
Abstract
Hepatocellular carcinoma (HCC) is a leading cause of cancer-related death globally, characterized by high morbidity and poor prognosis. The complex molecular and immune landscape of HCC makes accurate patient stratification and personalized treatment essential. In this study, we utilized large-scale gene expression data from TCGA and GSE54236, alongside eQTL GWAS data, to identify key genes that influence HCC prognosis. Machine learning analysis was performed on the genes identified through Mendelian randomization (MR) and survival association analysis, using 101 algorithms to construct a robust prognostic model. A novel riskScore model was developed by integrating genetic, clinical, and immune cell infiltration data. The prognostic performance of model was validated through survival analysis, and its association with chemotherapy and immunotherapy sensitivity. The impact of key genes on the proliferation and invasion capabilities of HCC cells was assessed through Western blot (WB), EdU, and invasion assays. A total of 27 candidate genes associated with HCC survival were identified, with 16 genes categorized as high-risk. The riskScore model demonstrated excellent performance in stratifying patients into high-risk and low-risk groups, with C-index exceeding 0.7 for both TCGA and GSE54236 datasets. High-risk patients exhibited poorer prognosis and higher immune cell infiltration, particularly T cells and neutrophils. The model also predicted drug sensitivity, with high-risk patients showing greater sensitivity to chemotherapy agents like 5-Fluorouracil and Paclitaxel. Mutation analysis revealed that TP53 and MUC16 mutations were prevalent in high-risk groups, highlighting their role in HCC progression and therapeutic response. And the key gene SLC16A3 and STRBP can significantly promote the proliferation and invasion ability of HCC cells. Our riskScore model, integrating genetic and immune factors, provides a robust prognostic tool with potential clinical application in patient stratification and chemotherapy decision-making for HCC patients.
Similar content being viewed by others

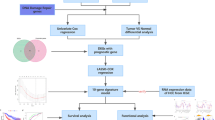

Introduction
HCC, the sixth most common cancer and the third leading cause of cancer-related deaths, is prevalent and deadly worldwide, with nearly 900,000 new cases diagnosed annually1,2. The incidence of HCC is rising, particularly in regions with widespread risk factors like chronic hepatitis B and C, liver cirrhosis, and aflatoxin exposure3. Despite advancements in detection and treatment, the prognosis remains poor due to its aggressive nature, high recurrence rates, and difficulties in treatment selection for advanced stages4. Furthermore, molecular and clinical heterogeneity of HCC complicates diagnosis and treatment, emphasizing the need for improved biomarkers and personalized therapies. Chemotherapy and immunotherapy are key treatment options for advanced HCC, but challenges such as drug resistance, side effects, and lack of specificity limit their efficacy. Sorafenib, the first FDA-approved systemic treatment, offers modest survival benefits but is hindered by drug resistance and tumor progression5. Recently, immune checkpoint inhibitors like nivolumab and pembrolizumab have shown promise, but their overall response rate remains low, and reliable biomarkers for predicting response are still lacking6. The immunosuppressive tumor microenvironment in HCC further complicates treatment outcomes, underscoring the urgent need for personalized, patient-tailored therapeutic strategies7. Consequently, there is an increasing need for personalized treatment approaches that integrate genetic, molecular, and immunological characteristics to better predict patient responses and guide therapeutic decisions.
Traditional observational studies are challenged by issues such as confounding bias and reverse causality, which limit the ability to make causal inferences. MR, which uses genetic variations as instrumental variables, has emerged as a crucial tool for revealing causal relationships in the pathogenesis of liver cancer and for identifying therapeutic targets. For example, an MR analysis conducted by Cao et al. demonstrated that low-density lipoprotein cholesterol (LDL-C) has a protective effect on liver cancer risk (OR 0.703, 95% CI 0.508–0.973), partly through coronary artery disease (58.52%), highlighting the role of metabolic pathways in liver cancer development8. Similarly, existing studies have established causal relationships between antidiabetic drug targets (such as ABCC8/KCNJ11) and gastrointestinal cancers, including liver cancer, underscoring the value of MR in drug target validation9. Furthermore, MR studies have identified sarcopenia-related traits, such as low limb lean mass and grip strength, as independent risk factors for hepatocellular carcinoma (HCC) in European populations, with C-reactive protein (CRP) mediating 7.4 to 9.1% of the effect10. These findings not only clarify biological mechanisms but also highlight actionable intervention targets. Additionally, a comprehensive MR analysis by Zhu et al. identified 10 plasma proteins (including ALDH1A1, TFPI2 etc.) as potential therapeutic biomarkers, which were validated through colocalization and single-cell analysis11. Overall, MR analysis fills gaps in causal inference and provides valuable insights for the prevention, early detection, and targeted treatment of hepatocellular carcinoma.
Many prognostic features identified in previous studies have limitations in clinical applications, primarily due to unclear causal relationships with cancer. To address this, we applied MR to identify key genes. MR leverages the theoretical random allocation of alleles to avoid reverse causality and unmeasured confounders, thus enabling the identification of genes that influence HCC progression. By intersecting genes selected through MR with prognostic genes from the TCGA and GSE54236 datasets, we developed a novel risk model, riskScore, that integrates both genetic and clinical factors. This model is designed not only to predict the overall survival of HCC patients but also to provide insights into the tumor immune landscape. Our findings underscore the crucial role of immune cell infiltration, particularly T cells, B cells, and macrophages in determining the invasiveness of HCC and its response to treatment. Furthermore, this model offers valuable predictions of chemotherapy and immunotherapy sensitivity, supporting the adoption of more personalized treatment strategies. By incorporating genetic variations that affect immune cell infiltration and tumor progression, this study provides a comprehensive framework for improving patient stratification and optimizing HCC treatment strategies, ultimately contributing to better clinical outcomes.
Materials and methods
Data collection and preprocessing
Gene expression data and clinical information for HCC were primarily sourced from TCGA data portal (https://portal.gdc.cancer.gov/), which includes 374 HCC samples and their corresponding clinical data. Additionally, we retrieved the GSE54236 HCC dataset from the GEO database, which contains gene expression data and clinical information for 80 HCC samples. To ensure consistency and comparability across datasets, batch effects between datasets were corrected using the ComBat function from the sva R package. Furthermore, we extracted eQTL GWAS data from the IEU OpenGWAS project (https://gwas.mrcieu.ac.uk/), covering 19,942 genes, and selected single nucleotide polymorphisms (SNPs) with a significance threshold of P < 5e−6 for further analysis. In addition, we integrated HCC-related GWAS data from the FinnGen R12 dataset (https://www.finngen.fi/fi) and the GWAS Catalog, specifically the GCST90013702 dataset. The FinnGen dataset includes data from 947 HCC patients and 378,749 healthy controls, while the GCST90013702 dataset includes data from 1,866 HCC patients and 195,745 healthy controls, providing valuable genetic association information for subsequent in-depth analysis.
MR analysis
In this study, a two-sample MR framework was implemented using the “TwoSampleMR” R package (v0.5.6) to infer the causal relationship between gene expression (eQTLs) and HCC risk. We selected independent instrumental variables (IVs) by applying linkage disequilibrium pruning (r2 = 0.1, kb = 10,000), and palindromic SNPs were excluded to avoid strand ambiguity. The inverse variance weighted (IVW) method was employed as the primary analysis strategy due to its superior statistical power under the assumption of valid instruments. To account for potential violations of the MR assumptions—such as horizontal pleiotropy—we conducted sensitivity analyses using multiple complementary MR methods, including MR-Egger regression, weighted median, and mode-based estimation. These methods provided robustness checks under different model assumptions and helped assess the consistency of causal estimates. The MR-Egger intercept test was used to detect directional pleiotropy, while heterogeneity was assessed via Cochran’s Q statistic. Additionally, we performed leave-one-out analysis to evaluate the influence of individual SNPs on the causal estimate. These sensitivity analyses confirmed the robustness of the MR results and minimized the likelihood of bias due to invalid instruments.
