
Abstract
Genetic causes of familial isolated pituitary adenomas (FIPAs) remain mostly elusive. A cohort of 20 FIPA cases from 12 different geographical regions of Türkiye was included to characterize clinical and genetic features. Whole exome sequencing (WES) was performed on genomic DNA of index cases, followed by confirmation through Sanger sequencing utilizing indexes and their relatives to interpret disease associated variants. Index cases among homogeneous (n = 10) and heterogeneous (n = 10) FIPA groups (45% female /55% male), age at diagnosis was 36.3 ± 11.98 years, median follow-up was 103 months. GH-secreting adenomas dominated homogeneous group (60% vs. 30% of heterogeneous group). Two predefined AIP variants [p.(Arg304Ter) and p.(Arg81Ter)] and a novel AIP variant at splice acceptor site [(c.646-1G > C)] were detected in three families (15%). Syndromic heterozygous novel NF1 [p.(Thr1295Ala)], TSC1 [p.(Arg517Gln)], SDHB [p.(Glu176Gly)] and CDH23 [p.(Ala765Val)] variants were detected in four FIPA families, along with novel candidate genes in the remaining patients of the cohort. Among all detected variants, three [p.(Arg81Ter) and (c.646-1G > C) in AIP, and p.(Glu216GlysfsTer61) in TINF2] were classified as pathogenic according to ACMG. AIP mutation frequency was 15% in our cohort. A novel AIP variant, and novel variations in syndromic genes were identified, along with the introduction of candidate genes. WES method is a crucial approach to identify new rare genetic variants in familial settings, and it will pave the way for future studies on targeted therapies.
Similar content being viewed by others

Introduction
Pituitary neuroendocrine tumours (PitNETs) occur sporadically, with 5% are believed to be of familial origin. Familial PitNETs may present as a part of isolated familial pituitary adenomas (FIPA) or may be a component of various syndromic diseases, such as classical MEN1 syndrome or Carney complex or hereditary paraganglioma syndromes (3PAs), MEN4, Tuberous Sclerosis-TCS, Neurofibromatosis type 1, DICER1 syndrome, Lynch syndrome, CDH23 syndrome1,2,3,4,5. FIPA is an autosomal-dominant disease defined by the presence of pituitary adenoma (PA) in two or more relatives. FIPA families can be heterogeneous (when more than one type of PA is present in the same family) or homogeneous. Most AIP mutation-positive FIPA patients present with somatotrophinomas or somatolactotrophinomas, while some exhibit lactotrophinomas, and non-functioning adenomas and corticotropinomas are rare6,7. FIPA related tumors are generally large, aggressive, occur at younger ages, and are resistant to treatment with first-generation somatostatin receptor ligands (SRLs)6. Although germline AIP mutations account for ∼20% of all FIPA cases, the genetic profile remains unknown in many instances8,9,10,11,12,13. Identifying potential new genetic variants across different geographical regions of the same country is essential for uncovering the hereditary causes of FIPAs. Therefore, we aimed to investigate the etiopathogenesis of PAs utilizing whole exome sequencing (WES) in affected individuals from FIPA families in different geographical regions of Türkiye.
Methods
In this multicenter study, clinical data and medical history of 45 FIPA families were followed-up for 2 to 276 months in 12 pituitary clinics located in different geographical regions of Turkiye, which were re-evaluated from the records by two senior endocrinologists. Functional PA diagnosis was based on clinical signs and symptoms, laboratory tests demonstrating elevated basal hormone levels and/or abnormal dynamic tests. Results were interpreted in accordance with the Endocrine Society’s current guidelines14,15,16. In patients with GH-secreting PA, acromegaly is diagnosed if typical clinical acral features are accompanied by IGF-1 levels above the upper normal range of age-sex matched range (14). In PRL-secreting adenomas, serum PRL levels are usually above 250 ng/ml and the presence of galactorrhea and sexual dysfunction makes the diagnosis of prolactinoma (15). Definitive diagnosis of Cushing’s disease due to hypercortisolemia caused by ACTH-secreting adenoma is made by abnormal circadian rhythm with failure to suppress hypercortisolemia with overnight 1 mg dexamethasone suppression test (DST) and low-dose 2-day dexamethasone suppression test (LDDT), and elevation of 24-hour urinary free cortisol (UFC) (16).
Pituitary MRI reports were provided by experienced neuroradiologists. The degree of cavernous sinus invasion of PAs was determined by Knosp classification, where grades 0–2 are considered non-invasive, and grades 3 and 4 indicated invasive characteristics17. The diagnosis was supported histopathologically in operated GH- and ACTH-secreting and nonfunctional PAs.
