and R: GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG) added to the internal primer attached to the 5′ end compatible with Illumina’s recommendations to prepare ITS libraries20. The obtained amplicons were used to prepare a genomic library for NGS sequencing in the MiSeq platform (Illumina, San Diego, California, United States) according to the Illumina protocol with modifications described by Sroka‑Oleksiak et al.21.
Bioinformatics and statistical analysis
Raw sequencing reads were controlled for quality using FastQC software and included into QIIME222 analysis pipeline. In this pipeline, initial reads quality control, filtering, denoising and feature table generation was done using DADA223 software. Reads taxonomic classification was done using a pre-trained Naive Bayes classifier (sklern) and the q2-feature-classifier plugin. This classifier was trained on the Unite v9.0 (18.07.2023) database24 which collects the eukaryotic nuclear ribosomal ITS region and clusters reference sequences at similarity levels ranging from 97 to 99%, based on expert curation of ITS sequence variability. A confidence threshold of 0.7 was applied for taxonomic assignments. The obtained output was filtered to retain only reads that matched Fungi kingdom classification.
Before further analysis, low abundance features (less than 4 reads in all samples and prevalence in < 20%) were removed. Alpha (Observed number of taxa, Chao1, Shannon and Simpson indexes) and beta (Jensen-Shannon and Jaccard) diversity parameters analysis for remaining data was done using MicorbiomeAnalyst software25. Permutational Multivariate Analysis of Variance (PERMANOVA) based on Bray–Curtis dissimilarity matrices was used to evaluate the differences in community composition between groups.
For differential microbiome analysis, a Linear Discriminant Analysis Effect Size (LEfSe) was performed using MicorbiomeAnalyst software. LefSe determines the operational taxonomic units most likely to explain differences between classes by coupling standard tests for statistical significance with additional tests encoding biological consistency and effect relevance26. As significantly altered taxa, ones with false discovery rate FDR < 0.05 and LDA score |x|> 2 were considered.
In the analysis of dental indices association with mycobiome composition, initially, the Shapiro–Wilk test was used to assess data distribution normality. Since most the data deviated from a normal distribution, the Spearman rank correlation coefficient was employed for correlation analysis between quantitative traits and selected fungi levels. For categorical variables with two levels (e.g. sex), the Mann–Whitney U test was applied. For categorical variables with multiple levels (such as tongue type), the nonparametric Kruskal–Wallis ANOVA test and Dunn’s post-hoc tests were used.
To enable the correlation of fungal and bacterial data from saliva samples, the results of the current ITS-1 analysis were compared with the findings of the 16S rRNA analysis, which was the subject of our team’s previous publication11.
Parameters such as DMFT, PI, BOP, as well as tongue type and WTCI were compared among groups using Kruskal–Wallis or ANOVA/PERMANOVA tests.
All methods were performed in accordance with the relevant guidelines and regulations. The study was performed in accordance with the Declaration of Helsinki.
Results
Demographic and oral health profiles of study participants
Generally, 45 patients (15 women and 30 men, aged 26–77 years, M ± SD: 51.31 ± 14.26) were included for the analysis. In terms of gender (p = 0.886; Chi square test), age (p = 0.316; ANOVA test) and number of teeth (p = 0.869), the groups studied in our research were well-matched, with no statistically significant differences in these parameters (Fig. 1; Table 1S).

Characteristics of study groups: demographics, oral health status, and a schematic representation of COVID-19 and its impact on the oral microbiome composition.
Statistically significant differences were observed in dental indices: PI (p < 0.001; ANOVA test), BOP index (p < 0.001; ANOVA test) and WTCI (p = 0.004; ANOVA test result with Welch’s correction) when comparing I versus III and II and III groups, respectively. Regarding the DMFT group I was statistically significantly different from group III (p = 0.01; ANOVA test result with Welch’s correction). Additionally, statistically significant differences were observed in tongue type (p = 0.001; Fisher’s exact test). In both group I (56.3%) and group II (47.1%), the presence of a coated tongue was the most frequently observed condition compared to the control group. Furthermore, a statistically significant difference was found when comparing group I with group III (p = 0.001). Conversely, the normal tongue type was most prevalent in group III (91.7%), with statistically significant differences observed when comparing group I with group III and group II with group III (p = 0.001) (Fig. 1; Table 1S).
Analysis of salivary microbiota composition and diversity
The raw data obtained by sequencing consisted of 2.6 M reads, from 17 to 92 K per sample. After initial filtering and denoising, from 2,7 to 72 K non—chimeric reads per sample (with an average number of reads of 22.1 K) was used for taxonomic classification. The taxonomic diversity profiling was performed at phylum (L2) and genus (L6) level.
Briefly, the alpha biodiversity of oral fungal microbiota expressed by the Shannon and Simpson indexes was statistically significant (p = 0.009 and p = 0.032, respectively) at the L2 level, taking into account comparisons among groups I, II, and III altogether. At the L6 level, alpha diversity was statistically significant in all tested indexes (Fig. 2).