To address heterogeneity in the HCC GWAS data, we first integrated the FinnGen R12 (947 HCC cases/378,749 controls) and GCST90013702 (1866 HCC cases/195,745 controls) datasets using Metal software12, corrected for batch effects, and generated summary statistics, which were then used for the final MR analysis.
Feature selection and riskScore model construction
In this study, a prognostic prediction model for HCC was developed by integrating multiple analytical approaches. Univariate Cox regression analysis (coxph function) was performed using the “survival” R package (v3.5.7) and the “survminer” R package (v0.4.9) to identify candidate genes significantly associated with overall survival in HCC patients (selection criterion: P < 0.05). These candidate genes were cross-validated with eQTL genes identified through Mendelian randomization analysis. A Venn diagram was used to determine a feature gene set with both genetic association and clinical prognostic significance. Based on the expression profiles of these feature genes, the classification performance of 101 machine learning algorithms (including Lasso + StepCox, survival-SVM, etc.) was systematically evaluated. The 101 methods used by the machine learning model and their corresponding hyperparameters are presented in Table S9. The optimal algorithm for constructing the riskScore model was selected through cross-validation and C-index. Patients were stratified into high-risk and low-risk groups using the median riskScore as the threshold. Kaplan–Meier curves and Log-rank tests were employed to assess the prognostic discriminative power of this stratification.
To mitigate overfitting and ensure model generalizability, we implemented a rigorous tenfold cross-validation strategy for all machine learning algorithms during model training. Specifically, in the RunEnet function (used for Lasso, and Ridge), cross-validation was performed using the cv.glmnet function with nfolds = 10, where the dataset was randomly partitioned into 10 equally sized subsets. In each iteration, one subset was held out for validation while the remaining nine were used for training. This process was repeated across all folds, and the optimal penalty parameter (lambda) was selected based on the minimum cross-validated error. For other algorithms such as CoxBoost, GBM, and SuperPC, internal cross-validation procedures (cv.CoxBoost, cv.gbm, superpc.cv) with K = 10 folds were applied to select hyperparameters and assess performance stability. These consistent tenfold cross-validation frameworks were used to reduce the risk of overfitting, improve model robustness, and select the best-performing model across 101 algorithmic configurations.
Survival analysis and model validation
This study employed a multi-dimensional validation framework to assess the prognostic performance of the risk model. First, time-dependent ROC curves for the risk model and clinical indicators (such as age and stage) were generated using the “timeROC” R package (v0.4.3) to evaluate 1-year, 3-year, and 5-year survival rates. The “rms” R package (v6.7-0) and “pec” R package were then used to calculate C-index, quantifying the model’s predictive accuracy. The parameters for the C-index calculation were set as splitMethod = “bootcv” and B = 1000. Subsequently, univariate and multivariate Cox regression models (using the “survival” R package) were applied to evaluate the statistical significance of riskScore as an independent prognostic factor (hazard ratios and 95% confidence intervals). Finally, Kaplan–Meier survival curves were constructed based on the median riskScore to stratify patients into high-risk and low-risk groups. The Log-rank test was employed to assess the significance of survival differences between the two groups (P < 0.05).
Functional enrichment analysis
GSEA Pathway Enrichment Analysis: Differential gene expression analysis was performed using the “limma” R package (v3.58.1). Pathway enrichment analysis was conducted with the “clusterProfiler” R package (v4.12.0) on gene sets from KEGG (c2.cp.kegg_legacy.v2024.1.Hs.symbols.gmt), Gene Ontology (c5.go.v2024.1.Hs.symbols.gmt), and Hallmark (h.all.v2024.1.Hs.symbols.gmt), selecting significant pathways with an FDR < 0.05.
Immune-Related Pathway Analysis: The single-sample Gene Set Enrichment Analysis (ssGSEA) method was used to quantify the activity of immune-related pathways. Additionally, algorithms such as CIBERSORT were employed to calculate the abundance of 28 immune cell subtypes. Spearman correlation analysis was performed to evaluate the association between the riskScore and immune infiltration (P < 0.05).
Immune cell infiltration assessment
This study utilized multiple methods to estimate immune cell infiltration, including Quantiseq, Timer, Mcp_counter, Xcell, CIBERSORT, and EPIC algorithms, for a comprehensive assessment of immune cell distribution patterns. Tumor purity and the composition of stromal immune cells were precisely estimated using the “estimate” R package to quantify the immune infiltration status of the tumor microenvironment. Furthermore, the TIDE (Tumor Immune Dysfunction and Exclusion) online tool (http://tide.dfci.harvard.edu/) was employed to analyze the immune evasion potential13. Differences in immune therapy response between the high-risk and low-risk groups were compared to investigate the potential link between riskScore and immune therapy prognosis.
Genetic variation characteristic
This study employed a multi-dimensional approach to investigate the genetic variation features associated with HCC. First, somatic mutation annotation and mutation feature analysis were conducted on the TCGA-LIHC dataset using the “maftools” R package. SNV waterfall plots were created to systematically illustrate the heterogeneity of gene mutation profiles across samples. Next, a gene co-mutation heatmap was used to identify co-occurrence and mutually exclusive patterns of high-frequency mutated genes (e.g., TP53, CTNNB1). Fisher’s exact test was applied to identify significant co-mutation events (P < 0.05).
Drug sensitivity prediction
This study integrated genomic-based drug sensitivity prediction models to systematically evaluate potential targeted therapies for HCC. Drug sensitivity prediction matrices were constructed using the “oncopredict” R package and the “pRRophetic” R package14,15. Spearman correlation analysis was employed to quantify the association between the drug half-maximal inhibitory concentration (IC50) and the riskScore. Chemotherapeutic drugs significantly correlated with the riskScore were identified based on Spearman correlation analysis (P < 0.001).
Statistical analysis
All statistical analyses were performed using R software (https://www.r-project.org/). Statistical significance was defined as a P value < 0.05. Correlation analyses between variables were conducted using Pearson’s correlation coefficient (for normally distributed data) and Spearman’s correlation coefficient (for non-parametric data). For continuous variables, independent sample t-tests (for normally distributed data) or Mann–Whitney U tests (for non-normally distributed data) were applied for pairwise comparisons, based on the distribution characteristics. Differences among multiple groups were analyzed using the Kruskal–Wallis test (non-parametric) or ANOVA (for normally distributed data with homogeneous variance). Survival analysis was conducted using the Kaplan–Meier method to visualize the prognostic differences between high- and low-risk groups, and the Log-rank test was used to assess the significance of survival differences.
Reagents and antibodies
The siRNAs targeting SLC16A3 and STRBP were constructed by Shanghai Genechem Co., Ltd. (Shanghai, China). All antibodies, including anti-MCT4 (SLC16A3) (22,787-1-AP), anti-STRBP (17,362-1-AP), anti-α-Tubulin (11,224-1-AP), and anti-GAPDH (10,494-1-AP), were purchased from Proteintech Group (Wuhan, China).