Among the 45 families meeting the FIPA criteria recruited from various centers, 20 index cases were identified as eligible for the study. These cases fulfilled the clinical, laboratory, and radiological criteria for inclusion and had histopathological diagnosis reports documented in their medical records. Of these families, 20 index cases and 15 relatives (alive and reachable) with proven history of pituitary adenoma were included in the study.
This human study was conducted in compliance with the principles in Declaration of Helsinki and was performed upon the approval of Clinical Ethics Committee of Baskent University Faculty of Medicine (project #KA21/247). All procedures strictly adhered to current guidelines and regulations. Peripheral blood samples were obtained following written informed consents of the participants.
Genetic analyses
WES was performed on DNA obtained from the peripheral blood of 20 index cases at the service of Medical Genetic Diagnostic Center in Izmir, Türkiye that utilized llumina NextSeq 550 system18. Variant annotations and subsequent filtering were achieved using NGS Cloud (www.ngscloud.com; Pairend Biotechnology LLC, Manisa, Türkiye), which is a cloud-based genetic data analysis platform supported through artificial intelligence19. Sequence reads were visualized through IGV 2.9.4 program, where minor allele frequencies (MAFs) were obtained from GnomAD, dbSNP, 1000 Genomes Project, Exome Sequencing Project, TopMED, Greater Middle East Variome Project and in house allele frequency of the NGS Cloud.
WES data was initially filtered for PA-related genetic variants using a gene panel developed from the literature. This panel comprised a comprehensive list of genes that are relevant to the pathogenesis of PAs and associated symptoms. It also included key molecular pathways, genes involved in epigenetic mechanisms, and additional genes identified through bioinformatics analyses (Supplementary Table S1). Variant interpretation and pathogenicity scores were achieved under the provisions of American College of Medical Genetics (ACMG) guidelines along with ClinVar database for PA-associated genes. This was followed by filtering for rare variants (MAF ≤ 0.01), in which in silico prediction tools have been a guide for plausible impact of the variant. The prioritization among novel genetic variants was achieved through functional relevance of the genes to disease pathogenesis. In this way, an initial draft of candidate genes was compiled and validated through Sanger sequencing, first in the index cases, and then in their relatives, in an effort to eliminate unrelated genes in PA pathogenesis. Sanger sequences were analysed by CLC Main Workbench. A list of primers utilized both in PCR amplification and Sanger sequencing are listed in Supplementary Table S2.
Results
Among 20 index cases, 9 were females (45%), 11 were males (55%). There were 10 homogeneous FIPA families; 6 somatotrophinomas/ 4 prolactinomas. Index cases of 10 heterogeneous FIPA families were those with somatotrophinomas (n = 3), prolactinomas (n = 4) and corticotropinomas (n = 3). The median age at diagnosis of index cases was 36.3 ± 11.98 years and median follow-up period was 103 months (range 2-276).
The frequency of GH-secreting adenomas in the homogeneous FIPA group was twice as high (60%) as in the heterogeneous group (30%). The mean delay in diagnosis of index cases with acromegaly was 24 months (range 24–132). 25% of index cases had micro (≤ 10 mm), 65% had macro (> 10 mm), and 10% had giant (≥ 40 mm) adenomas. Adenomas were classified as 65% Knosp 0–2 and 35% Knops 3–4 according to their degree of invasion.
Among relatives; there were 3 GH- and 4 PRL-secreting adenomas in homogeneous group (n = 7/20) and 2 PRL-, 3 GH-secreting and 3 apparently non-functional (clinically and hormonally inactive) adenomas in heterogeneous group (n = 8/10).
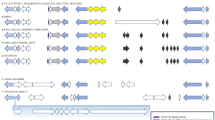
Table 1 summarizes the clinical findings of index cases with genetic variants confirmed in cases and available affected relatives. Figure 1 shows family pedigrees, highlighting the affected members and available clinical histories, as well as indicating which patients provided genetic materials. Accordingly, genetic material from the relatives of five index cases (2, 7, 10, 11 and 15) were not available. In 3 FIPA families, two known [p.(Arg304Ter) and p.(Arg81Ter)] and one novel [splice acceptor site (c.646-1G > C)] AIP variants were detected (15%). Moreover, (c.646-1G > C) and p.(Arg81Ter) AIP variants in cases 12 and 13, respectively, along with p.(Glu216GlysfsTer61) in TINF2 in case 2 were determined as pathogenic by ACMG. In addition, variants in RUNX2 p.(Gln68_Gln71dup) in case 9 and NF1 p.(Thr1295Ala) in case 17 were reported as “conflicting classifications of pathogenicity” in the ClinVar database. All these variants, except for the one in TINF2, were shared among the relatives in pertinent families.

