
Abstract
Observational studies have suggested a connection between mitochondrial DNA copy number (mtDNA-CN) and osteoporosis, but the causal role of mtDNA-CN remains uncertain. This study aimed to elucidate the causal association between mtDNA-CN and osteoporosis using a two-sample Mendelian randomization (MR) approach. Genome-wide association studies (GWAS) summary data were utilized. Four MR methods—inverse-variance weighted (IVW), weighted median, weighted mode, and MR-Egger—were applied, with IVW as the primary analysis. Sensitivity analyses included the MR-PRESSO test to assess pleiotropy and heterogeneity. Outcomes comprised bone mineral density (BMD) measurements at total body, lumbar spine, femoral neck, and forearm. No significant causal relationship was observed between mtDNA-CN and osteoporosis. IVW results showed odds ratios (ORs) of 0.97 (95% CI 0.89–1.05, P = 0.427) for total BMD, 1.00 (95% CI 0.87–1.15, P = 0.977) for lumbar spine BMD, 1.00 (95% CI 0.89–1.12, P = 0.988) for femoral neck BMD, and 0.90 (95% CI 0.70–1.16, P = 0.421) for forearm BMD. Sensitivity analyses confirmed robustness, with no evidence of horizontal pleiotropy (MR-Egger intercept P > 0.05) or outliers (MR-PRESSO P > 0.05). This MR study found no causal effect of mtDNA-CN on osteoporosis risk across multiple skeletal sites. The null association challenges prior observational hypotheses, suggesting mtDNA-CN may not be a primary determinant of osteoporosis. Further research is warranted to explore alternative mechanisms.
Similar content being viewed by others

Introduction
Osteoporosis, a skeletal disease, is marked by bone tissue’s microarchitectural degeneration and a reduction in bone mineral density (BMD), leading to heightened bone fragility and an increased risk of fractures1. Fractures secondary to osteoporosis are prevalent among older adults, affecting women over 55 and men over 65, and entail substantial morbidity, mortality, and considerable healthcare costs associated with their management2. BMD, a main contributor to osteoporosis, is known to be responsive to mechanical loading, with its variation attributable to not only environmental such as oxidative stress3 and inflammation4 and but also genetic factors such as family history of osteoporosis5 and copy number variation (CNV) of the human genome6, notably, genetic components explain a significant portion of the normal variance in bone mass, estimated to range from 50 to 85%2. Genome-wide association studies (GWAS) have uncovered approximately 100 genomic loci linked to bone density and related traits influencing bone strength and fracture susceptibility, however, the causal pathways for a majority of these loci remain to be elucidated7.
Mitochondria are pivotal in energy metabolism, primarily through the electron transport chain, a process that inherently generates reactive oxygen species (ROS) as a consequence8. Notably, mitochondrial DNA (mtDNA) is recognized for its heightened susceptibility to assault by free radicals8. Animal studies have demonstrated that elevated levels of mtDNA mutations in somatic cells of mtDNA-mutator mice precipitate the early manifestation of multiple age-associated phenotypes, including osteoporosis, alopecia, and decreased body weight9. In addition, a particular deletion in mtDNA has been reported to be linked to heightened oxidative stress observed in peripheral blood mononuclear cells (PBMCs) of men diagnosed with advanced osteoporosis10. Variants in mtDNA within PBMCs have been implicated in the pathogenesis of osteoporosis11. These findings collectively imply a potential connection between osteoporosis and dysfunctions in mitochondrial activity. Notably, research exploring the causal correlation between mitochondrial function and osteoporosis in humans remains limited. It has been postulated that mtDNA content, quantified through copy number, serves as an indicator of mitochondrial genomic stability and biogenesis, thereby offering insight into mitochondrial functionality12. In this viewer, further studies are warranted to elucidate the causal relationship between mitochondrial function, as inferred from mtDNA content, and osteoporosis.
Mendelian Randomization (MR) is an instrumental variable methodology that employs single nucleotide polymorphisms (SNPs) as natural instrumental variables to infer causal relationships between phenotypic traits13. This approach intrinsically minimizes biases introduced by confounding factors and circumvents the challenge of reverse causation14,15. This study used large-scale GWAS datasets to investigate mtDNA copy number (mtDNA-CN) potentially causally related to osteoporosis through a two-sample MR study. Findings of this study may help to illuminate the genetic underpinnings and biological pathways implicated in osteoporosis.