Cell culture and transfection
The human normal liver cell line MIHC and HCC cell lines Hep3B, PLC/PRF/5, Huh7, and MHCC97H were obtained from the American Type Culture Collection (ATCC). Cells were cultured in a 37 °C incubator with 5% CO₂, and cell morphology and confluency were monitored every 8–24 h. When cell confluency reached 80–95%, cells were passaged using 0.25% EDTA trypsin. After centrifugation (1000 rpm, 5 min), cells were resuspended in fresh complete medium for continued culture. Transfection was performed in 6-well plates: when the cell confluency reached 70–80%, siRNA was mixed with opt-MEM medium and transfected using the Lipofectamine 3000 reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. After 6–8 h, the medium was replaced, and subsequent experiments were conducted 48 h post-transfection.
WB analysis
Sample Preparation: Cells were lysed with pre-chilled RIPA lysis buffer (containing protease and phosphatase inhibitors) and incubated on ice for 30 min. The lysates were then centrifuged at 12,000 rpm for 15 min at 4 °C to collect the supernatant. Protein concentrations were measured using the BCA assay. The samples were mixed with 5 × loading buffer and boiled for denaturation.
Electrophoresis and Transfer: A 10% separating gel (acrylamide/Bis 29:1, Tris–HCl pH 8.8, 0.1% SDS, 0.05% APS, 0.1% TEMED) and a 5% stacking gel (acrylamide/Bis 29:1, Tris–HCl pH 6.8, 0.1% SDS, 0.05% APS, 0.1% TEMED) were prepared. The electrophoresis was carried out with an initial voltage of 80 V (stacking gel), followed by 120 V (separating gel). Proteins were transferred to methanol-activated PVDF membranes (Millipore) using a wet transfer system (Bio-Rad) at a constant current of 250 mA for 90 min.
Antibody Incubation and Detection: The PVDF membranes were blocked with 5% non-fat milk for 1 h, then incubated overnight at 4 °C with the primary antibody, followed by a 1-h incubation at room temperature with the HRP-conjugated secondary antibody. Protein bands were visualized using ECL chemiluminescence reagents, and the results were captured using a chemiluminescence imaging system. The images were saved for further analysis. GAPDH and α-Tubulin were used as internal controls in WB assays to normalize protein loading across samples.
EdU-488 cell proliferation assay and transwell invasion assay
EdU-488 Cell Proliferation Assay: Cells were seeded in 6-well plates and incubated until they reached 30%-40% confluence. The medium was then replaced with fresh medium containing 50 μM EdU reagent, and cells were incubated at 37 °C for 2 h. After removing the medium, the cells were fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.3% Triton X-100 for 15 min, and incubated with Click reaction solution (containing Alexa Fluor 488) for 30 min in the dark. The cells were subsequently counterstained with Hoechst for 10 min. Fluorescence images were acquired from random fields of view under a fluorescence microscope.
Transwell Invasion Assay: Transwell chambers (Corning, 8 μm pore size) were pre-chilled to 4 °C. Matrigel was diluted in serum-free medium at a 1:8 ratio and applied evenly to the upper chamber (40 μl per chamber), followed by incubation at 37 °C for 1 h to allow solidification. Log-phase cells were trypsinized, resuspended in serum-free medium (2 × 104 cells/200 μl), and seeded into the upper chamber. The lower chamber was filled with medium containing 20% fetal bovine serum. After 48 h of incubation at 37 °C, the cells were fixed with 4% paraformaldehyde for 30 min and stained with 0.1% crystal violet for 15 min. Non-invasive cells on the upper surface of the membrane were gently removed with a cotton swab, and images of random fields were captured under a microscope.
Results
Identification of HCC-related genes through eQTL MR analysis
The workflow diagram of this study is presented in Fig. S1. ID conversion was performed for the eQTL data from 19,942 genes obtained from blood samples (Table S1), retaining 15,307 eQTL data points that included gene names. Next, we compared the SNPs associated with each eQTL to HCC GWAS data, including datasets from the FinnGen and Japanese cohorts. SNPs were screened using the F-statistic and five MR methods, identifying those with significant associations to HCC-related genes (Tables S2, S3). To address cross-cohort heterogeneity, we conducted a meta-analysis using a fixed-effects model to obtain the adjusted effect sizes from both GWAS datasets. Based on this, we carried out a second-stage MR analysis, ultimately confirming a significant causal relationship between the eQTL levels of 239 genes and HCC risk (Table 1, Table S4, S5).
Identification of candidate genes and construction of machine learning models
To further investigate prognosis-related genes in HCC, we conducted univariate Cox regression analysis on RNA expression data from the TCGA and GSE54236 datasets, identifying 10,145 and 5752 genes, respectively, that were significantly associated with patient survival (Tables S6, S7). We then intersected the prognosis-related genes from both datasets with the MR risk genes, ultimately identifying 27 candidate genes (Fig. 1A, B, Table S8). Among these, 16 genes were found to be high-risk genes potentially contributing to HCC development, while 11 genes were identified as protective (Fig. 1C). To further classify these genes and clarify their roles, we used gene expression data from the TCGA and GSE54236 datasets and applied 101 different machine learning algorithms to analyze the expression data of the 27 risk genes (Tables S10, S11). The gene set showed C-indices greater than 0.7 in the majority of machine learning models, suggesting a significant impact on HCC prognosis. Based on the highest average C-index, we selected the Enet [α = 0.1] model for further analysis in our study (Fig. 1D).

Gene screening and machine learning model construction. (A) The intersection of risk genes in MR and prognostic genes in TCGA and GSE54236. (B) The location of candidate genes on chromosomes. (C) The eQTL and HCC GWAS MR analysis results of 27 model genes. nsnp represents the number of valid SNPs. Only the results of the IVW method are shown. (D) The analysis results of 101 machine learning methods for candidate genes, showing the performance of different models in TCGA and GSE54236. The c-index value represents the performance of the model.
Risk model validation
Based on the riskScore calculated using the Enet [α = 0.1] model, patients were stratified into high-risk and low-risk groups according to the median riskScore value (Table S12). The results revealed that patients in the high-risk group from both the TCGA and GSE54236 datasets had significantly worse prognosis compared to those in the low-risk group (Fig. 2A, E). The model’s performance was then evaluated using ROC curves and C-index values. The AUC and C-index for predicting the 5-year survival rate of TCGA patients were significantly greater than 7. In addition, when compared to clinical staging, the machine learning model showed higher accuracy in predicting the 5-year survival rate for TCGA patients (Fig. 2B–D). Similarly, for GSE54236 patients, the AUC and C-index values for predicting the 3-year survival rate exceeded 8 (Fig. 2F, G). To further validate the predictive power of the model across various clinical variables, univariate and multivariate Cox regression analyses were performed. The results demonstrated that the riskScore remained strongly associated with prognosis, with high hazard ratio (HR) values even after adjusting for clinical variables in both datasets, confirming that riskScore is an independent prognostic factor for HCC (Fig. 2I–L). Validation analyses in the TCGA and GSE54236 datasets thus confirmed that the model offers robust accuracy and sensitivity in predicting HCC patient prognosis.

Validation of the riskScore model. (A, E) Kaplan–Meier survival curve analysis showed the survival difference between high-risk group and low-risk group in TCGA and GSE54236. (B, C, F, G) ROC curve was used to evaluate the predictive ability of different clinical factors and riskScore model at different time points. (D, H) The independent predictive ability of riskScore and clinical variables (such as age, gender, pathological stage) on patient prognosis was measured by C-index value. (I, K) Univariate Cox regression analysis results of riskScore between different clinical factors and risk groups. (J, L) Multivariate Cox regression analysis results of riskScore after adjusting clinical variables.