Family Pedigrees Depicted is the pedigrees for 20 index cases evaluated. Different colors indicate distinct PA types to illustrate the disease history in the families. The star sign indicates individuals who consented to provide DNA material and the arrows indicate the index cases.
Clinical characteristics and genetic variation(s) of both index cases and relatives with AIP mutations are summarized in Table 2. Among the AIP positive families, Case 12 and his daughter had macroadenoma with cavernous sinus invasion. Despite these tumour characteristics, the father is in drug-free remission following surgery and SRL treatments. Excess GH was detected in daughter diagnosed with prolactinoma. Her father had acromegaly and upon inspection her nose felt slightly enlarged, although she reported no complaints about it. Nevertheless, the additional diagnosis of clinically silent acromegaly was not overlooked in her. She initially preferred DA treatment, however, the adenoma did not shrink and medication could not be discontinued, despite clinical and hormonal response to the treatment. In the other heterogeneous FIPA family (Case 13 and his sister), who carried no genetic variant other than the known AIP p.(Arg81Ter) mutation, adenoma was larger and more invasive in younger affected member. The brother with acromegaly had his disease activity controlled through three surgeries and a combination of two medications. In contrast, his sister, who previously suffered from apoplexy, achieved remission from prolactinoma, and her DA treatment was discontinued one year later.
Although other components of NF1, TCS, 3PAs and Usher syndromes were absent, heterozygous novel NF1 [p.(Thr1295Ala)], TSC1 [p.(Arg517Gln)], SDHB [p.(Glu176Gly)] and CDH23 [p.(Ala765Val)] variants were detected in 4 FIPA families (Table 3). Candidate genes other than AIP and syndromic genes in our 13 FIPA families are summarized in Table 4.
Discussion
This study presents the first genetic analysis of a large cohort of FIPA families from various geographical regions of Turkiye, utilizing WES. We identified three AIP variants in three different families. Two pathogenic AIP variants previously described in the literature p.(Arg304Ter) and p.(Arg81Ter) were identified in two independent families from Western Black Sea Region, whereas the novel AIP variant (c.646-1G), located in intron 4–5 that potentially affects the splice acceptor site, was identified in a family from Eastern Anatolia region. Consistent with the literature, we determined AIP mutation frequency as 15% in our FIPA cohort6,7,8,20.
Among limited number of studies screening AIP variations in sporadic young-onset somatotropinomas from Turkiye, prevalence of AIP mutation was found to be 1%, 2.2% or 2.1%, where p.(Arg304Ter) has been the prominent mutation21,22,23. In this respect, Arg304Ter was identified by Sanger sequencing in 7 acromegaly patients in a large FIPA family24. However, in our previous study, we identified only two homozygous missense SNPs (rs641081 [Q228K] and rs4930195 [Q307R]) in AIP by Sanger sequencing 14 different FIPA families25. The Arg304Ter mutation, located in the hotspot region of AIP, was most frequently reported in FIPAs from Ireland, Romania, Britain, Italy, USA, India and Mexico, while Arg81Ter mutation had been reported in families from Brazil, USA, India and the UK10,12,26,27,28,29.
Interestingly, in homogeneous FIPA group, acromegalic index Case 6, who carried both pathogenic AIP p.(Arg304Ter) mutation and a novel variant p.(Gly334del) in Sequestosome 1 (SQSTM1) gene, responded positively to first-generation SRL and her adenoma shrank by 50%. This contradicts the findings that state acromegaly patients harbouring AIP variations are unresponsive to first-generation SRLs8,11,27,30. This provoked the hypothesis of a possible impact of accompanying novel variant in treatment response. Accordingly, Tulipano et al. reported that in rat pituitary GH3 tumor cells, octreotide treatment enhanced autophagic flux through SQSTM1/p62 protein downregulation31. Further studies are needed to clarify the impact of this novel variant in SQSTM1/p62 protein expression. Unfortunately, we did not have the chance to support the index case’s positive response to first-generation SRL, as no data could be obtained from the acromegalic sibling, who participated only with a blood sample and shared the same variations with the index.
In a heterogeneous FIPA family, we described a novel AIP (c.646-1G > C) variant for the first time in the literature. This novel AIP variant was accompanied by two different novel genetic variations as p.(Pro281Leu) in SUFU and p.(Ile240Thr) in LGALS3. Recently, it has been reported that PKA/SUFU/GLI1 signalling pathway, known as the Hedgehog signalling, is activated in primary PA cells, as well as in surgical PA samples, leading to inhibition of apoptosis and promotion of the cell cycle32. LGALS3 encodes Galectin-3, which has an important role in pituitary cell proliferation and tumor progression33. Galectin-3 is a well-recognized biomarker for the aggressive behaviour of PRL-secreting adenomas and their prognosis34. The co-occurrence of these variations may result in unexpected outcomes, which merits functional evaluation.