Materials and methods
Study design
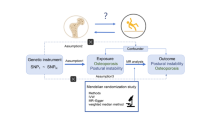
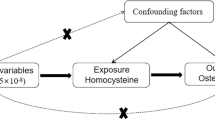
In our study, single nucleotide polymorphisms (SNPs) from GWAS were selected as genetic IVs. This MR study was performed based on the following three assumptions (Fig. 1): (1) Relevance assumption: the genetic variant is directly related to the exposure of interest; (2) Independence assumption: genetic variant is not related to any confounders between the exposure and outcome; (3) Exclusion restriction assumption: genetic variant does not affect outcome except through its association with the exposure16,17. The two-sample MR analysis was used to assess the causal association of mtDNA-CN (exposure) with the risk of osteoporosis and its subtypes. No further ethical approval was required because the present study was based on publicly available GWAS data. Our study was reported according to the “STrengthening the Reporting of OBservational studies in Epidemiology using Mendelian Randomization (STROBE-MR)” checklist17.

Description of the study design. (A) three fundamental MR assumptions. (B) Flow chart of the study design.
Data source
The summary-level data on mtDNA-CN were obtained from three GWAS meta-analyses18,19,20. The sample sizes for the three studies were 395,781, 465,809, and 295,150, respectively. All participants in the three meta-analyses were of European ancestry. Summary-level data for mtDNA-CN were obtained from three large-scale GWAS meta-analyses of European-ancestry populations (Study 1: N = 395,781; Study 2: N = 465,809; Study 3: N = 295,150) investigating mitochondrial biology18,19,20.
Summary data on total BMD were acquired from a GWAS meta-analysis of 30 GWASs of total body BMD including 66,628 European individuals21. Data on lumbar spine BMD, femoral neck BMD, and forearm BMD were from a GWAS meta-analysis investigating novel non-coding genetic variants with large effects on BMD (N = 53,236) and fracture (N = 508,253) in individuals of European ancestry from the general population22. All datasets included mixed sex distributions with age ranges representative of osteoporosis risk populations (adults > 40 years).
Utilizing GWAS summary data from three European-ancestry meta-analyses (total N = 1,156,740 across exposure datasets; outcome data from 66,628 individuals for total BMD and 53,236–508,253 for site-specific BMD/fractures), four MR analytical methods. Instrumental variables were selected using genome-wide significant SNPs (P < 5 × 10−8) after LD clumping (R2 < 0.001, 10,000kb window), MAF > 0.01, and F-statistics > 10 to ensure strength and independence.
Instrumental variables (IVs) selection
In order to uphold the validity and precision of outcomes regarding the causal relationship between mtDNA-CN and osteoporosis, quality control measures were employed in the selection of optimal IVs: (1) Genome-Wide Significance: SNPs showing a strong association with mtDNA-CN and osteoporosis, with a significance level of P < 5 × 10−8, were initially considered23. (2) Minor Allele Frequency: Only SNPs exhibiting a minor allele frequency (MAF) greater than 0.01 were retained to ensure common variant representation23. (3) Linkage Disequilibrium Management: To avoid multicollinearity, SNPs in high linkage disequilibrium (LD; R2 < 0.001) within a 10,000-kb window were excluded24. (4) Proxy SNP Inclusion: In instances where an instrumental variable was missing from the outcome dataset, high-LD proxies (R2 > 0.8) were sought and employed as substitutes25. (5) Instrument Strength Assessment: The F statistic was computed for each SNP in the instrumental variable set to gauge instrument strength and mitigate weak instrument bias. The calculation followed the formula F = [R2 * (N − 2)]/(1 − R2), where R2 represents the proportion of exposure variance explained by the SNP, and N denotes the sample size26. SNPs with an F statistic greater than 10 were retained to ensure adequate instrument power26. During the instrumental variable selection process, the strong instrumental effect of the selected SNPs was ensured through the F-statistic (mean value of 91.02), and horizontal pleiotropy interference was excluded by the MR-PRESSO test. For missing SNPs (such as rs8176645, etc.) in BMD data from different skeletal sites, proxy SNPs with high LD values (R2 > 0.8) were used as substitutes. SNPs without suitable proxies were excluded.
Mendelian randomization
The MR analysis was primarily conducted using the Inverse-Variance Weighted (IVW) approach, which presumes the absence of an average pleiotropic effect27. The IVW method is deemed the most efficacious strategy to assess the causal relationship between the mtDNA-CN and osteoporosis using odds ratios (ORs) and the corresponding 95% confidence intervals (CIs)27. The IVW method calculates a weighted mean of effect sizes, allocating weights inversely proportional to the variance of each SNP27. To ensure the resilience of the findings in this study, supplementary methodologies such as MR-Egger28, weighted median29, and weighted mode30 analyses were also implemented. The MR-Egger methodology accommodates an intercept term, thereby furnishing unbiased estimates of causal effects even amidst horizontal pleiotropy28. The weighted median approach rests on the premise that at least half of the instrumental variables accurately gauge the causal link between exposure and outcome29. Conversely, the weight mode strategy identifies the causal impact by concentrating on the SNP subset exhibiting the highest concentration, achieved through clustering SNPs into groups based on similarity in causal effects30. All statistical analyses were executed using the ‘TwoSampleMR’ package in R software, version 4.3.231. Multiple testing correction for association P-values was conducted using the Bonferroni method, with statistical significance set at P < 0.01 (0.05 divided by 5*1) to account for multiple comparisons32. A scatter plot of was used to depict the association between mtDNA-CN and osteoporosis33. A forest plot was used to show the MR estimate and 95% CI for each SNP33.