Alpha diversity box plots representing various indices across the studied groups (Group I vs. Group II vs. Group III).
There were no significant differences between the three groups either at the phylum level (L2) or the genus level (L6), further (Fig. 1S) analysis of beta diversity was performed, comparing differences between pairs of groups individually (Table 1). The detailed analysis revealed, that the beta diversity was similar in the convalescents who suffered from COVID-19 received antibiotics during hospitalization (group I) and those who had not received antibiotics (group II) (p > 0.05) at both the L2 and the L6 levels, considering all tested indexes (Table 1). No significant differences were found in comparisons between I group and III group (control) and group II and the III group (control), respectively.
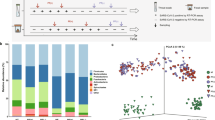
Afterwards, a systematic assessment of the fungal profile at the taxonomic level L2 and at L6 was conducted to provide a comprehensive overview and detailed analysis of differences in the oral fungal microbiota composition of the tested samples. At L2 level, we identified OTUs corresponding to three types (Basidiomycota, Ascomycota, and an unidentified phylum) (Fig. 3). In all three groups, Ascomycota fungi were the most prevalent in the saliva samples, accounting for 89.02% in I group, 80.86% in II group, and 64.00% in control group (III group). The second most abundant phylum was Basidiomycota which accounted for 10.81% versus 18.77% and 31.18% of the total in I, II and III group, respectively. An unidentified fungi phylum was in the minority (0.17%-I group vs. 0.37%-II group and 4.57% for control group). Additionally, fungi _phy_Incertae_sedis was found in the control group (0.25%).

Differences in the abundance of saliva samples at phylum taxonomic level (L2), taking into account whole study groups (A) and individual patients’ profiles (B). Legend: red circle—patients with an acrylic partial denture.
At the genus level (L6), OTUs corresponding to 21 genera were identified in healthy volunteers. Both groups of patients—those who received antibiotic (group I) and those who were not subjected to this treatment (group II) showed lower genus richness compared to the control group. Dominant genera were Candida, and Malassezia in all tested groups. However, at the same time, as the percentage of Candida (88.11% I group vs. 78.20% II group vs. 45.81% III group) in the group decreased, this phenomenon was accompanied by an increase in the genus of Malasezzia (9.67% I group vs. 17,82% II group vs. 21.45%- III group) and increase of Debaromyces (0.56% I group vs. 1.51% II group vs. 4.14% III group) (Fig. 4).

Differences in the abundance of saliva samples at genus taxonomic levels (L6), taking into account whole study groups (A) and individual patients’ profiles (B). Legend: red circle—patients with an acrylic partial denture.
Linear discriminant analysis effect size (LEfSe) for mycobiota comparison
LEfSe histograms were utilized to identify oral fungal genera that differed significantly in abundance among the three analyzed groups, highlighting the statistical and biological distinctions between them. Through linear discriminant analysis (LDA), five genera were found to be significantly more abundant in group I, while three genera were more prevalent in group II when all three study groups were compared together (Fig. 5A). Notably, in the comparisons between groups I and II, as well as groups I and III, the genus Candida remained the most significantly different for COVID-19 convalescents who had received antibiotics during hospitalization (Fig. 5B,C). It is also worth noting that the LEfSe found that Malassezia genus was associated with the COVID-19 convalescents who did not receive antibiotics during hospitalization (II group) when comparing with all three tested groups (Fig. 5A) and with I group alone (Fig. 5B). While comparing group II with control group (III), the Candida and Malassezia genera were most abundant (Fig. 5D).

The LEfSe analysis for the identification of the most differentially abundant genera of the oral mycobiome across three groups: group I—COVID-19 convalescents who received antibiotics during hospitalization, group II—COVID-19 convalescents who did not receive antibiotics during hospitalization, and group III—healthy individuals who have neither been infected with SARS-CoV-2 nor treated with antibiotics (control group).
Correlations between oral fungi, dental indices, and oral bacteria
The correlations between oral fungi and dental indices were analyzed in greater detail at the L6 taxonomic level (Table 2).
To evaluate the correlations between oral bacteria and fungi at the L6 taxonomic level, we focused on bacterial genera identified as indicators in our previous study11. In group I, correlation analyses showed a very strong and statistically significant negative correlation between Candida and Porphyromonas genera indicating an inverse relationship between genera from different biological kingdoms. In group I, correlation analyses revealed that the Candida coefficient was significantly negatively correlated with the coefficients for Porphyromonas (p < 0.001) In contrast, the Yarrowia genus was very strongly and positively associated with Aggregatibacter, indicating that as the abundance of Yarrowia increases, the levels of Aggregatibacter also tend to rise. In the control group of healthy individuals who had neither been infected with SARS-CoV-2 nor treated with antibiotics, a significant positive correlation was observed between Aspergillus and Cladosporium (p = 0.011) and Amyloporia and Cladosporium (Table 2).