In the other heterogeneous FIPA family (Case 13 and his sister), carrying AIP p.(Arg81Ter) mutation, prognosis of the cases were consistent with the literature stating that tumor apoplexy is more common in individuals with the AIP mutation compared to those without it8. Additionally, familial pituitary apoplexy has also been described in AIP mutation-positive families8,35. Although prolactinomas with AIP mutations are generally reported to be large, aggressive, and to manifest at younger ages, they have also been reported not to cause resistance to DA, as seen in our case of prolactinoma36,37.
Syndromic germline alterations in our FIPA cohort
Apart from the well-recognized FIPA-related AIP variations, we have detected heterozygous novel syndromic germline SDHB, NF1, TSC1 and CDH23 variations (Table 4). The “3PA” syndrome, which combines PAs with pheochromocytomas/paragangliomas, is associated with germline mutations in the SDHx genes, which are linked to the development of PAs38. Mutations in SDHB and SDHD have frequently been associated with PRL-secreting, GH-secreting and non-functioning adenomas in origin, which tend to be more aggressive, more resistant to SRLs, and often require surgical intervention20,39. In contrast to the literature, our cases carrying the same germline heterozygous SDHB variant showed distinct outcomes: the macroprolactinoma in Case 14 responded positively to CAB, while the macro-NFA in his father, who refused surgery, did not show an aggressive course.
Neurofibromatosis type-1 (NF1) gene encodes a large protein called neurofibromin, which functions as a negative regulator of the RAS proto-oncogene and mutations in NF1 are responsible for NF1 syndrome40. Rarely, NF1 patients develop acromegaly due to autonomic GH hypersecretion resulting solely from optic pathway gliomas41,42. While true PAs are extremely rare, Hozumi et al.43 reported for the first time a case of NF1 syndrome with acromegaly resulted from GH-secreting adenoma due to somatic GNAS p.(R201C) mutation. Recently, Hong et al.44 described two cases of PAs with rare somatic NF1 variants. In our study, within a FIPA family carrying a heterozygous germline NF1 variant, the young index patient (Case 17) was unable to discontinue DA treatment despite her macroprolactinoma disappearing under therapy. In contrast, her acromegalic relative achieved full recovery following surgery. This germline variant was thought to be a factor determining the tendency to develop PA in our FIPA family.
Tuberous sclerosis is an autosomal dominant neurocutaneous syndrome, caused by mutations in one of the two tumor suppressor genes, TCS1 and TCS245. A few case reports have documented PAs in patients with TSC; including one GH-secreting adenoma, two ACTH-secreting adenomas, and one silent gonadotroph tumor. However, the existence of genotype–phenotype correlations remains a subject of debate46,47,48. In our young acromegaly patient (Case 7), who presented with invasive macroadenoma and carried heterozygous germline TSC1 p.(Arg517Gln) variant, disease activity was ultimately controlled only after two surgeries, radiotherapy and a triple drug combination. In addition to novel TSC1 variant, Fibroblast Growth Factor Receptor-2 [FGFR2 p.(Ile504Leu)], Somatostatin Receptor Type 4 [SSTR4 p.(Phe321Ser)] and ATPase Plasma Membrane Ca2 + Transporting 3 [ATP2B3 p.(Asp641Asn)] gene variants were identified in this patient. Unfortunately, we could not compare the clinical data of the index case with his deceased cousin, who had acromegaly.
Cadherin-related 23 (CDH23) is a member of the cadherin superfamily, which comprises calcium-dependent cell-cell adhesion glycoproteins49. Germline mutations in CDH23 have been identified in people with Usher syndrome and nonsyndromic autosomal-recessive deafness50. Recently, mutations in CDH23, which is involved in cAMP-related pathways, are linked to familial and sporadic PAs and proposed to play important roles in PA pathogenesis51,52. Zhang et al.51 identified a heterozygous missense p.(Arg1379Leu) mutation in CHD23 in 33% of familial PAs and reported this gene as a risk factor for FIPAs. Our index patient (Case 20) and her brother harboured a heterozygous novel CDH23 p.(Ala765Val) variant in one of the 20 FIPA families (5%). We additionally identified a novel variant p.(Ala486Thr) in KCNQ1 in the affected individuals of this family. Both of these genes have been described to have roles in PA development51,53. The PA in our index case was ACTH-secreting type. To date, while many new mutations have been identified, approximately 12–28% of ACTH-secreting adenomas still have no known mutations54. The new variants in these cases may likely have triggered the onset of PAs, but they have not yet led to a relapse after surgery.