Sensitivity analysis
Sensitivity analyses were conducted to examine the robustness of MR findings against potential pleiotropic influences. Heterogeneity among the instrumental variables (IVs) was tested using the Cochran’s Q test, where a P value < 0.05 signified significant heterogeneity34. Furthermore, the MR-Egger regression methodology was invoked to probe for horizontal pleiotropy, with an intercept near zero or statistically non-significant in the MR-Egger regression indicative of an absence of substantial pleiotropic interference35. A funnel plot was employed to visually depict horizontal pleiotropy31. Besides, the MR Pleiotropy Residual Sum and Outlier (MR-PRESSO) method was applied to assess horizontal pleiotropy and identify and exclude outliers36. Additionally, following the exclusion of outlying IVs, we assessed whether there was a statistically significant discrepancy from those prior to their removal. The leave-one-out analysis was applied to identify whether each SNP influenced the main causal association37.
Results
IVs selection
For MR analysis with mtDNA-CN as the exposure, 70 IVs with no LD were selected. The mean F statics was 91.02. The min and max of F statics were 29.54 and 473.58. For total BMD, lumbar spine BMD, femoral neck BMD, and forearm BMD as outcomes, the numbers of selected SNPs were 67, 62, 62, and 62 respectively. The respective numbers of SNPs that did not match the summary data for total BMD, lumbar spine BMD, femoral neck BMD, and forearm BMD were 1, 6, 6, and 6. In the analysis with total BMD as the outcome, one SNP (rs8176645) lacked corresponding information in the summary data, proxy SNPs were identified for rs5745582 (represented by rs75080135) and rs28665408 (substituted by rs1790964). For the MR analysis with femoral neck BMD as the endpoint, six SNPs (“rs111701081”, “rs1613662”, “rs73250616”, “rs392020”, “rs2977608”, “rs3002416”) were not matched in the summary data, and no proxy SNPs were found for these. When analyzing lumbar spine BMD, the same six SNPs were not located in the summary dataset, and no alternative proxy SNPs could be assigned. In the MR analysis with forearm BMD as the outcome, the absence of information persisted for the same set of six SNPs in the summary data, and no suitable proxy SNPs were identified for them. Details on matching information in the summary data were shown in Supplementary file (Tables S1–S4).
Mendelian randomization
MR findings revealed no statistically significant associations between mtDNA-CN and BMD (Table 1 and Figs. 2, 3). IVW showed an insignificant association between mtDNA-CN and total BMD (OR = 0.97, 95% CI 0.89–1.05, P = 0.427), lumbar spine BMD (OR = 1.00, 95% CI 0.87–1.15, P = 0.977), femoral neck BMD (OR = 1.00, 95% CI 0.89–1.12, P = 0.988), and forearm BMD (OR = 0.90, 95% CI 0.70–1.16, P = 0.421). Results of MR-Egger, Weighted Median, and Weighted Mode were consistent with the IVW method.

Scatter plots of the association between mtDNA-CN and BMD. (A) mtDNA-CN and total BMD. (B) mtDNA-CN and lumbar spine BMD. (C) mtDNA-CN and femoral neck BMD. (D) mtDNA-CN and forearm BMD.

Forest plots of the association between mtDNA-CN and BMD. (A) mtDNA-CN and total BMD. (B) mtDNA-CN and lumbar spine BMD. (C) mtDNA-CN and femoral neck BMD. (D) mtDNA-CN and forearm BMD.
Sensitivity analyses
The results of Cochran’s tests indicated significant heterogeneity in the association between mtDNA-CN and lumbar spine BMD (Q = 96.36, P = 0.002), femoral neck BMD (Q = 91.42, P = 0.007), and forearm BMD (Q = 101.18, P = 0.001) (Table 2 and Fig. 4). Results of MR-Egge tests indicated insignificant pleiotropy in the relationship between mtDNA-CN and total BMD (P = 0.16), lumbar spine BMD (P = 0.39), femoral neck BMD (P = 0.58), and forearm BMD (P = 0.32) (Table 2). Results of the MR-PRESSO test showed no potential pleiotropy and outliers for all the MR estimates in this study (Table 3). Leave-one-out analysis suggested that the causal estimates of mtDNA-CN and BMD were not influenced by any single SNP. The leave-one-out analysis plots are depicted in Fig. 5.

Funnel plots of the association between mtDNA-CN and BMD. (A) mtDNA-CN and total BMD. (B) mtDNA-CN and lumbar spine BMD. (C) mtDNA-CN and femoral neck BMD. (D) mtDNA-CN and forearm BMD.