In addition to quantitative characteristics, we also examined the association between a qualitative feature—tongue morphology type—and fungi at the L6 taxonomic level. Despite differences in tongue morphology among the groups studied (Fig. 1; Table 1S), no statistically significant associations were found between tongue morphology types and the types of fungi in any group.
Patient-specific analysis of salivary mycobiota composition
Considering individual patient profiles, we observed that in patients wearing acrylic partial dentures, the fungal communities were notably similar. Specifically, OTU’s sample no. 2793 from a patient in group II and OTU sample no. 2812 from a patient in group I both exhibited similar fungal genera, including Debaromyces and Candida, with Malassezia additionally present in sample 2812. Furthermore, a high percentage of the Debaromyces genus was identified in sample no. 2799 from a volunteer (with Penicillium also present). In five out of 16 patients (no. 2785, 2806, 2848, 2886, 2871) who underwent antibiotic therapy (group I) and 3 out of 17 (no. 2789, 2828, 2852) those who were not subjected to this treatment (group II) Candida genus was found more than 98% abundant. Furthermore, a high percentage of the Malassezia genus was observed in four samples from the first study group, including one of Malasseziaceae_gen_Incertae_sedis (no. 2856), and in four samples from the second study group, compared to only two samples from healthy individuals. The Kazachstania genus was characteristic of two samples (no. 2815, 2816) from group II. Rhodotorula was most abundant in patient 2839, who was treated with Biotraxon. Meanwhile, the Yarrowia genus was found in higher abundance in samples no. 2783 and no. 2862 from healthy controls, while Hanseniaspora genus was characteristic in one sample (no. 2853) from a volunteer group (III group) (Fig. 4B).
Discussion
The condition of the oral cavity and its resident microbiota are interrelated elements, the balance or disruption of which may lead to the onset of various diseases. Therefore, maintaining the health of the oral cavity—including the soft tissues and teeth—is crucial for the overall well-being of the human body. Earlier research has demonstrated that periodontal pockets create a conducive environment for the development of microbiota abundant in bacterial and viral species, which may invade the tissues through the frequently ulcerated pocket epithelium. This could enable the SARS-CoV-2 virus to enter the body either directly through these damaged epithelial layers or indirectly by enhancing the expression of angiotensin-converting enzyme 2 (ACE-2) receptors27. In the study we conducted, people who were infected with SARS-CoV-2 had significantly higher values of dental indicators (DMFT, PI, BOP, WTCI) than those who were not infected with the virus. Elevated DMFT, PI, and BOP indices among COVID-19 convalescents reflect chronic structural changes (such as carious lesions and loss of tooth attachment) and increased periodontal inflammation, likely attributable to the prolonged disease course. Our findings align with previous findings that local gingival inflammation may promote early and long-term SARS-CoV-2 colonization8. However, in contrast to previous reports8, we did not observe the increased prevalence of geographic tongue as a characteristic manifestations of COVID-19. Additionally, we did not identify any statistically significant correlations between the morphologically distinct tongue types and the types of fungi in any of the study group. A critical aspect of our study is that it was conducted during the COVID-19 pandemic, requiring strict adherence to hospital sanitary protocols. Performing dental examinations and collecting samples under these conditions posed a significant challenge, as ensuring patient and examiner safety while maintaining the highest standards of medical practice was of utmost importance. Opposite to our research, Larvine et al. based on results self-reported by patients such as bleeding gums, painful gums, and loose teeth which were used as proxies for diagnosing periodontal disease. Larvine et al. assumed that painful and bleeding gums were associated with mild to moderate periodontal disease, while loose teeth indicated severe periodontal disease. In contrast, participants who did not report these symptoms were classified as the control group, representing individuals without periodontal disease28. Another approach to assessing oral hygiene and health during the SARS-CoV-2 pandemic was to perform radiological examinations29. Among the teams conducting dental examinations on COVID-19 patients, DMFT, PI, and BOP indexes were comparable to ours. In the study by Costa et al.30 (DMFT index = 18), which assessed oral health status and adverse COVID-19 outcomes in hospitalized patients, we gained similar findings (DMFT index = 20.00 ± 3.18 for group I and DMFT = 17.65 ± 6.60 for group II, respectively). Moreover, in Anand P et al. research the participants with COVID-19 had significantly higher mean values of plaque scores (0.77 ± 0.50 vs. 0.29 ± 0.30, respectively, p < 0.001) and gingival bleeding scores (0.62 ± 0.24 vs. 0.29 ± 0.20, respectively; p < 0.001) compared to the controls, which is similar to our findings for I and II group31. Based on the observations mentioned above, it can be concluded that there was an association between COVID-19 and periodontitis severity. Gingival bleeding and dental plaque accumulation were also more frequent among COVID-19 patients.