Other germline alterations in the remaining FIPA families
WES revealed additional candidate genes among which SQSTM1, LGALS3 and SUFU accompanied AIP variations; ATP2B3, SSTR4 and FGFR2 accompanied TSC1, and KCNQ1 accompanied CDH23. Our findings in four different Cases (3, 4, 9 and 12) have demonstrated the strength of our study, highlighting the interactions between SUFU, LGALS3 and RUNX2 genes. The protein-protein interactions were established between SUFU and Galectin 3, both by an in vivo study in mice and in STING and Cytoscape based analyses utilizing human microarray data. This interaction was postulated to mediate global mRNA maturation55. Moreover, Galectin 3 was also determined to interact with RUNX2 protein, in which RUNX2 as a transcription factor was depicted to upregulate the expression of Galectin 3 in human pituitary tumors through initiating tumor progression56. Moreover, shared genes among index cases and relatives, as well as the presence of the same genes in different families prioritized them as candidates. In this respect, NR5A1(SF1), PKD1, NPR2, BRAF, PTTG1 were implicated to play roles in PA development57,58,59,60,61. Since other genes identified in this study were common in different cases in our cohort, we can emphasize that they are involved in PA pathogenesis as new genes that need to be replicated by independent studies.
In this context, studies utilizing WES on human subjects face challenges in variant interpretation, as each individual’s data may accommodate around 20,000 variants. While WES is effective for identifying novel and rare variants, many are not documented in literature or genetic databases, necessitating the use of in silico prediction tools to estimate the potential impact of these variants on protein function and their conservation across species. However, varying algorithms can yield conflicting results, making it crucial to correlate these findings with existing gene function information to validate candidate genes. In our study, we addressed these challenges by leveraging familial cases. After filtering for candidate variants from WES data, we performed familial segregation analysis with Sanger sequencing, which, along with the presence of novel genes across cases, highlighted their potential role in PA pathogenesis. Thus, our research suggested novel variants and candidate genes within a cohort of FIPA families that received definitive diagnoses.
In conclusion, our study highlights the potential of WES in identifying possible common candidate genes other than AIP among affected relatives in FIPAs. In this respect, among the detected variants, p.(Arg81Ter) and (c.646-1G > C) in AIP, along with p.(Glu216GlysfsTer61) in TINF2, were reported as pathogenic in accordance with ACMG guidelines. Moreover, using in silico prediction tools GERP++, DANN, SIFT, LIST-S2 and AlphaMissense (a new machine-learning based tool), several novel variants were identified as potentially pathogenic. These findings were supported by AlphaMissense and at least one other tool. In this respect, NR5A1 p.(Gly178Arg), RUNX2 p.(Pro390Leu), LGALS3 p.(Pro46Arg) and p.(Ile240Thr), SDHB p.(Glu176Gly), NPR2 p.(Tyr338Cys) and CDH23 p.(Ala765Val) variants were determined as strong candidates involved in PA pathogenesis. The advancement of next-generation sequencing will increase knowledge about the pathogenesis, invasiveness, recurrence, and prognosis of FIPAs and will contribute significantly to the development of targeted therapies.
Data availability
The data that support the findings of this study are available in Zenodo repository [https://doi.org/10.5281/zenodo.15697716 and https://doi.org/10.5281/zenodo.15704233], but restrictions apply to the availability of these data due to the Personal Data Protection Law (KVKK) in Turkiye. Data are however available from the authors upon reasonable request through the repository.
References
Caimari, F. & Korbonits, M. Novel genetic causes of pituitary adenomas. Clin. Cancer Res. https://doi.org/10.1158/1078-0432.CCR-16-0452 (2016).
Akkus, G. & Korbonits, M. Genetic testing in hereditary pituitary tumors. Arch. Med. Res. https://doi.org/10.1016/j.arcmed.2023.102920 (2023).
Wermer, P. Genetic aspects of adenomatosis of endocrine glands. Am. J. Med. https://doi.org/10.1016/0002-9343(54)90353-8 (1954).
Thakker, R. V. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol. Cell. Endocrinol. https://doi.org/10.1016/j.mce.2013.08.002 (2014).
Mouchtouris, N. et al. A review of multiomics platforms in pituitary adenoma pathogenesis. Front. Biosci. (Landmark Ed. https://doi.org/10.31083/j.fbl2703077 (2022).
Daly, A. F. et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: an international collaborative study. J. Clin. Endocrinol. Metab. https://doi.org/10.1210/jc.2009-2556 (2010).