Leave-one-out plots of the association between mtDNA-CN and BMD. (A) mtDNA-CN and total BMD. (B) mtDNA-CN and lumbar spine BMD. (C) mtDNA-CN and femoral neck BMD. (D) mtDNA-CN and forearm BMD.
Discussion
This was the first study to investigate the causal association between mtDNA-CN and osteoporosis by conducting MR estimates. Results of this two-sample MR analyses did not show significantly causal relationship between mtDNA-CN and osteoporosis.
The possible reasons for the insignificant relationship between mtDNA-CN and osteoporosis may be that the pathogenesis of osteoporosis is intricate, influenced by a multitude of factors including genetics, environment, nutrition, hormonal elements, lifestyles, and the interaction between these factors, while mitochondrial function is pivotal to bone metabolism, mtDNA-CN may represent just one among several influencing factors, and its isolated effect may not be substantial enough to significantly contribute to the onset of osteoporosis38,39,40. Mitochondria regulate osteoblast differentiation and osteoclast activity via energy production and ROS signaling. While mtDNA-CN reflects mitochondrial biogenesis, it may not fully capture functional aspects (e.g., oxidative phosphorylation efficiency, mtDNA mutations) critical for bone homeostasis. It remains unclear whether the mtDNA-CN detected in peripheral blood can truly reflect the mitochondrial status within the bone microenvironment. Mitochondria in bone tissue may exhibit distinct biological characteristics from circulating cells due to microenvironment specificity, such as mechanical stress and local redox status. This tissue heterogeneity could potentially weaken the association between peripheral blood mtDNA-CN and bone phenotypes. Mitochondria regulate osteoblast differentiation and osteoclast activity via energy production and ROS signaling. While mtDNA-CN reflects mitochondrial biogenesis, it may not fully capture functional aspects (e.g., oxidative phosphorylation efficiency, mtDNA mutations) critical for bone homeostasis. It remains unclear whether the mtDNA-CN detected in peripheral blood can truly reflect the mitochondrial status within the bone microenvironment. Mitochondria in bone tissue may exhibit distinct biological characteristics from circulating cells due to microenvironment specificity, such as mechanical stress and local redox status. This tissue heterogeneity could potentially weaken the association between peripheral blood mtDNA-CN and bone phenotypes. Notably, existing evidence supports this tissue-specific mitochondrial regulation. In SAMP6 accelerated aging mice, ultrastructural mitochondrial abnormalities in bone tissue were independent of systemic changes41. Furthermore, studies demonstrate that PGC-1α/β-driven mitochondrial biogenesis in bone cells directly regulates osteoblast-osteoclast balance through energy metabolism and redox signaling46,47, while Nrf2 deficiency specifically impairs mitochondrial antioxidant capacity in bone, leading to trabecular bone loss49. These findings collectively suggest that bone-specific mitochondrial dysfunction may occur independently of peripheral blood mtDNA-CN measurements.
Furthermore, as a quantitative indicator, mtDNA-CN is unable to capture critical abnormalities in mitochondrial function, such as large-scale mtDNA deletions, heteroplasmic mutations, or defects in respiratory chain complex activity. While observational studies suggest associations, they cannot disentangle whether mtDNA-CN alterations precede bone loss or merely represent epiphenomena of aging-related metabolic dysfunction. These functional alterations may independently affect bone metabolism, regardless of changes in copy number. It is worth noting that the energy metabolism of bone cells, especially osteoblasts, demonstrates significant plasticity. Studies have shown that even in the presence of mitochondrial dysfunction, bone cells can maintain energy supply by enhancing glycolysis. This metabolic compensatory mechanism may decouple the association between mitochondrial genome stability (represented by mtDNA-CN) and bone phenotypes. For instance, exercise enhances mitochondrial biogenesis, potentially overriding genetic influences on mtDNA-CN. More studies are needed to verify the mechanism for this finding.