The statistically significant differences in alpha diversity observed between COVID-19 convalescents who received antibiotic treatment during hospitalization and those who recovered from COVID-19 without antibiotic therapy, as well as individuals who were not infected with the virus, were unexpected. We hypothesize that the observed outcomes can be attributed to the significant interindividual variability in microbiota composition, which varies widely among individual convalescents and volunteers included in our research (Fig. 4B). In contrast to our findings, three other studies found no significant differences in alpha diversity among the patients studied6,10,18 although it should be noted that each of these studies analysed different types of biological samples, including mouthwashes (Gupta et al.), throat swabs (Wei N. et al.) and tongue coating samples (Xiaobo Hu et al.). However, one of the study included patients during SARS-CoV-2-infection and healthy controls who were not treated with antibiotics18. In the study by Wei et al., recovered COVID-19 patients were examined 1 year after infection but were also not treated with antibiotics for 6 months prior to the examination6. The study analyzed 71 COVID-19 patients, 22 recovered patients, 36 suspected cases, 36 recovered suspected cases, and 132 controls10.
In turn, our study did not observe a clearly distinct mycobiota composition for the individual groups measured with the beta diversity, which indicates similarity between them (Fig. 1S). We hypothesize that the absence of significant differences among our patients’ mycobiota could be attributed to the mild nature of their COVID-19 cases. The relatively mild severity of the disease in these patients may not have been a sufficient factor to induce notable alterations in the mycobiota. Results similar to ours were obtained by Wei et al. studying differences in the oral fungal community of throat swabs in recovered COVID-19 and healthy controls. The above observations are explained by some authors as being due to the significantly lower variability of fungi compared to the higher variability of bacteria comunity6.
Nevertheless, we have observed the fungal quantitative composition in COVID-19 recovered patients, both those who received antibiotics during hospitalization and those who did not, differ from that of healthy individuals. The phyla Ascomycota and Basidiomycota were predominant in both COVID-19 patients and healthy individuals, but the percentage share of Ascomycota to Basidiomycota was highest in patients who took antibiotics during COVID-19 treatment. This can be explained, among other things, by the fact that the high prevalence of the Candida genus, which not only exhibited the highest relative abundance in Group I but was also identified as a key differentiating taxon in the Linear Discriminant Analysis (LDA).
The Candida genus is well known as the common opportunistic fungal genus in the oral cavity32. The overgrowth of Candida can lead to thrush or invasive candidiasis and candidemia33 and even promoting oral cancer34 and colon cancer35. Candida pathogenicity can be promoted via immunosuppression, the prolonged use of antimicrobials and corticosteroids, and direct cell damage by SARS-CoV-233. In healthy people bacteria in the oral cavity colonize the mucus membrane and can limit the invasion of fungi and viruses36. Our previous studies on the oral microbiome in patients who recovered from COVID-19 showed that SARS-CoV-2 colonization and the administration of antibacterial drugs limited bacterial growth11. It might contributed to the loss of mechanisms inhibiting the development of opportunistic fungi and initiated Candida invasion what we observed in our present research. Genus Candida may destroying immune cells and promote inflammation itself having virulence factors such as production of phospholipases, proteases, hemolysin, candidalysin, and biofilm formation responsible for adherence and invasion37. The overrepresentation of Candida genus in both the I and II groups compared to the control group, as demonstrated by percentage composition and the LefSE method, suggests a potential risk for the development of oral candidiasis in these patients. Previous studies have reported the occurrence of candidiasis in COVID-19 patients, particularly in those who experienced severe cases or were immunocompromised16,38. However, our study cohort consisted of patients who were in good health at the time of examination and sampling, which was conducted on the day of discharge following hospitalization for COVID-19. These patients did not exhibit impaired immunity and were not admitted to the intensive care unit, conditions that are often associated with secondary infections, including oral candidiasis, post-COVID-1916. In the study by Singulani et al. fungal infections were diagnosed after more than two weeks of hospitalization16. Therefore, fungal infections often develop later, progress more slowly than bacterial infections, and are influenced by various risk factors, for example antibiotic administration. Our patients were mainly treated by ceftriaxone as the many other patients hospitalized from COVID-19 during the first phase of the SARS-CoV-2 pandemic39. Maeda et al. used azithromycin and levofloxacin (narrow-spectrum antibiotics) for the mild COVID-19 group, and meropenem, and tazobactam/piperacillin (broad-spectrum antibiotics), for the severe group infected by SARS-CoV-2 virus. They found that the composition of Candida species and the fungal beta diversity showed no significant difference between the severe and mild study groups. Moreover, Maeda et al. reported the alpha diversity of mycobiota in the severe group was reduced, but no difference was observed between the groups with and without meropenem. They stated the effect of antibiotic use on mycobiota alterations is not significant in the gut40. We did not find any studies describing this phenomenon in the oral cavity. Another factor considered to be conducive to the occurrence of oral candydosis are dental prostheses, which is confirmed by the results of studies reporting the occurrence of denture stomatitis in 60% of patients with COVID-19 using dentures, most often caused by Candida albicans41. In our study, only two patients reported the use of dentures. These patients had a characteristic quantitative composition of fungi, in which we observed an overrepresentation of Candida genus. Surprisingly, in the saliva from patients recovered from COVID-19 wearing the dentures were fungi from the Debaromyces genus. Previous studies reported an increase in the number of Debaromyces spp. in inflammatory bowel diseases in children and mouse model42,43. Fungi from the genus Debaromyces can maintain inflammation, which undoubtedly occurs in patients wearing dentures and suffering from COVID-19, which we confirmed by the increased dental coefficients observed in these patients. However, the observations we made based on these two patients should be repeated in a larger study group.