Igreja, S., International FIPA consortium et al. International FIPA consortium.: Characterization of Aryl hydrocarbon receptor interacting protein (AIP) mutations in Familial isolated pituitary adenoma families. Hum. Mutat. https://doi.org/10.1002/humu.21292 (2010).
Daly, A. F. et al. Clinical characterization of Familial isolated pituitary adenomas. J. Clin. Endocrinol. Metab. https://doi.org/10.1210/jc.2005-2671 (2006).
Vierimaa, O. et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science https://doi.org/10.1126/science.1126100 (2006).
Beckers, A., Aaltonen, L. A., Daly, A. F. & Karhu, A. Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the Aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr. Rev. https://doi.org/10.1210/er.2012-1013 (2013).
Daly, A. F. et al. Aryl hydrocarbon receptor-interacting protein gene mutations in Familial isolated pituitary adenomas: analysis in 73 families. J. Clin. Endocrinol. Metab. https://doi.org/10.1210/jc.2006-2513 (2007).
Hernández-Ramírez, L. C., International FIPA consortium et al. International FIPA consortium.: landscape of Familial isolated and young-onset pituitary adenomas: prospective diagnosis in AIP mutation carriers. J. Clin. Endocrinol. Metab. https://doi.org/10.1210/jc.2015-1869 (2015).
Coopmans, E. C. & Korbonits, M. Molecular genetic testing in the management of pituitary disease. Clin. Endocrinol. (Oxf). https://doi.org/10.1111/cen.14706 (2022).
Katznelson, L. et al. Endocrine society.acromegaly: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. https://doi.org/10.1210/jc.2014-2700 (2014).
Melmed, S. et al. Endocrine society. Diagnosis and treatment of hyperprolactinemia: an endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. (2011).
Fleseriu, M. et al. B.M.K.: Consensus on diagnosis and management of Cushing’s disease: a guideline update. Lancet Diabetes Endocrinol. (2021). https://doi.org/10.1016/S2213-8587(21)00235-7
Knosp, E., Steiner, E., Kitz, K. & Matula, C. Pituitary adenomas with invasion of the cavernous sinus space: A magnetic resonance imaging classification compared with surgical findings. Neurosurgery https://doi.org/10.1227/00006123-199310000-00008 (1993).
Kina, B. G. et al. Whole exome sequencing reveals novel candidate variants for endometriosis utilizing multiple affected members in a single family. Mol. Genet. Genomic Med. https://doi.org/10.1002/mgg3.2312 (2024).
Torun, D., Arslan, M. & Yüksel, Z. Coexistence of severe developmental delay, epilepsy, and hemangioma in Snijders Blok-Fisher syndrome suggests the presence of a POU3F3-related SNIBFIS endophenotype: A case report. Am. J. Med. Genet. Part. A. https://doi.org/10.1002/ajmg.a.62135 (2021).
Tatsi, C. & Stratakis, C. A. The genetics of pituitary adenomas. J. Clin. Med. https://doi.org/10.3390/jcm9010030 (2020).
Ozkaya, H. M. et al. Germline mutations of Aryl hydrocarbon receptor-interacting protein (AIP) gene and somatostatin receptor 1–5 and AIP immunostaining in patients with sporadic acromegaly with poor versus good response to somatostatin analogues. Pituitary https://doi.org/10.1007/s11102-018-0876-4 (2018).
Karaca, Z., Taheri, S., Tanriverdi, F., Unluhizarci, K. & Kelestimur, F. Prevalence of AIP mutations in a series of Turkish acromegalic patients: Are synonymous AIP mutations relevant? Pituitary https://doi.org/10.1007/s11102-015-0659-0 (2015).
Tuncer, F. N. et al. Screening of AIP gene variations in a cohort of Turkish patients with young-onset sporadic hormone-secreting pituitary adenomas. Genet. Test. Mol. Biomarkers. https://doi.org/10.1089/gtmb.2018.0133 (2018).
Niyazoglu, M. et al. Familial acromegaly due to Aryl hydrocarbon receptor-interacting protein (AIP) gene mutation in a Turkish cohort. Pituitary https://doi.org/10.1007/s11102-013-0493-1 (2014).
Yarman, S., Ogret, Y. D. & Oguz, F. S. Do the Aryl hydrocarbon receptor interacting protein variants (Q228K and Q307R) play a role in patients with Familial and sporadic Hormone-Secreting pituitary adenomas?? Genet. Test. Mol. Biomarkers. https://doi.org/10.1089/gtmb.2014.0333 (2015).