Studies on accelerated aging mice (SAMP6) have uncovered ultrastructural alterations in bone tissue, further implicating mitochondrial changes as a significant factor in the development of senile osteoporosis41. A cross-sectional investigation of postmenopausal women revealed a substantial, independent positive correlation between mtDNA-CN in peripheral blood and femoral neck BMD, even after adjusting for variables such as age, body mass index as a measure of obesity, levels of inflammatory markers, indices of bone metabolism, and exercise engagement8. However, the participants in this cross-sectional study were postmenopausal women, postmenopause has been proved to be associated with a heightened risk of fractures, with postmenopausal women constituting the predominant group affected by fracture incidents42. The participants in our study were not limited to postmenopausal women. Interestingly, mtDNA-CN was not connected with lumbar BMD in the aforementioned cross-sectional study, which was consistent with the current MR estimates. The underlying mechanisms are not elucidated and further evidence is needed to address the differential findings between mtDNA-CN and BMD in specific part of human body. Moreover, a recent MR study by He et al. pinpointed four mtDNA loci exhibiting indicative associations with heel BMD, notably including the variant m.16356T>C located in the MT-DLOOP region (P = 1.50 × 10−3) when considering the total sample cohort43. Of note, He et al.’s study focused on heel BMD, while the outcomes in our study were total BMD, lumbar spine BMD, femoral neck BMD, and forearm BMD. This may explain the different results between the current study and He et al.’s. To date, the existing evidence suggests that cells of the osteoblast lineage generate adenosine triphosphate (ATP) from glucose, employing both oxidative phosphorylation and glycolytic metabolic pathways, when mitochondrial function is impaired and cellular energy supply diminishes, cells can compensate for reduced ATP production by upregulating the activity of the glycolytic pathway44. Glycolysis, occurring in the cytoplasm independently of mitochondria, swiftly yields ATP albeit at a lower efficiency, proving particularly efficacious during periods of acute energy demand44. Furthermore, Peroxisome proliferator-activated receptor (PPAR)-γ coactivator-1 (PGC-1) enhances mitochondrial biogenesis and respiratory capacity through direct coactivation of multiple transcription factors central to cellular metabolic regulation, notably nuclear respiratory factor-1 (NRF-1) and nuclear respiratory factor-2 (NRF-2), elevated expression of PGC-1 provokes the transcription of both NRF-1 and NRF-2, which in turn bind to the promoter region of the nuclear-encoded mitochondrial transcription factor A (TFAM)45. This process initiates a regulatory cascade enhancing the expression of key mitochondrial genes encoding ATP synthase, Cytochrome C, and Cytochrome c oxidase subunit I, thereby augmenting mitochondrial biogenesis, this upregulation is crucial for meeting the heightened energy requirements during osteoblast and osteoclast formation46. Studies have demonstrated that the deletion of ERRα or ASXL2, both pivotal targets of PGC-1β, results in severe osteopetrosis, a condition characterized by impaired osteoclast differentiation and a consequential reduction in bone resorption47,48. Another possible mechanism for the insignificant causal relationship between mtDNA-CN and osteoporosis may be that under conditions of oxidative stress, Nrf2 is activated, thereby amplifying the expression of antioxidant genes and safeguarding osteocytes against oxidative damage49. A previous animal study has illustrated that mice lacking Nrf2 exhibit diminished trabecular bone mineral density in their femurs and a decrease in cortical bone area within their vertebrae, highlighting the critical role of Nrf2 in maintaining bone integrity49. Although mtDNA-CN reflects mitochondrial biosynthesis, it may not fully align with key parameters of mitochondrial function, such as oxidative phosphorylation efficiency and mtDNA mutation load. For instance, large deletions in mtDNA or defects in complex I/III activity can significantly impact ROS generation and apoptotic signaling, yet these functional abnormalities may occur within the normal range of mtDNA-CN, thus escaping detection by the genetic instrumental variables designed in this study.
There are strengths for our study. The findings of this study may provide evidence for the research on pathogenesis of osteoporosis. This study, for the first time, explored the causal relationship between mitochondrial DNA copy number (mtDNA-CN) and osteoporosis through Mendelian randomization, addressing the limitations of observational studies that cannot exclude confounding factors. Although the results showed no significant causal association, this conclusion suggests that caution should be exercised in clinical practice when considering mtDNA-CN as a biomarker for osteoporosis. Future research should focus on other mitochondrial functional indicators or genetic factors. Besides, based on the current evidence, mtDNA-CN may not serve as a reliable marker for the detection or prediction of risk for osteoporosis in the clinical setting. Meanwhile, there are limitations in this study. Firstly, the Mendelian randomization (MR) analysis was confined to a cohort of European ancestry, which restricts our ability to generalize these findings to more diverse populations. Secondly, while the possibility of participant overlap between exposure and outcome datasets cannot be excluded, accurately measuring the degree of this overlap presents a challenge. Nonetheless, the study’s use of highly robust instrumental variables, as evidenced by F statistics markedly exceeding 10, serves to effectively minimize bias that might arise from such overlaps50. Thirdly, the restricted number of single nucleotide polymorphisms (SNPs) employed as instrumental variables potentially captures only a small fraction of the exposure’s variability, possibly diminishing the statistical power of the inferred causal relationships. Consequently, given these constraints, interpretations of the study’s results should be made with caution. While mtDNA-CN is sensitive to oxidative stress and inflammation, these factors may act as confounders or effect modifiers. For example, chronic inflammation (e.g., elevated IL-6) accelerates mitochondrial dysfunction and bone loss independently of mtDNA-CN. Similarly, estrogen deficiency in postmenopausal women alters mitochondrial ROS production, which may overshadow mtDNA-CN’s role in BMD regulation. Our MR design, which isolates genetic effects, may not capture these dynamic interactions, necessitating future studies integrating environmental mediators.