Going deeper, we analyzed the mycobiome in parallel with the bacteriobiome from our previous research11 which will help understand synergism and antagonism between genera of fungal and bacterial kingdoms, which may promote or prevent COVID-19 infection and outcomes. Microorganisms such as bacteria and fungi release various signaling molecules and chemicals that can either inhibit or support each other’s growth and survival, thereby influencing the host’s immune response and overall health4,10. A well-documented fungal-bacterial interaction in the oral microbiome is Candida albicans colonization linked to Streptococcus spp. and Porphyromonas gingivalis, which alters microbial composition, worsens carious lesions, and accelerates tooth demineralization44,45. Sztukowska et al. described that the InlJ protein from P. gingivalis binds to the Als3 protein on C. albicans hives, which promotes the formation of common biofilms and potentially increases the virulence of these microorganisms45. Bartnicka et al. found that in biofilms C. albicans creates a protective environment for P. gingivalis under aerobic conditions, which is possible due to the expression of adhesion proteins such as Als3 and aspartyl proteases Sap6 and Sap946. In our study, Candida genus was strongly negatively correlated with Porphyromonas, in group I. Our findings suggest that Candida in saliva may act as a limiting factor for Porphyromonas colonization. One possible explanation is the impact of antibiotic treatment administered during the hospitalization of COVID-19 patients, which may have affected Porphyromonas populations. Additionally, in saliva, the biofilm formed by Candida and Porphyromonas is less structured compared to dental plaque. As a result, an increase in Candida may exert an opposite effect, restricting the growth of P. gingivalis rather than promoting its survival, as observed in plaque-associated biofilms. Another potential mechanism at play in convalescent patients involves the immune response triggered by Candida. This fungal species can stimulate host immunity, leading to the production of defensins, cytokines, and other antimicrobial factors that may suppress P. gingivalis proliferation. Furthermore, Candida produces various metabolites, including organic acids and hydrogen peroxide, which could influence the growth and viability of anaerobic bacteria such as Porphyromonas. Furthermore, heme competition may explain the antagonism between Candida albicans and Porphyromonas gingivalis in saliva. Both microorganisms rely on heme for growth, so Candida may outcompete Porphyromonas limiting its proliferation. Unlike in subgingival biofilms, where C. albicans enhances P. gingivalis virulence under heme scarcity, in saliva, weaker biofilm formation and transient interactions may favor Candida genus dominance47. Our findings, along with those of other researchers, highlight the context-dependent nature of the Candida-Porphyromonas relationship, underscoring the need for further investigation. In the group of COVID-19 convalescents who received antibiotic treatment during hospitalization, we also observed a strong positive correlation between fungi of the Yarrowia genus and bacteria of the Aggregatibacter genus. Unlike Candida, the presence of Yarrowia in the oral cavity is poorly documented, and its pathogenic potential remains largely unknown48. We suspect that in this case, Yarrowia genus might serve as a physical scaffold for Aggregatibacter, facilitating its adhesion to oral surfaces. In oral biofilms, interactions between bacteria and fungi often contribute to mutual protection against the host immune system. Our other results, which show weaker correlations with various microorganisms (Table 2), suggest that the biological roles of Yarrowia are not uniform and may depend on the surrounding environment as well as the fungal and bacterial communities with which the Yarrowia genus coexists.
We discovered that Malassezia was overrepresented in COVID-19 convalescents who did not receive antibiotics during hospitalization (group II). Dupuy et al. identified the Malassezia genus as a dominant fungal genus in the oral microbiome. Moreover, their research demonstrates that Malassezia is consistently present in healthy individuals, suggesting a potential role in oral homeostasis. Notably, their findings align closely with our results, further supporting the significance of Malassezia in the oral environment49. Additionally, Malassezia has been implicated in cases of systemic inflammation during the progression of COVID-1913.