Differential signaling pathways between high-risk and low-risk groups
Differential expression analysis of gene expression data from the TCGA dataset revealed significantly differentially expressed genes between the high-risk and low-risk groups, allowing us to identify distinct signaling pathways. In the GO pathway analysis, the high-risk group was primarily enriched in pathways such as “DNA repair” and “cell cycle regulation,” which are closely linked to HCC progression and malignant transformation (Fig. 3A). Conversely, the low-risk group showed significant enrichment in pathways associated with HCC metabolism, including “lipid metabolism” and “amino acid metabolism” (Fig. 3A). In the KEGG pathway analysis, the high-risk group was significantly enriched in pathways related to cell cycle regulation and DNA repair, such as the “cell cycle” and “mismatch repair” pathways, suggesting that these pathways play a crucial role in HCC progression in high-risk patients (Fig. 3B). Meanwhile, the low-risk group exhibited notable enrichment in metabolic pathways, particularly “fatty acid metabolism” and “primary bile acid biosynthesis,” which may contribute to a better prognosis and higher metabolic adaptability in these patients (Fig. 3B). Additionally, Hallmark pathway analysis revealed significant enrichment in the “KRAS signaling down,” “G2M checkpoint,” and “angiogenesis” pathways in the high-risk group (Fig. 3E), which are associated with tumor progression and cell cycle changes. These findings further elucidate the differences in tumor progression mechanisms between the high-risk and low-risk groups in HCC.

Differential pathway analysis between high-risk and low-risk groups. Differential signal pathway analysis between high-risk and low-risk groups, (A) GO; (B) KEGG; (C) Hallmark.
Differences in immune-related signaling pathways between high-risk and low-risk groups
SsGSEA analysis of immune signaling pathways in the high-risk and low-risk group samples (Table S13) revealed distinct differences in immune signal expression between the two groups. In pathways related to immune cell infiltration and tumor progression, the high-risk group was predominantly enriched in DNA homologous recombination and cell cycle pathways, which aligns with the previous analysis. Additionally, the high-risk group showed significant enrichment in the FGFR3-related gene set, suggesting a higher likelihood of recognizing patients with FGFR3 overexpression. In contrast, the low-risk group was primarily enriched in the Antigen Processing Machinery pathway (Fig. 4A). Immune-related gene enrichment analysis further indicated that the low-risk group was primarily associated with the IFN response and cytolytic activity (Fig. 4B). In terms of immune response-related signaling pathways, the low-risk group was mainly enriched in inflammatory and adaptive immune responses (Fig. 4C). Finally, analysis of the seven-step immune response characteristics revealed that the high-risk group was predominantly associated with “infiltration of immune cells into tumors” (Fig. 4D). These results suggest that HCC in the high-risk group is more closely linked to tumor progression and immune cell infiltration, while the low-risk group is primarily associated with basic and initial immune responses.

Differences in immune signaling pathways between high-risk and low-risk groups. Enrichment of high-risk and low-risk groups in different tumor and immune cell gene sets, (A) Tumor progression-related pathways; (B) Immune-related gene sets; (C) Immune response-related pathways; (D) Signature gene sets for the 7 steps of immune response.
Immune microenvironment differences between high-risk and low-risk groups
To better understand the immune microenvironment in the high-risk and low-risk groups, we evaluated the immune cell composition of HCC samples using six tumor immune cell infiltration scoring methods. In most methods, a higher riskScore was associated with increased infiltration of B cells, CD4 + T cells, CD8 + T cells, and neutrophils. In the Quantiseq method, riskScore was positively correlated with M2 macrophage infiltration, while in the xCell method, riskScore showed a negative correlation with M2 macrophage infiltration, suggesting that riskScore alone may not effectively distinguish M2 macrophage infiltration levels in HCC patients (Fig. 5A). In the ssGSEA immune cell enrichment analysis, the infiltration of activated CD4 T cells was positively correlated with riskScore, while the infiltration of CD56bright NK cells showed a negative correlation with riskScore (Fig. 5B). TIDE analysis revealed that riskScore was positively correlated with both the TIDE score and the infiltration levels of Exclusion and MDSCs, indicating that higher riskScores are associated with an increased likelihood of T-cell exclusion and potentially poorer immunotherapy outcomes (Fig. 5C).

Relationship between riskScore and immune microenvironment. (A) Correlation analysis between riskScore and the level of tumor immune cell infiltration using 6 methods. (B) Correlation analysis between riskScore and the results of ssGSEA analysis of immune gene sets. (C) Correlation analysis between riskScore and TIDE analysis results. (D, E) Expression of immune co-inhibitory checkpoints and immune co-stimulatory checkpoints in high-risk and low-risk groups. (F, H) Differences in StromalScore and ESTIMATEScore values between high-risk and low-risk groups. (G, I) Correlation analysis between riskScore and StromalScore and ESTIMATEScore.
Analysis of immune checkpoint expression differences between the high-risk and low-risk groups showed that the expression of most immune checkpoint inhibitors (including PDCD1, CD274, and CTLA4) was significantly higher in the high-risk group compared to the low-risk group (Fig. 5D, E). Using the ESTIMATE tool, we found that the StromalScore was significantly lower in the high-risk group and negatively correlated with riskScore, while the Estimate score also demonstrated a negative correlation with riskScore (Fig. 5F–I). These results suggest that the low-risk group may have a higher proportion of stromal components and non-tumor cells. Overall, our comprehensive analysis of the immune microenvironment reveals a strong association between HCC riskScores and immune cell infiltration levels, indicating that immune microenvironment changes may significantly influence the prognosis of HCC patients.
Clinical and pathological characteristics of high-risk and low-risk groups
We next analyzed the differences in clinical features between the high-risk and low-risk groups. Sankey diagrams revealed that the high-risk group had a greater proportion of patients who were unresponsive to immunotherapy, while a higher number of patients who responded to immunotherapy and survived were also in the high-risk group. This observation is consistent with the higher expression of immune checkpoint inhibitors in the high-risk group (Fig. 6A). Regarding T stage, the high-risk group had more patients in stages T2 and T3, whereas the low-risk group had a higher proportion of patients in stage T1 (Fig. 6B). Moreover, patients in stage T1 had significantly lower riskScores compared to those in stages T2 and T3 (Fig. 6C). Similar findings were observed in the stage analysis, which was consistent with the T stage results (Fig. 6D, E). The high-risk group also had a higher number of patients who died, and the riskScore of the deceased patients was significantly higher than that of the survivors (Fig. 6F, G). Additionally, the high-risk group exhibited higher tumor mutational burden (TMB) and maximum variant allele frequency (MaxVAF), both of which were positively correlated with riskScore (Figs. 6H-K). In summary, riskScore effectively differentiates the clinical and pathological features between the high-risk and low-risk groups.

Relationship between riskScore and clinical pathological characteristics of HCC. (A) Sankey diagram showing the proportion of high-risk and low-risk groups and TIDE immune response and patient survival and clinical stage. (B, D, F) The proportion of T stage, stage, and survival status between high-risk and low-risk groups. (C, E, G) Differences in riskScores of patients with different T and Stage, and differences in riskScores of patients who survive and die. (H, J) Analysis of differences in TMB and MaxVAF values of patients in high-risk and low-risk groups. (I, K) Correlation analysis between riskScore and TMB and MaxVAF values.