While mtDNA-CN may not directly drive osteoporosis, our findings urge exploration of alternative mitochondrial mechanisms (e.g., mtDNA mutations, electron transport chain efficiency) and their interplay with lifestyle factors. Clinically, interventions targeting mitochondrial health (e.g., antioxidants, exercise) may still benefit bone metabolism via pathways beyond mtDNA-CN. Future research should prioritize gene-environment interactions and multi-omics approaches to unravel the complexity of osteoporosis etiology. The discrepancies between the findings of this study and previous observational research may stem from two aspects: First, mtDNA-CN solely reflects mitochondrial genome stability and cannot comprehensively characterize the functional status of mitochondria. Second, compensatory mechanisms exist in the energy supply for bone metabolism—when oxidative phosphorylation is impaired, cells may enhance the glycolytic pathway to sustain ATP production, which might attenuate the causal effect of mtDNA-CN on bone mineral density.
The findings from this two-sample MR analysis do not provide evidence for a genetically inferred causal link between mtDNA-CN and osteoporosis. Therefore, the correlation previously observed in observational studies between mtDNA-CN and osteoporosis might be attributed to the impact of confounding factors. The study population is limited to samples of European ancestry, while mitochondrial genome characteristics exhibit variations across different ethnic groups. This genetically informed approach allows us to circumvent the 'exposure measurement fallacy’, where blood mtDNA-CN inaccurately proxies bone mitochondrial status, by anchoring the analysis in germline genetic variants that systemically regulate mitochondrial biogenesis. For instance, Asian-specific mtDNA haplotypes may influence the association patterns between copy number and bone metabolism, which necessitates validation through cross-ethnic genome-wide association studies (GWAS) data. To solidify these study outcomes, future analyses should employ larger GWAS summary datasets based on multiple populations and an expanded repertoire of genetic instruments. Given the potential heterogeneity in frequency and effect size of the genome across different ethnic groups, the conclusions of this study should be cautiously generalized to other populations. We call for future studies to utilize multi-ethnic cohorts and cross-racial GWAS data to validate the association between mtDNA-CN and osteoporosis, in order to elucidate the potential impact of genetic background and environmental interactions.
Data availability
All data generated or analyzed during this study are included in this article and supplementary information files.
References
Clynes, M. A. et al. The epidemiology of osteoporosis. Br. Med. Bull. 133(1), 105–117. https://doi.org/10.1093/bmb/ldaa005 (2020).
Compston, J. E., McClung, M. R. & Leslie, W. D. Osteoporosis. Lancet 393(10169), 364–376. https://doi.org/10.1016/s0140-6736(18)32112-3 (2019).
Sánchez-Rodríguez, M. A., Ruiz-Ramos, M., Correa-Muñoz, E. & Mendoza-Núñez, V. M. Oxidative stress as a risk factor for osteoporosis in elderly Mexicans as characterized by antioxidant enzymes. BMC Musculoskelet. Disord. 8, 124. https://doi.org/10.1186/1471-2474-8-124 (2007).
Koh, J. M. et al. Higher circulating hsCRP levels are associated with lower bone mineral density in healthy pre- and postmenopausal women: Evidence for a link between systemic inflammation and osteoporosis. Osteoporos. Int. 16(10), 1263–1271. https://doi.org/10.1007/s00198-005-1840-5 (2005).
Li, W. F. et al. Genetics of osteoporosis: Accelerating pace in gene identification and validation. Hum. Genet. 127(3), 249–285. https://doi.org/10.1007/s00439-009-0773-z (2010).
Yang, T. L. et al. Genome-wide copy-number-variation study identified a susceptibility gene, UGT2B17, for osteoporosis. Am. J. Hum. Genet. 83(6), 663–674. https://doi.org/10.1016/j.ajhg.2008.10.006 (2008).
Boudin, E. & Van Hul, W. Mechanisms in endocrinology: Genetics of human bone formation. Eur. J. Endocrinol. 177(2), R69-r83. https://doi.org/10.1530/eje-16-0990 (2017).
Kim, J. H. & Lee, D. C. Mitochondrial DNA copy number in peripheral blood is associated with femoral neck bone mineral density in postmenopausal women. J. Rheumatol. 39(7), 1465–1472. https://doi.org/10.3899/jrheum.111444 (2012).
Trifunovic, A. et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429(6990), 417–423. https://doi.org/10.1038/nature02517 (2004).
Varanasi, S. S., Francis, R. M., Berger, C. E., Papiha, S. S. & Datta, H. K. Mitochondrial DNA deletion associated oxidative stress and severe male osteoporosis. Osteoporos. Int. 10(2), 143–149. https://doi.org/10.1007/s001980050209 (1999).