At the same time, we noticed the overrepresentaion of molds such as Aspergillus and Penicillium in the saliva of our control group (Fig. 4C,D). Furthermore, we showed a strong positive correlation of the genus Aspergillus with the genus Cladosporium and Amyloporia and Cladosporium. For example, Aspergillus and Cladosporium have previously been identified as genus-level constituents of the oral mycobiome32,49. Due to their ubiquitous nature, the presence of these fungi in the oral cavities of healthy individuals was interpreted as most likely of environmental origin, from food and breathing49. Wei et al.6 reported results similar to ours regarding the Aspergillus genus, observing a prevalence of 1.34% in COVID-19 recovered patients compared to 2.49% in healthy controls. We would also like to point out that we have demonstrated fungi in individual patients that are not yet well characterised as residents or oral pathogens. Nevertheless, they were present in the saliva of the patients we examined or of the control subjects. For example, we noticed characteristic overrepresentation of Kazachstania genus due to the high percentage of this fungus in two patients in particular. The genus Kazachstania is rarely described as a human pathogen to date50. In our patients, one of them when the Kazachstania genus was present, there was no Candida genus (no. 2815, Fig. 3B), but in the second one, there was the co-presence of Kazachstania and Candida genera. It should be investigated in the larger study group in the future. In the study designed by Kralova et al. Kazachstania heterogenica var. weizmannii was discovered as murine commensal that antagonizes C. albicans colonization and significantly reduces its abundance in animal model51. This suggests that Kazachstania may play a protective role in reducing Candida proliferation, potentially mitigating fungal overgrowth in the oral cavity.
We made significant efforts to elucidate the alterations and roles of oral fungi in patients who have recovered from COVID-19; however, certain limitations remain in this study. One of the limitations of our research was the challenges associated with existing fungal taxonomy databases. Enhancing these databases through a more comprehensive classification of taxa would significantly aid in advancing our understanding of oral fungal ecology in mycobiome research. Another aspect to consider is that our findings are based on samples collected from a specific geographic area, so the observed changes in composition might not be representative of COVID-19 patients from different regions or countries. Similar limitations were noted in the studies by Rizzello et al.13.
Additional large-scale prospective studies are necessary to compare the mycobiome of COVID-19 patients with that of contemporary healthy individuals. Such research could provide further validation of our findings and contribute to corroborating existing data. Despite some shortcomings of our study, we achieved our goal, which was to provide a broader perspective on SARS-CoV-2 infection and to evaluate the interaction between oral health and microbiota, with a particular focus on the mycobiome.
Conclusions
The LDA analysis highlights the Candida genus as more prevalent among convalescents who were treated with antibiotics during their hospitalization for COVID-19. Based on the LDA analysis, it can be concluded that antibiotic use promotes the growth of Candida fungi. This, in turn, reinforces the well-established fact that antibiotic use should be justified by the suspicion of mixed viral-bacterial infections or bacterial infections secondary to viral infection.
Based on the LDA method, we identified fungi from the Malassezia genus as a characteristic marker in the group of COVID-19 convalescents who were not treated with antibiotics.
Analysis of the patients’ microbiome indicates possible connections between fungi and bacteria and their mutual influence on the disruption of homeostasis after COVID-19 and the use of antibiotic therapy. This is another argument strengthening the hypothesis that the microbiome dynamically interacts with the host and its disorders affect homeostasis.
Data availability
Raw sequencing data from this study are available in the NCBI Sequence Read Archive (SRA) under BioProject accession number PRJNA1191366. The data can be accessed using the following link: https://www.ncbi.nlm.nih.gov/sra/PRJNA1191366.
References
Łobacz, M. et al. The bloody crossroads: Interactions between periodontitis and hematologic diseases. Int. J. Mol. Sci. 25, 6115. https://doi.org/10.3390/ijms25116115 (2024).
Iyer, P. Oral cavity is the gateway to the body: Role of oral health professionals: A narrative review. J. Calif. Dent. Assoc. 51, 1. https://doi.org/10.1080/19424396.2023.2193372 (2023).
Brandini, D. A. et al. Covid-19 and oral diseases: Crosstalk, synergy or association?. Rev. Med. Virol. 31, 2226. https://doi.org/10.1002/rmv.2226 (2021).
Cannon, R. D. Oral fungal infections: Past, present, and future. Front. Oral Health 3, 838639. https://doi.org/10.3389/froh.2022.838639 (2022).
Boyapati, R., Dhulipalla, R., Kolaparthy, L. K. & Bodduru, R. COVID-19 and oral implications: An updated review. J. Oral Maxillofac. Pathol. 25, 400–403. https://doi.org/10.4103/jomfp.jomfp_198_21 (2021).
Wei, N. et al. Characterization of oral bacterial and fungal microbiome in recovered COVID-19 patients. BMC Microbiol. 23, 123. https://doi.org/10.1186/s12866-023-02872-3 (2023).
Talukdar, D. et al. Association of gut microbial dysbiosis with disease severity, response to therapy and disease outcomes in Indian patients with COVID-19. Gut Pathog. 15, 22. https://doi.org/10.1186/s13099-023-00546-z (2023).
Huang, N. et al. SARS-CoV-2 infection of the oral cavity and saliva. Nat. Med. 27, 892–903. https://doi.org/10.1038/s41591-021-01296-8 (2021).