Genetic mutation characteristics in high-risk and low-risk HCC groups
To investigate the differences in genetic mutation characteristics between high-risk and low-risk groups in HCC, we used the “maftools” R package to examine the mutation frequencies and patterns between the two groups in TCGA HCC samples. The results revealed that in the high-risk group, genes such as TP53 and MUC16 had significantly higher mutation frequencies compared to the low-risk group. TP53 mutations were particularly prominent in the high-risk group, with a mutation frequency exceeding 45%, while the mutation frequency of TP53 was notably lower in the low-risk group. Additionally, the mutation frequency of CTNNB1 was higher in the low-risk group compared to the high-risk group (Fig. 7A, B). Co-mutation analysis showed that TP53 mutations in the high-risk group were frequently co-mutated with genes such as OBSCN and PCLO, while in the low-risk group, CTNNB1 mutations were often co-mutated with ALB and ARID2 (Fig. 7C, D). These results suggest that the co-mutation patterns of genes in different risk groups may reveal distinct molecular mechanisms underlying HCC. Furthermore, variant allele frequency (VAF) analysis was performed to assess the clonal status of mutations in both risk groups. The VAF analysis indicated a significant difference between the high-risk and low-risk groups. In the high-risk group, high VAF values were primarily associated with RB1 and TP53 mutations, while in the low-risk group, high VAF values were mainly observed for AXIN1 and ARID1A mutations. This suggests that the two groups exhibit different clonal states for their genetic mutations. In summary, our findings highlight significant differences in the genetic mutation characteristics between the high-risk and low-risk groups in HCC, particularly in the mutation frequency and co-mutation patterns of key cancer-related genes.

Genetic mutation characteristics of high-risk and low-risk groups. (A, B) show the gene mutation characteristics of the top 20 in the TCGA dataset for the high-risk and low-risk groups. (C, D) Gene co-mutation analysis shows the differences in the co-occurrence patterns of gene mutations between the high-risk and low-risk groups. (E, F) Box plots show the differences in the clonal status of gene mutations between the high-risk and low-risk groups.
Chemotherapy drug sensitivity prediction in HCC based on a risk model
To evaluate chemotherapy drug sensitivity in high-risk and low-risk groups of HCC patients, we utilized the “oncoPredict” and “pRRophetic” packages to assess drug responses. Initially, we predicted the drug sensitivity of the high-risk and low-risk groups using GDSC2 data and compared the IC50 values across various chemotherapy drugs. The results demonstrated that patients in the high-risk group had lower IC50 values for 5-Fluorouracil, Paclitaxel, Vinorelbine, and Bortezomib, indicating higher sensitivity to these drugs. In contrast, the low-risk group exhibited higher IC50 values, suggesting lower sensitivity to these chemotherapy agents. Moreover, the “pRRophetic” package confirmed a negative correlation between the riskScore and IC50 values for these drugs, further validating our prediction results (Figs. 8B–E, H–K). For Gemcitabine and Lapatinib, the predicted IC50 values exhibited the opposite trend, indicating that riskScore-based predictions for these drugs were less accurate (Fig. 8A, F, G, L). These findings suggest that the model can effectively predict chemotherapy drug responsiveness in HCC patients for certain drugs.

Relationship between riskScore and chemotherapy drug sensitivity. (A–F) Differences in IC50 values of common chemotherapy drugs between high-risk group and low-risk group predicted by oncopredict in GDSC2 data. (G–L) Correlation analysis between riskScore and IC50 values of different chemotherapy drugs using “pRRophetic” package.
Prognostic prediction for HCC patients based on riskScore
To assess the prognosis of HCC patients, we conducted a multivariate Cox regression analysis incorporating the riskScore along with clinical variables (age, gender, pathological stage, and TMB). The results indicated that the riskScore provided an independent and robust prognostic prediction, with HR greater than 3, regardless of the clinical features (Fig. 9A). Survival analysis, based on stratification by high and low-risk groups and high and low TMB expression, revealed significant prognostic differences between these groups (Fig. 9B). Furthermore, we constructed a comprehensive prognostic model using TMB, stage, and riskScore, resulting in a Nomogram score (Fig. 9C, Table S14). This Nomogram demonstrated high accuracy in predicting the 1-, 3-, and 5-year survival rates for HCC patients (Fig. 9D). ROC analysis of TMB, stage, riskScore, and the Nomogram further confirmed that the Nomogram exhibited the highest AUC value, highlighting its superior predictive performance when integrating clinical indicators and the riskScore (Fig. 9E).

Comprehensive model of riskScore and patient prognosis prediction. (A) Multivariate Cox regression analysis results combining age, gender, pathological stage, TMB and riskScore. (B) TCGA liver cancer samples were divided into four groups according to TMB and riskScore, and the Kaplan–Meier survival curve showed the survival between the groups. (C) Nomogram model based on pathological stage, TMB and riskScore for prognosis evaluation of liver cancer patients. (D) Calibration curve to evaluate the accuracy and specificity of the Nomogram model in 1-year, 3-year and 5-year survival prediction. (E) The AUC values of the Nomogram model, riskScore, Stage and TMB.
SLC16A3 and STRBP significantly enhance proliferation and invasion in HCC cells
Through the analysis of the expression of key model genes, SLC16A3 and STRBP, in HCC cell lines and normal liver cell lines, we observed that the protein expression levels of SLC16A3 and STRBP were significantly higher in HCC cell lines compared to the normal liver cell line MIHA (Fig. 10A). The HCC cell lines PLC/PRF/5 and MHCC97H, which exhibited the highest expression of these genes, were selected for further study. Si-RNA targeting each gene effectively reduced the protein expression of SLC16A3 and STRBP (Fig. 10B, C), demonstrating efficient gene knockdown. EdU assays showed that knockdown of SLC16A3 and STRBP markedly inhibited the proliferative capacity of PLC/PRF/5 and MHCC97H cells (Fig. 10D, E). Furthermore, invasion assays revealed that silencing SLC16A3 and STRBP decreased the invasive ability of PLC/PRF/5 and MHCC97H cells (Fig. 10F, G). These results suggest that the key genes SLC16A3 and STRBP play crucial roles in regulating the proliferation and invasion of HCC cells.

Expression of SLC16A3 and STRBP and their proliferation and invasion abilities. (A) Protein expression of SLC16A3 and STRBP in normal liver cell lines and tumor cell lines. (B) Protein expression of SLC16A3 in PLC/PRF/5 after knockdown of SLC16A3. (C) Protein expression of STRBP in MHCC97H after knockdown of STRBP. (D, E) The edu-488 levels in the nuclei of PLC/PRF/5 and MHCC97H cells after knockdown of SLC16A3 and STRBP. (F, G) The invasion of PLC/PRF/5 and MHCC97H cells after knockdown of SLC16A3 and STRBP.
Discussion
Although significant progress has been made in the diagnosis and treatment of HCC in recent years, the prognosis for HCC patients remains poor due to its complex biological features and inherent heterogeneity. As a result, there is an urgent need to delve deeper into the pathogenesis of HCC, identify novel prognostic biomarkers, and develop more effective, individualized therapeutic approaches.