Guo, Y. et al. Mitochondria-wide association study of common variants in osteoporosis. Ann. Hum. Genet. 75(5), 569–574. https://doi.org/10.1111/j.1469-1809.2011.00663.x (2011).
Clay Montier, L. L., Deng, J. J. & Bai, Y. Number matters: Control of mammalian mitochondrial DNA copy number. J. Genet. Genom. 36(3), 125–131. https://doi.org/10.1016/s1673-8527(08)60099-5 (2009).
Lawlor, D. A., Harbord, R. M., Sterne, J. A. C., Timpson, N. & Davey, S. G. Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Stat. Med. 27(8), 1133–1163. https://doi.org/10.1002/sim.3034 (2008).
Emdin, C. A., Khera, A. V. & Kathiresan, S. Mendelian randomization. JAMA 318(19), 1925–1926. https://doi.org/10.1001/jama.2017.17219 (2017).
Davey Smith, G. & Hemani, G. Mendelian randomization: Genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 23(R1), R89–R98. https://doi.org/10.1093/hmg/ddu328 (2014).
Burgess, S. et al. Guidelines for performing Mendelian randomization investigations: Update for summer 2023. Wellcome Open Res. 4, 186. https://doi.org/10.12688/wellcomeopenres.15555.3 (2019).
Skrivankova, V. W. et al. Strengthening the reporting of observational studies in epidemiology using Mendelian randomization: The STROBE-MR statement. JAMA 326(16), 1614–1621. https://doi.org/10.1001/jama.2021.18236 (2021).
Chong M, Mohammadi-Shemirani P, Perrot N, et al. GWAS and ExWAS of blood mitochondrial DNA copy number identifies 71 loci and highlights a potential causal role in dementia. Elife. (2022). https://doi.org/10.7554/eLife.70382
Longchamps, R. J. et al. Genome-wide analysis of mitochondrial DNA copy number reveals loci implicated in nucleotide metabolism, platelet activation, and megakaryocyte proliferation. Hum. Genet. 141(1), 127–146. https://doi.org/10.1007/s00439-021-02394-w (2022).
Hägg, S., Jylhävä, J., Wang, Y., Czene, K. & Grassmann, F. Deciphering the genetic and epidemiological landscape of mitochondrial DNA abundance. Hum. Genet. 140(6), 849–861. https://doi.org/10.1007/s00439-020-02249-w (2021).
Medina-Gomez, C. et al. Life-course genome-wide association study meta-analysis of total body BMD and assessment of age-specific effects. Am. J. Hum. Genet. 102(1), 88–102. https://doi.org/10.1016/j.ajhg.2017.12.005 (2018).
Zheng, H. F. et al. Whole-genome sequencing identifies EN1 as a determinant of bone density and fracture. Nature 526(7571), 112–117. https://doi.org/10.1038/nature14878 (2015).
Long, Y., Tang, L., Zhou, Y., Zhao, S. & Zhu, H. Causal relationship between gut microbiota and cancers: A two-sample Mendelian randomisation study. BMC Med. 21(1), 66. https://doi.org/10.1186/s12916-023-02761-6 (2023).
Abecasis, G. R. et al. A map of human genome variation from population-scale sequencing. Nature 467(7319), 1061–1073. https://doi.org/10.1038/nature09534 (2010).
Deng, M. G. et al. Association between frailty and depression: A bidirectional Mendelian randomization study. Sci. Adv. 9(38), eadi3902. https://doi.org/10.1126/sciadv.adi3902 (2023).
Burgess, S. & Thompson, S. G. Avoiding bias from weak instruments in Mendelian randomization studies. Int. J. Epidemiol. 40(3), 755–764. https://doi.org/10.1093/ije/dyr036 (2011).
Burgess, S., Butterworth, A. & Thompson, S. G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 37(7), 658–665. https://doi.org/10.1002/gepi.21758 (2013).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44(2), 512–525. https://doi.org/10.1093/ije/dyv080 (2015).
Bowden, J., Davey Smith, G., Haycock, P. C. & Burgess, S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40(4), 304–314. https://doi.org/10.1002/gepi.21965 (2016).
Hartwig, F. P., Davey Smith, G. & Bowden, J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 46(6), 1985–1998. https://doi.org/10.1093/ije/dyx102 (2017).
Hemani G, Zheng J, Elsworth B, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. (2018). https://doi.org/10.7554/eLife.34408
Curtin, F. & Schulz, P. Multiple correlations and Bonferroni’s correction. Biol. Psychiatry. 44(8), 775–777. https://doi.org/10.1016/s0006-3223(98)00043-2 (1998).
Huang, D., Lin, S., He, J., Wang, Q. & Zhan, Y. Association between COVID-19 and telomere length: A bidirectional Mendelian randomization study. J. Med. Virol. 94(11), 5345–5353. https://doi.org/10.1002/jmv.28008 (2022).