Wu, Y. et al. Altered oral and gut microbiota and its association with SARS-CoV-2 viral load in COVID-19 patients during hospitalization. NPJ Biofilms Microbiomes 7, 61. https://doi.org/10.1038/s41522-021-00232-5 (2021).
Hu, X. et al. Oral fungal alterations in patients with COVID-19 and recovered patients. Adv. Sci. (Weinh) 10, e2205058. https://doi.org/10.1002/advs.202205058 (2023).
Brzychczy-Sroka, B. et al. Oral microbiota study of the patients after hospitalisation for COVID-19, considering selected dental indices and antibiotic therapy using the next generation sequencing method (NGS). J. Oral Microbiol. 15, 2264591. https://doi.org/10.1080/20002297.2023.2264591 (2023).
Brzychczy-Sroka, B. et al. Standardization of the protocol for oral cavity examination and collecting of the biological samples for microbiome research using the next-generation sequencing (NGS): own experience with the COVID-19 patients. Sci. Rep. 14, 3717. https://doi.org/10.1038/s41598-024-53992-3 (2024).
Rizzello, F. et al. Signatures of disease outcome severity in the intestinal fungal and bacterial microbiome of COVID-19 patients. Front. Cell Infect. Microbiol. 14, 1352202. https://doi.org/10.3389/fcimb.2024.1352202 (2024).
Talaga-Ćwiertnia, K. et al. New insights into diversity of the upper respiratory tract microbiota and its relationship with SARS-CoV-2 viral load in the nasopharyngeal epithelial cells in patients with COVID-19. Pol. Arch. Intern. Med. 133, 16442. https://doi.org/10.20452/pamw.16442 (2023).
Pisano, M. et al. Oral candidiasis in adult and pediatric patients with COVID-19. Biomedicines 11, 846. https://doi.org/10.3390/biomedicines11030846 (2023).
Singulani, J. L. et al. The impact of COVID-19 on antimicrobial prescription and drug resistance in fungi and bacteria. Braz. J. Microbiol. 53, 1925–1935. https://doi.org/10.1007/s42770-022-00818-x (2022).
Chin, V. K. et al. Mycobiome in the gut: A multiperspective review. Mediators Inflamm. 2020, 9560684. https://doi.org/10.1155/2020/9560684 (2020).
Gupta, A. et al. Oral dysbiosis and its linkage with SARS-CoV-2 infection. Microbiol. Res. 261, 127055. https://doi.org/10.1016/j.micres.2022.127055 (2022).
World Health Organization. Therapeutics and COVID-19: Living guideline. World Health Organization https://www.who.int/publications/i/item/WHO-2019-nCoV-therapeutics-2022.5 (2023).
Strati, F. et al. Altered gut microbiota in Rett syndrome. Microbiome 4, 41 (2016).
Sroka-Oleksiak, A. et al. Metagenomic analysis of duodenal microbiota reveals a potential biomarker of dysbiosis in the course of obesity and type 2 diabetes: A pilot study. J. Clin. Med. 9, 369 (2020).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Abarenkov, K. et al. The UNITE database for molecular identification and taxonomic communication of fungi and other eukaryotes: sequences, taxa and classifications reconsidered. Nucleic Acids Res. https://doi.org/10.1093/nar/gkad1039 (2024).
Dhariwal, A. et al. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 45, W180–W188 (2017).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60 (2011).
Tamimi, F., Altigani, S. & Sanz, M. Periodontitis and coronavirus disease 2019. Periodontol. 2000(89), 207–214 (2022).
Larvin, H., Wilmott, S., Wu, J. & Kang, J. The impact of periodontal disease on hospital admission and mortality during COVID-19 pandemic. Front. Med. (Lausanne) 7, 604980 (2020).
Sirin, D. A. & Ozcelik, F. The relationship between COVID-19 and the dental damage stage determined by radiological examination. Oral Radiol. 37, 600–609 (2021).
Costa, C. A. et al. Poor oral health status and adverse COVID-19 outcomes: A preliminary study in hospitalized patients. J. Periodontol. 93, 1889–1901 (2022).
Anand, P. S. et al. A case-control study on the association between periodontitis and coronavirus disease (COVID-19). J. Periodontol. 93, 584–590 (2022).
Ghannoum, M. A. et al. Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog. 6, e1000713 (2010).
Nambiar, M. et al. Mycotic infections—mucormycosis and oral candidiasis associated with COVID-19: A significant and challenging association. J. Oral Microbiol. 13, 1967699 (2021).
Vadovics, M. et al. Candida albicans enhances the progression of oral squamous cell carcinoma in vitro and in vivo. MBio 13, e0314421 (2021).
Zhu, Y. et al. Fungal-induced glycolysis in macrophages promotes colon cancer by enhancing innate lymphoid cell secretion of IL-22. EMBO J. 40, e105320 (2021).
Li, X. et al. The oral microbiota: community composition, influencing factors, pathogenesis, and interventions. Front. Microbiol. 13, 895537 (2022).