In HCC, the role of neutrophils is primarily mediated through the infiltration of tumor-associated neutrophils (TANs), particularly the pro-tumorigenic N2 subtype. These N2 neutrophils promote tumor growth, metastasis, and immune evasion by secreting factors such as CXCL5 and TGF-β, which enhance cancer stem cell-like properties and mediate immunosuppression16,17,18,19,20. CD4⁺ T cells play a central immunoregulatory role within the tumor microenvironment. Subsets such as Th1, Th2, Th17, and regulatory T cells (Tregs) influence tumor progression through a delicate balance of cytokine signaling, with Tregs facilitating immune tolerance21. The functional exhaustion of both CD4⁺ and CD8⁺ T cells further contributes to immune tolerance and is closely associated with tumor progression22. B cells exhibit dual roles in HCC. Tumor-infiltrating B cells (TIBs), such as CD20⁺ cells, are associated with favorable prognosis in HBV-related HCC by activating T and NK cells to suppress tumor growth23,24. Conversely, regulatory B cells can promote tumor progression by suppressing CD8⁺ T cell function via cytokines such as IL-1025,26. Dysfunction of natural killer (NK) cells is typically characterized by an expansion of the CD56^bright subset alongside a reduction of the cytotoxic CD56^dim subset, with checkpoint molecules such as PD-1 and NKG2A impairing their anti-tumor surveillance capabilities27,28. Among macrophages, the M2-polarized tumor-associated macrophages (TAMs) drive malignant progression through immunosuppression, promotion of metastasis, angiogenesis, and drug resistance29,30,31,32, making TAMs an emerging target for therapeutic intervention33,34,35.
In our study, the constructed riskScore effectively stratified HCC patients based on neutrophil infiltration levels, with the high-risk group exhibiting elevated neutrophil infiltration. The score also distinguished between high- and low-risk groups based on CD4⁺ and CD8⁺ T cell infiltration, with the high-risk group showing features of T cell exclusion and poorer prognosis, indicative of T cell dysfunction. Furthermore, the riskScore was negatively correlated with CD56^bright NK cell infiltration. Although overall B cell infiltration was higher in the high-risk group, specific B cell subsets were not distinguished in this analysis. Additionally, the model could not capture the infiltration status of macrophages, highlighting the need for more refined predictive tools. In summary, the riskScore provides significant predictive value for assessing the infiltration and functional status of immune cells in HCC, offering a basis for patient prognosis evaluation.
HCC is characterized by a complex and immunosuppressive tumor microenvironment, accompanied by pronounced immune evasion7. In this study, we observed significantly elevated expression of immune checkpoint molecules PD-L1 (CD274), PD-1 (PDCD1), and CTLA-4 in the high-risk group, which carries multiple biological implications. Traditionally, high PD-L1 expression has been regarded as a hallmark of enhanced adaptive immune resistance in tumors. However, in conjunction with the high TIDE scores, pronounced T cell exclusion, and increased infiltration of myeloid-derived suppressor cells (MDSCs) observed in our analysis, the upregulation of PD-L1 in the high-risk group more likely reflects a state of global immune suppression within the tumor microenvironment. Recent studies support this notion, indicating that in HCC, PD-L1 is not only expressed by tumor cells but is also enriched in immunosuppressive myeloid populations such as M2 macrophages. These cells cooperate with MDSCs to shape a “cold tumor” phenotype that hinders effective T cell infiltration36. Additionally, the significantly lower stromal scores (StromalScore) observed in the high-risk group suggest another layer of complexity. A low stromal score indicates aberrant tumor stroma remodeling, which may impair the formation of tertiary lymphoid structures (TLSs)—structures that have been identified as key predictors of response to anti-PD-1 therapy37. Moreover, MDSCs contribute to T cell exhaustion and Treg expansion through immunosuppressive cytokines such as IL-10 and TGF-β. These mechanisms not only upregulate PD-L1 expression but also promote epithelial-mesenchymal transition (EMT), thereby enhancing tumor immune evasion and aggressiveness38. While high PD-L1 expression is often considered a potential biomarker of immunotherapy responsiveness, its co-occurrence with low stromal scores, high MDSC infiltration, and T cell exclusion in the high-risk group indicates a more complex immune landscape. Thus, our model is capable of identifying intricate immunological features within the HCC tumor microenvironment.
TP53 mutations in HCC are primarily associated with the regulation of the cell cycle and stress responses. Phosphorylation analyses have shown that TP53 mutations are linked to alterations in the phosphorylation of key proteins involved in the cell cycle, lipid metabolism, and protein synthesis, which may drive increased proliferation and higher histological grades in HCC cells39. The TP53 mutation rate in our high-risk group (> 45%) aligns with the TCGA-LIHC cohort, where TP53 mutations are present in 28% of patients40. Our findings reveal that the frequency of TP53 mutations is significantly higher in the high-risk group compared to the low-risk group, which is accompanied by poorer prognosis and a higher proportion of patients in stages II and III, consistent with previous studies. The role of CTNNB1 mutations in HCC mainly involves the activation of β-catenin, which promotes tumorigenesis, especially when collaborating with other oncogenes to induce liver tumor formation41. HCC tumors with CTNNB1 mutations are generally well-differentiated, low-proliferation tumors that exhibit lower malignancy, fewer genetic mutations, and are associated with cholestasis and metabolic abnormalities42. In our study, the low-risk group showed greater enrichment of metabolic signaling pathways, with a higher frequency of CTNNB1 mutations and a better prognosis. This is in line with previous findings that CTNNB1 mutations in HCC are linked to lower malignancy and metabolic dysfunction. FGFR3 contributes to HCC progression through its involvement with FGFR4 and KLB in regulating the FGF19 signaling pathway, thereby promoting tumor growth. Studies suggest that the loss of FGFR3 enhances the anticancer effects of FGFR4 inhibitors, while the redundancy between FGFR3 and FGFR4 limits the efficacy of single FGFR4 inhibitors. This indicates that the cooperative action of multiple FGFRs plays a critical role in the development and progression of FGF19-positive HCC43. In our study, the high-risk group exhibited higher enrichment scores for FGFR3-related genes, which was associated with a poorer prognosis, supporting previous research that FGFR3 promotes tumor growth in HCC.
Several drugs play pivotal roles in the chemotherapy and targeted therapy of HCC. 5-FU exerts its effect by disrupting DNA/RNA synthesis and inhibiting thymidylate synthase (TS). The combination of FOLFOX (5-FU + leucovorin + oxaliplatin) has demonstrated certain advantages in Asian populations, and the 2021 NCCN guidelines recommend it as a first-line treatment 44,45. Cisplatin works by inducing DNA cross-linking to inhibit tumor growth, with an overall response rate (ORR) ranging from 16–27% in combination therapies; however, it is associated with significant nephrotoxicity46. Gemcitabine, a pyrimidine metabolism inhibitor, specifically targets cells during the DNA synthesis phase. Although its efficacy as a monotherapy is limited, combining it with liposomal doxorubicin (PLD) or oxaliplatin improves the ORR (e.g., GEMOX regimen ORR 18%)47,48. When combined with PLD, the ORR reaches 25%, and its toxicity is manageable49. Among molecular targeted therapies, sorafenib, a multi-kinase inhibitor, shows a median overall survival (OS) of 10.7 months in monotherapy50, though its efficacy is lower in Asian populations (OS 6.5 months)51. Its combination with DOX or GEMOX does not significantly improve survival52,53. Lenvatinib, targeting VEGFR/FGFR, offers a comparable OS to sorafenib (13.6 months vs. 12.3 months), but at a more affordable cost49,54. Using the riskScore, we can predict the sensitivity of HCC patients to 5-fluorouracil, as well as to paclitaxel and vinorelbine for HCC treatment. Although paclitaxel and vinorelbine are not first-line chemotherapy agents for HCC, they can serve as adjunctive therapies for HCC patients.