Chahal, H. S., Chapple, J. P., Frohman, L. A., Grossman, A. B. & Korbonits, M. Clinical, genetic and molecular characterization of patients with Familial isolated pituitary adenomas (FIPA). Trends Endocrinol. Metab. https://doi.org/10.1016/j.tem.2010.02.007 (2010).
Leontiou, C. A. et al. The role of the Aryl hydrocarbon receptor-interacting protein gene in Familial and sporadic pituitary adenomas. J. Clin. Endocrinol. Metab. https://doi.org/10.1210/jc.2007-2611 (2008).
Occhi, G. et al. The R304X mutation of the Aryl hydrocarbon receptor interacting protein gene in Familial isolated pituitary adenomas: mutational hot-spot or founder effect? J. Endocrinol. Invest. https://doi.org/10.1007/BF03350345 (2010).
Chahal, H. S. et al. AIP mutation in pituitary adenomas in the 18th century and today. N Engl. J. Med. https://doi.org/10.1056/NEJMoa1008020 (2011).
Bogner, E. M. et al. miR-34a is upregulated in AIP-mutated somatotropinomas and promotes octreotide resistance. Int. J. Cancer. https://doi.org/10.1002/ijc.33268 (2020).
Tulipano, G. & Giustina, A. Effects of octreotide on autophagy markers and cell viability markers related to metabolic activity in rat pituitary tumor cells. Pituitary https://doi.org/10.1007/s11102-020-01028-0 (2020).
Ding, X. et al. SCP2-mediated cholesterol membrane trafficking promotes the growth of pituitary adenomas via Hedgehog signaling activation. J. Exp. Clin. Cancer Res. https://doi.org/10.1186/s13046-019-1411-9 (2019).
Righi, A. et al. Galectin-3 expression in pituitary adenomas as a marker of aggressive behavior. Hum. Pathol. https://doi.org/10.1016/j.humpath.2013.05.020 (2013).
Papadimitriou, E. et al. Prognostic biomarkers in pituitary tumours: A systematic review. TouchREV Endocrinol. https://doi.org/10.17925/EE.2023.19.2.12 (2023).
Xekouki, P. et al. Familial pituitary apoplexy as the only presentation of a novel AIP mutation. Endocr. Relat. Cancer. https://doi.org/10.1530/ERC-13-0218 (2013).
Vroonen, L. et al. The clinical and therapeutic profiles of prolactinomas associated with germline pathogenic variants in the Aryl hydrocarbon receptor interacting protein (AIP) gene. Front. Endocrinol. (Lausanne). https://doi.org/10.3389/fendo.2023.1242588 (2023).
Salenave, S. et al. Macroprolactinomas in children and adolescents: factors associated with the response to treatment in 77 patients. J. Clin. Endocrinol. Metab. https://doi.org/10.1210/jc.2014-3670 (2015).
Xekouki, P. et al. C.A.: Pituitary adenoma with paraganglioma/pheochromocytoma (3PAs) and succinate dehydrogenase defects in humans and mice. J Clin Endocrinol Metab. (2015). https://doi.org/10.1210/jc.2014-4297
Xekouki, P., Brennand, A., Whitelaw, B., Pacak, K. & Stratakis, C. A. The 3PAs: an update on the association of pheochromocytomas, paragangliomas, and pituitary tumors. Horm. Metab. Res. https://doi.org/10.1055/a-0661-0341 (2019).
DeBella, K., Szudek, J. & Friedman, J. M. Use of the National institutes of health criteria for diagnosis of neuro-fibromatosis 1 in children. Pediatrics https://doi.org/10.1542/peds.105.3.608 (2000).
Cambiaso, P. et al. Growth hormone excess in children with neurofibromatosis type-1 and optic glioma. Am. J. Med. Genet. A. https://doi.org/10.1002/ajmg.a.38308 (2017).
Hannah-Shmouni, F., Trivellin, G. & Beckers, P. Neurofibromatosis type 1 has a wide spectrum of growth hormone excess. J. Clin. Med. https://doi.org/10.3390/jcm11082168 (2022).
Hozumi, K. et al. Acromegaly caused by a somatotroph adenoma in patient with neurofibromatosis type 1. Endocr. J. https://doi.org/10.1507/endocrj.EJ19-0035 (2019).
Hong, C. S. et al. Somatic NF1 mutations in pituitary adenomas: report of two cases. Cancer Genet. https://doi.org/10.1016/j.cancergen.2021.03.004 (2021).
European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell https://doi.org/10.1016/0092-8674(93)90618-z (1993).
Dworakowska, D. & Grossman, A. B. Are neuroendocrine tumours a feature of tuberous sclerosis? A systematic review. Endocr. Relat. Cancer. https://doi.org/10.1677/ERC-08-0142 (2009).