Allert, S. et al. Candida albicans-induced epithelial damage mediates translocation through intestinal barriers. MBio 9, e00915–e00918 (2018).
Macauley, P. & Epelbaum, O. Epidemiology and mycology of candidaemia in non-oncological medical intensive care unit patients in a tertiary center in the United States: Overall analysis and comparison between non-COVID-19 and COVID-19 cases. Mycoses 64, 634–640 (2021).
Khan, S. et al. Antimicrobial consumption in patients with COVID-19: a systematic review and meta-analysis. Expert Rev. Anti Infect. Ther. 20, 749–772 (2022).
Maeda, Y. et al. Longitudinal alterations of the gut mycobiota and microbiota on COVID-19 severity. BMC Infect. Dis. 22, 572 (2022).
Jerônimo, L. S. et al. Oral candidiasis and COVID-19 in users of removable dentures: Is special oral care needed?. Gerontology 68, 80–85 (2022).
Krawczyk, A. et al. Changes in the gut mycobiome in pediatric patients in relation to the clinical activity of Crohn’s disease. World J. Gastroenterol. 29, 2172–2187 (2023).
Jain, U. et al. Debaryomyces is enriched in Crohn’s disease intestinal tissue and impairs healing in mice. Science 371, 1154–1159 (2021).
Hwang, G. et al. Candida albicans mannans mediate Streptococcus mutans exoenzyme GtfB binding to modulate cross-kingdom biofilm development in vivo. PLoS Pathog. 13, e1006407 (2017).
Sztukowska, M. N. et al. Community development between Porphyromonas gingivalis and Candida albicans mediated by InlJ and Als3. MBio 9, e00202–e002018 (2018).
Bartnicka, D. et al. Adhesive protein-mediated cross-talk between Candida albicans and Porphyromonas gingivalis in dual species biofilm protects the anaerobic bacterium in unfavorable oxic environment. Sci. Rep. 9, 4376 (2019).
Guo, Y. et al. Heme competition triggers an increase in the pathogenic potential of Porphyromonas gingivalis in P. gingivalis–C. albicans mixed biofilm. Front. Microbiol. 11, 596459 (2020).
Desnos-Ollivier, M. et al. Yarrowia lipolytica causes sporadic cases and local outbreaks of infections and colonisation. Mycoses 63, 737–745 (2020).
Dupuy, A. K. et al. Redefining the human oral mycobiome with improved practices in amplicon-based taxonomy: Discovery of Malassezia as a prominent commensal. PLoS ONE 9, e90899 (2014).
Gallotti, A. C. et al. Kazachstania slooffiae, an emerging pathogen to watch for in humans?. Med. Mycol. Case Rep. 42, 100604 (2023).
Sekeresova Kralova, J. et al. Competitive fungal commensalism mitigates candidiasis pathology. J. Exp. Med. 221, e20231686 (2024).
Acknowledgements
The purchase of the ball homogenizer used in this study was supported by a grant from the Priority Research Area qLIFE under the Strategic Programme Excellence Initiative at Jagiellonian University. Figure 1 has been prepared using Canva free graphics. The engraving created by the authors is their original concept. To correlate fungal and bacterial data from saliva samples, the results of the current ITS-1 analysis were compared with the findings of the 16S analysis, which was the focus of our team’s previous publication11.
Funding
This publication was supported by the National Center for Research and Development CRACoV-HHS project (Model of multi-specialist hospital and non-hospital care for patients with SARS-CoV-2 infection) through the initiative “Support for specialist hospitals in fighting the spread of SARS-CoV-2 infection and in treating COVID-19” (contract number—SZPITALE-JEDNOIMIENNE/18/2020). The described research was implemented by a consortium of the University Hospital in Cracow and the Jagiellonian University Medical College.
Author information
Authors and Affiliations
Contributions
J.Z. and M.B.W. designed research; B.B.S., E.Z.F., W.O. and J.K. collected materials; K.T.Ć., A.S.O. and A.K. performed the research; K.T.Ć., A.G. and A.S.O. analyzed data; K.T.Ć. wrote the main manuscript text; K.T.Ć., B.B.S. and M.B.W. prepared figures; A.S.O., A.G., M.B.S., T.G. and J.Z. reviewed and edited the text; supervision: T.G. and M.B.W.; All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This research has been approved by the Jagiellonian University Ethical Committee (no. 1072.6120.333.2020 of 7 December 2020).
Informed consent
Informed consent was obtained in writing from each study participant.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Talaga-Ćwiertnia, K., Sroka-Oleksiak, A., Brzychczy-Sroka, B. et al. Variations in oral health outcomes and mycobiome composition among COVID-19 convalescents. Sci Rep 15, 21638 (2025). https://doi.org/10.1038/s41598-025-05078-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-05078-x
Keywords
This article is cited by
-
Dynamic alterations of oral fungal microbiota in Omicron infected patients
Scientific Reports (2025)