Finally, through in vivo cellular experiments, we validated the key genes SLC16A3 and STRBP, which were found to be highly expressed in HCC cell lines. Reducing the expression of SLC16A3 and STRBP significantly inhibited HCC cell proliferation and invasion. This confirms the biological role of SLC16A3 and STRBP in promoting HCC cell proliferation and invasion.
To evaluate the relative performance of our model, we compared its riskScore with several established prognostic models for HCC. Using evaluation metrics such as C-index and AUC, we conducted comparative analyses on both the TCGA and GSE54236 datasets. In both cohorts, the 3-year ROC AUC of our riskScore exceeded 0.79, and the 3-year C-index was consistently above 0.75, demonstrating markedly superior performance compared to other models55,56,57. These findings indicate that our riskScore model offers enhanced discriminatory power and greater robustness in stratifying HCC patients. This model not only predicts prognosis but also reveals the molecular characteristics distinguishing high- and low-risk groups through functional enrichment analysis. The high-risk group is enriched in pathways related to the cell cycle, DNA repair, and KRAS signaling, whereas the low-risk group is predominantly associated with metabolic pathways and anti-tumor immune responses. Additionally, the riskScore can assess the infiltration levels of CD4 + , CD8 + T cells, B cells, and neutrophils, providing molecular evidence for personalized treatment strategies. The model also revealed significant differences in mutation frequencies and co-mutation patterns of key genes, such as TP53 and CTNNB1, between high- and low-risk groups. Sankey diagram analysis demonstrated a strong correlation between the high-risk group and advanced clinical stages (T3/T4, Stage III/IV) as well as poor immune therapy response, offering multi-dimensional evidence for precise subtyping. Furthermore, the chemotherapy drug sensitivity predictions based on riskScore further supported the biological rationale of the model. Lastly, phenotypic experiments in HCC cell lines validated the biological effects of key model genes, such as SLC16A3 and STRBP, confirming their in vitro role.
This study integrates multi-dimensional data, including genomics, transcriptomics, and MR analysis, to construct a more comprehensive and accurate prognostic risk model. Moreover, The clinical relevance of the riskScore model lies in its dual ability to predict prognosis and guide treatment strategies in HCC. By stratifying patients into high- and low-risk groups, the model identifies individuals who are more likely to benefit from specific therapeutic interventions. For chemotherapy, the model predicted that high-risk patients are more sensitive to agents such as 5-Fluorouracil and Paclitaxel, which may justify a more aggressive chemotherapy regimen in this subgroup. In contrast, low-risk patients may benefit from conservative strategies or targeted therapies. Regarding immunotherapy, high-risk patients exhibited elevated expression of immune checkpoints (PDCD1, CD274, CTLA4), and higher TIDE scores, suggesting potential resistance to immune checkpoint inhibitors. These findings underscore the utility of the model in identifying patients less likely to respond to monotherapy, and who may benefit from combination therapies or novel immunomodulators. Thus, the riskScore has the potential to support individualized treatment decisions and improve clinical outcomes. Through validation with large sample sizes and evaluation using multiple machine learning algorithms, the stability and reliability of the model were ensured. However, several limitations should be acknowledged. First, the model was primarily developed and internally validated using retrospective data from public databases, which may introduce selection bias and limit generalizability. Second, most datasets originated from single-center studies, lacking geographic and clinical diversity. Third, no prospective or real-world external cohort was available to confirm the clinical applicability of the model. To address this limitation, we plan to conduct prospective validation using clinical samples collected from multi-center cohorts in future studies. These will include transcriptomic and genomic profiling of HCC patients undergoing standardized treatment and follow-up. This approach will allow us to assess predictive accuracy, clinical utility, and generalizability of the model in actual applications.
Data availability
Data sources: TCGA data (https://portal.gdc.cancer.gov/); GSE54236 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE54236); IEU OpenGWAS project (https://gwas.mrcieu.ac.uk/); FinnGen R12 dataset (https://www.finngen.fi/fi); GWAS Catalog (https://www.ebi.ac.uk/gwas/studies/GCST90013702). Additional analysis data can be found in the supplementary tables.
References
Jemal, A. et al. Annual report to the Nation on the Status of Cancer, 1975–2014, featuring survival. J. Natl. Cancer Inst 109, (9) djx030 (2017).
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71 (3), 209–249 (2021).
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68 (6), 394–424 (2018).
Hou, Z. et al. Use of chemotherapy to treat hepatocellular carcinoma. Biosci. Trends. 16 (1), 31–45 (2022).
Llovet, J. M. et al. Immunotherapies for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 19 (3), 151–172 (2022).
Li, X. et al. The immunological and metabolic landscape in primary and metastatic liver cancer. Nat. Rev. Cancer. 21 (9), 541–557 (2021).
Donne, R. & Lujambio, A. The liver cancer immune microenvironment: Therapeutic implications for hepatocellular carcinoma. Hepatology 77 (5), 1773–1796 (2023).
Cao, J. et al. Low-density lipoprotein cholesterol and risk of hepatocellular carcinoma: A Mendelian randomization and mediation analysis. Lipids Health Dis. 22 (1), 110 (2023).
Yang, Y. et al. Association of glucose-lowering drug target and risk of Gastrointestinal cancer: A Mendelian randomization study. Cell. Biosci. 14 (1), 36 (2024).
Cao, J. et al. Causal association of sarcopenia with hepatocellular carcinoma risk in European population: A Mendelian randomization study. Front. Nutr. 11, 1292834 (2024).
Zhu, W. et al. Therapeutic targets for hepatocellular carcinoma identified using proteomics and Mendelian randomization. J. Gastroenterol. Hepatol. 40 (1), 282–293 (2025).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26 (17), 2190–2191 (2010).
Fu, J. et al. Large-scale public data reuse to model immunotherapy response and resistance. Genome Med. 12 (1), 21 (2020).
Maeser, D., Gruener, R. F. & Huang, R. S. OncoPredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief Bioinform 22 (6), bbab260 (2021).
Geeleher, P., Cox, N. & Huang, R. S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS One. 9 (9), e107468 (2014).
Song, M. et al. Cancer-associated fibroblast-mediated cellular crosstalk supports hepatocellular carcinoma progression. Hepatology 73 (5), 1717–1735 (2021).
Zhou, S-L. et al. A positive feedback loop between cancer stem-like cells and tumor-associated neutrophils controls hepatocellular carcinoma progression. Hepatology 70 (4), 1214–1230 (2019).
Zhou, S-L. et al. Overexpression of CXCL5 mediates neutrophil infiltration and indicates poor prognosis for hepatocellular carcinoma. Hepatology 56 (6), 2242–2254 (2012).
Haider, C. et al. Transforming growth Factor-β and Axl induce CXCL5 and neutrophil recruitment in hepatocellular carcinoma. Hepatology 69 (1), 222–236 (2019).
Zhou, S-L. et al. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology 150 (7), 1646 (2016).
McGeachy, M. J. & Cua, D. J. T cells doing it for themselves: TGF-beta regulation of Th1 and Th17 cells. Immunity 26 (5), 547–549 (2007).
Zheng, C. et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 169 (7) 1342, (2017).