Regazzo, D. et al. Silent gonadotroph pituitary neuroendocrine tumor in a patient with tuberous sclerosis complex: evaluation of a possible molecular link. Endocrinol. Diabetes Metab. Case Rep. https://doi.org/10.1530/EDM-18-0086 (2018).
Barry, S. & Korbonits, M. Update on the genetics of pituitary tumors. Endocrinol. Metab. Clin. North. Am. https://doi.org/10.1016/j.ecl.2020.05.005 (2020).
Sotomayor, M., Gaudet, R. & Corey, D. P. Sorting out a promiscuous superfamily: Towards Cadherin connectomics. Trends Cell. Biol. https://doi.org/10.1016/j.tcb.2014.03.007 (2014).
Schultz, J. M. et al. Allelic hierarchy of CDH23 mutations causing non-syndromic deafness DFNB12 or Usher syndrome USH1D in compound heterozygotes. J. Med. Genet. https://doi.org/10.1136/jmedgenet-2011-100262 (2011).
Zhang, Q. et al. Germline mutations in CDH23, encoding Cadherin-Related23 are associated with both Familial and sporadic pituitary adenomas. Am. J. Hum. Genet. https://doi.org/10.1016/j.ajhg.2017.03.011 (2017).
Chang, M., Yang, C., Bao, X. & Wang, R. Genetic and epigenetic causes of pituitary adenomas. Front. Endocrinol. (Lausanne). https://doi.org/10.3389/fendo.2020.596554 (2021).
Tommiska, J. et al. Two missense mutations in KCNQ1 cause pituitary hormone deficiency and maternally inherited gingival fibromatosis. Nat. Commun. https://doi.org/10.1038/s41467-017-01429-z (2017).
Sbiera, S. et al. The new genetic landscape of cushing’s disease: Deubiquitinases in the spotlight. Cancers (Basel). https://doi.org/10.3390/cancers11111761 (2019).
Paces-Fessy, M., Boucher, D., Petit, E., Paute-Briand, S. & Blanchet-Tournier, M. F. The negative regulator of gli, suppressor of fused (Sufu), interacts with SAP18, Galectin3 and other nuclear proteins. Biochem. J. https://doi.org/10.1042/BJ20030786 (2004).
Zhang, H. Y. et al. RUNX1 and RUNX2 upregulate Galectin-3 expression in human pituitary tumors. Endocrine https://doi.org/10.1007/s12020-008-9129-z (2009).
Asa, S. L., Mete, O., Riddle, N. D. & Perry, A. Multilineage Pituitary Neuroendocrine Tumors (PitNETs) Expressing PIT1 and SF1. Endocr Pathol. (2023). https://doi.org/10.1007/s12022-023-09777-x
Syro, L. V. et al. Somatotroph pituitary adenoma with acromegaly and autosomal dominant polycystic kidney disease: SSTR5 polymorphism and PKD1 mutation. Pituitary https://doi.org/10.1007/s11102-011-0325-0 (2012).
Thompson, I. R. et al. Expression of Guanylyl cyclase-B (GC-B/NPR2) receptors in normal human fetal pituitaries and human pituitary adenomas implicates a regulatory role for C-type natriuretic peptide. Endocr. Relat. Cancer. https://doi.org/10.1530/ERC-12-0129 (2012).
De Martino, I. et al. B-RAF mutations are a rare event in pituitary adenomas. J. Endocrinol. Invest. https://doi.org/10.1007/BF03347386 (2007).
Jia, W., Lu, R., Jia, G., Ni, M. & Xu, Z. Expression of pituitary tumor transforming gene (PTTG) in human pituitary macroadenomas. Tumour Biol. https://doi.org/10.1007/s13277-013-0686-2 (2013).
Funding
This project has been funded by the Society of Endocrinology and Metabolism of Turkiye (SEMT) (2021/P03).
Author information
Authors and Affiliations
Contributions
S.Y., M.E.E. designed the study, collected data from centers, reviewed the eligibility of relevant data, performed literature search and wrote the paper. F.N.T. led the laboratory work and interpreted and wrote the genetic part of the paper. S.A. contributed to laboratory work and performed literature search S.C., S.T., O.S.S.,O.T., M.E., P.K., B.C.,C.S., Z.P.,G.G.O.,B.C. provided relevant data from their centers.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ertorer, M.E., Tuncer, F.N., Ciftci, S. et al. Aryl hydrocarbon receptor interacting protein and syndromic gene variants detected in Turkish isolated pituitary adenoma families by whole exome sequencing. Sci Rep 15, 24279 (2025). https://doi.org/10.1038/s41598-025-08610-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-08610-1