Greco, M. F., Minelli, C., Sheehan, N. A. & Thompson, J. R. Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat. Med. 34(21), 2926–2940. https://doi.org/10.1002/sim.6522 (2015).
Burgess, S. & Thompson, S. G. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur. J. Epidemiol. 32(5), 377–389. https://doi.org/10.1007/s10654-017-0255-x (2017).
Verbanck, M., Chen, C. Y., Neale, B. & Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50(5), 693–698. https://doi.org/10.1038/s41588-018-0099-7 (2018).
Burgess, S., Bowden, J., Fall, T., Ingelsson, E. & Thompson, S. G. Sensitivity analyses for robust causal inference from mendelian randomization analyses with multiple genetic variants. Epidemiology 28(1), 30–42. https://doi.org/10.1097/ede.0000000000000559 (2017).
Choi, C. K. et al. Association between alcohol and bone mineral density in a Mendelian randomization study: the Dong-gu study. J. Bone Miner. Metab. 40(1), 167–173. https://doi.org/10.1007/s00774-021-01275-6 (2022).
Chandra A, Rajawat J. Skeletal aging and osteoporosis: mechanisms and therapeutics. Int. J. Mol. Sci. (2021). https://doi.org/10.3390/ijms22073553
Föger-Samwald, U., Dovjak, P., Azizi-Semrad, U., Kerschan-Schindl, K. & Pietschmann, P. Osteoporosis: Pathophysiology and therapeutic options. Excli. J. 19, 1017–1037. https://doi.org/10.17179/excli2020-2591 (2020).
Chen, H., Shoumura, S. & Emura, S. Ultrastructural changes in bones of the senescence-accelerated mouse (SAMP6): A murine model for senile osteoporosis. Histol. Histopathol. 19(3), 677–685. https://doi.org/10.14670/hh-19.677 (2004).
Kanis, J. A. et al. Long-term risk of osteoporotic fracture in Malmö. Osteoporos. Int. 11(8), 669–674. https://doi.org/10.1007/s001980070064 (2000).
He D, Liu H, Zhao Y, et al. Assessing the interaction effects of mitochondrial DNA polymorphisms and lifestyle on heel bone mineral density. J. Clin. Endocrinol. Metab. (2024). https://doi.org/10.1210/clinem/dgae195
Riddle, R. C. & Clemens, T. L. Bone cell bioenergetics and skeletal energy homeostasis. Physiol. Rev. 97(2), 667–698. https://doi.org/10.1152/physrev.00022.2016 (2017).
Chen, H., Fan, W., He, H. & Huang, F. PGC-1: A key regulator in bone homeostasis. J. Bone Miner. Metab. 40(1), 1–8. https://doi.org/10.1007/s00774-021-01263-w (2022).
Yu, B. et al. PGC-1α controls skeletal stem cell fate and bone-fat balance in osteoporosis and skeletal aging by inducing TAZ. Cell Stem Cell 23(2), 193-209.e5. https://doi.org/10.1016/j.stem.2018.06.009 (2018).
Izawa, T. et al. ASXL2 regulates glucose, lipid, and skeletal homeostasis. Cell Rep. 11(10), 1625–1637. https://doi.org/10.1016/j.celrep.2015.05.019 (2015).
Yang, D. & Wan, Y. Molecular determinants for the polarization of macrophage and osteoclast. Semin. Immunopathol. 41(5), 551–563. https://doi.org/10.1007/s00281-019-00754-3 (2019).
Ibáñez, L. et al. Effects of Nrf2 deficiency on bone microarchitecture in an experimental model of osteoporosis. Oxid. Med. Cell Longev. 2014, 726590. https://doi.org/10.1155/2014/726590 (2014).
Pierce, B. L. & Burgess, S. Efficient design for Mendelian randomization studies: Subsample and 2-sample instrumental variable estimators. Am. J. Epidemiol. 178(7), 1177–1184. https://doi.org/10.1093/aje/kwt084 (2013).
Funding
This study was supported by General Project of National Natural Science Foundation of China (82074063, 82274151), Youth Science and Technology Innovation Studio of Shanghai Jiao Tong University School of Medicine and Shanghai Sixth People’s hospital Medical Service Level Improvement project (20250103).
Author information
Authors and Affiliations
Contributions
Misu Xiao performed the statistical analysis and carried out the studies, participated in collecting data, and drafted the manuscript. Quanjun Yang participated in its design. Professor Cheng Guo conducted quality control on the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This article is a Mendelian randomization study. The data for this study were obtained from publicly available databases and published literature data and does not require ethical approval.
Patient consent
This article is a Mendelian randomization study. The data for this study were obtained from publicly available databases and published literature data and does not require written informed consent.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiao, M., Guo, C. & Yang, Q. MR analysis reveals no causal association between mitochondrial DNA copy number and osteoporosis. Sci Rep 15, 22286 (2025). https://doi.org/10.1038/s41598-025-05287-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-05287-4