Shi, J-Y. et al. Margin-infiltrating CD20(+) B cells display an atypical memory phenotype and correlate with favorable prognosis in hepatocellular carcinoma. Clin. Cancer Res. 19 (21), 5994–6005 (2013).
Garnelo, M. et al. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut 66 (2), 342–351 (2017).
Blair, P. A. et al. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity 32 (1), 129–140 (2010).
Xue, H. et al. Overrepresentation of IL-10-Expressing B cells suppresses cytotoxic CD4 + T cell activity in HBV-Induced hepatocellular carcinoma. PLoS One. 11 (5), e0154815 (2016).
Chiossone, L., Dumas, P-Y., Vienne, M. & Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 18 (11), 671–688 (2018).
Liu, Y. et al. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene 36 (44), 6143–6153 (2017).
Krenkel, O. & Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 17 (5), 306–321 (2017).
Huang, Y. et al. The role of tumor associated macrophages in hepatocellular carcinoma. J. Cancer. 12 (5), 1284–1294 (2021).
Zhang, Q-B. et al. High-mobility group protein box1 expression correlates with peritumoral macrophage infiltration and unfavorable prognosis in patients with hepatocellular carcinoma and cirrhosis. BMC Cancer. 16 (1), 880 (2016).
Dong, P. et al. CD86⁺/CD206⁺, diametrically polarized tumor-associated macrophages, predict hepatocellular carcinoma patient prognosis. Int. J. Mol. Sci. 17 (3), 320 (2016).
Li, X. et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut 66 (1), 157–167 (2017).
Zhu, Y. et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 Blockade. Gut 68 (9), 1653–1666 (2019).
Meng, Y-M. et al. Monocytes/macrophages promote vascular CXCR4 expression via the ERK pathway in hepatocellular carcinoma. Oncoimmunology 7 (3), e1408745 (2018).
Lemaitre, L. et al. Spatial analysis reveals targetable macrophage-mediated mechanisms of immune evasion in hepatocellular carcinoma minimal residual disease. Nat. Cancer. 5 (10), 1534–1556 (2024).
Huang, Y. et al. Single-nucleus and spatial transcriptome profiling delineates the multicellular ecosystem in hepatocellular carcinoma after hepatic arterial infusion chemotherapy. Adv. Sci. (Weinh). 12 (5), e2405749 (2025).
Liu, X., Kang, X., Kang, H. & Yan, H. The immunosuppressive role of MDSCs in HCC: Mechanisms and therapeutic opportunities. Cell. Commun. Signal. 23 (1), 155 (2025).
Ng, C. K. Y. et al. Integrative proteogenomic characterization of hepatocellular carcinoma across etiologies and stages. Nat. Commun. 13 (1), 2436 (2022).
Pulakuntla, S., Syed, K. & Reddy, V. D. Analysis of somatic mutations in the TCGA-LIHC whole exome sequence to identify the neoantigen for immunotherapy in hepatocellular carcinoma. Curr. Issues Mol. Biol. 46 (1), 106–120 (2023).
Molina-Sánchez, P. et al. Cooperation between distinct Cancer driver genes underlies intertumor heterogeneity in hepatocellular carcinoma. Gastroenterology 159 (6), 2203 (2020).
Liang, B. et al. TBX3 functions as a tumor suppressor downstream of activated CTNNB1 mutants during hepatocarcinogenesis. J. Hepatol. 75 (1), 120–131 (2021).
Tao, Z., Cui, Y., Xu, X. & Han, T. FGFR redundancy limits the efficacy of FGFR4-selective inhibitors in hepatocellular carcinoma. Proc. Natl. Acad. Sci. U S A. 119 (40), e2208844119 (2022).
Shah, M. H. et al. Neuroendocrine and adrenal tumors, version 2.2021, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Canc Netw. 19 (7), 839–868 (2021).
Qin, S. et al. Randomized, multicenter, open-label study of oxaliplatin plus fluorouracil/leucovorin versus doxorubicin as palliative chemotherapy in patients with advanced hepatocellular carcinoma from Asia. J. Clin. Oncol. 31 (28), 3501–3508 (2013).
Lee, J. O. et al. Combination chemotherapy with capecitabine and cisplatin for patients with metastatic hepatocellular carcinoma. Ann. Oncol. 20 (8), 1402–1407 (2009).
Lombardi, G. et al. Pegylated liposomal doxorubicin and gemcitabine in patients with advanced hepatocellular carcinoma: Results of a phase 2 study. Cancer 117 (1), 125–133 (2011).
Louafi, S. et al. Gemcitabine plus oxaliplatin (GEMOX) in patients with advanced hepatocellular carcinoma (HCC): Results of a phase II study. Cancer 109 (7), 1384–1390 (2007).
Kim, J. J., McFarlane, T., Tully, S. & Wong, W. W. L. Lenvatinib versus Sorafenib as First-Line treatment of unresectable hepatocellular carcinoma: A Cost-Utility analysis. Oncologist 25 (3), e512–e519 (2020).
Llovet, J. M. et al. Sorafenib in advanced hepatocellular carcinoma. N Engl. J. Med. 359 (4), 378–390 (2008).
Cheng, A-L. et al. Efficacy and safety of Sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 10 (1), 25–34 (2009).
Lee, S. et al. Sorafenib versus cytotoxic chemotherapy for patients with advanced hepatocellular carcinoma: A retrospective, single-institution study. Invest. New. Drugs. 30 (3), 1150–1157 (2012).
Assenat, E. et al. Sorafenib alone vs. sorafenib plus GEMOX as 1st-line treatment for advanced HCC: The phase II randomised PRODIGE 10 trial. Br. J. Cancer. 120 (9), 896–902 (2019).
Kudo, M. et al. Lenvatinib versus Sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 391 (10126), 1163–1173 (2018).
Wu, J. et al. A cuproptosis-related LncRNA signature for predicting prognosis and immune response in hepatocellular carcinoma. Heliyon 9 (9), e19352 (2023).
Cao, H., Huang, P., Qiu, J., Gong, X. & Cao, H. Immune landscape of hepatocellular carcinoma tumor microenvironment identifies a prognostic relevant model. Heliyon 10 (3), e24861 (2024).
Cao, L. et al. Constructing an immune-related prognostic signature for predicting prognosis and immune response in hepatocellular carcinoma. Heliyon 10 (13), e34012 (2024).
Acknowledgements
We acknowledge the TCGA, GEO, and FinnGen databases and their contributors.
Funding
This work was supported by grants from the WU JIEPING MEDICAL FOUNDATION (320.6750.2022–19-37).
Author information
Authors and Affiliations
Contributions
Conceptualization, QK; methodology, QK and ZW; investigation, QK, ZW, WZ, ZL; writing—original drafts, QK and ZW; writing—review & editing, XZ and SQ; funding acquisition, QM and XZ; supervision, XZ.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
All authors informed and consent.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kou, Q., Wu, Z., Zhao, W. et al. Multi-omics analysis identifies SNP-associated immune-related signatures by integrating Mendelian randomization and machine learning in hepatocellular carcinoma. Sci Rep 15, 23930 (2025). https://doi.org/10.1038/s41598-025-09010-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-09010-1
Keywords
This article is cited by
-
A demethylation-driven gene signature predicts prognosis and therapeutic vulnerability in hepatocellular carcinoma
Scientific Reports (2026)
-
Young-onset hepatocellular carcinoma following double outlet right ventricle surgery without liver congestion: a case report with genetic analysis
Clinical Journal of Gastroenterology (2026)
