
Abstract
The inherent tradeoff between gravimetric and volumetric hydrogen storage capacities in metal-organic frameworks (MOFs) limits their commercial viability. While benchmarked MOFs like MOF-5, IRMOF-20, and PCN-610 perform well at 77 K, maintaining their performance at elevated temperatures (298 K) remains challenging. To address this, a multi-objective particle swarm optimization framework was developed to identify promising MOFs for hydrogen storage. The optimization was guided by predictions from the bootstrapped-random forest trees. This optimization yielded 152 theoretical MOF feature combinations, which were matched with 733,792 existing structures. A nearest neighbor search identified 43 promising MOFs, with Zn-based MOF-2087 emerging as the global best, exhibiting consistent hydrogen storage performance across temperatures. Grand Canonical Monte Carlo simulations confirmed its high hydrogen uptake (5.3 wt% and 7.4 gH2 L− 1 at 298 K). Molecular dynamics simulations further revealed C-clusters and metal sites as key adsorption centers, supporting the enhanced hydrogen storage behavior of MOF-2087. These findings highlight MOF-2087 as a computationally promising MOF for hydrogen storage up to 298 K and demonstrate the effectiveness of the optimization-driven screening strategy.
Similar content being viewed by others

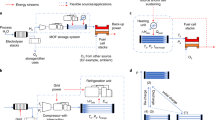
Introduction
Designing an efficient and reliable hydrogen storage system is crucial for sustainable transportation. Unlike commercially available high-pressure Type-IV cylinders that store hydrogen at 700 bar, metal-organic frameworks (MOFs) offer an effective alternative for storing hydrogen at low pressures due to their exceptional surface areas, tunable properties, and reversible gas storage properties1,2,3,4,5. Several factors influence hydrogen storage in MOFs, including storage temperature, pressure, pore geometry, crystal density, heat of adsorption6,7,8,9. Identifying commercially viable MOFs with the desired features remains a challenge. Computational screening methods like high-throughput density functional theory10,11,12,13 and high-throughput grand canonical Monte Carlo (HT-GCMC) simulations14,15,16,17 have been widely used to address this complexity. Due to their computational cost, machine learning (ML) approaches12,16,17,18,19,20,21,22 have emerged as an efficient alternatives for predicting MOF performance. Supplementary Table 1 summarizes recent ML models applied in this domain. These models can rapidly evaluate the hydrogen storage performance of MOF candidates. However, they require classical benchmark structures like MOF-5 to compare performance and identify best performing candidates.
Ahamed et al. compared the hydrogen storage performance of MOFs from various databases with MOF-5 at 77 K and identified IRMOF-20 and PCN-610 as the best-performing MOFs23,24. Table 1 summarizes the gravimetric and volumetric hydrogen uptake of benchmarked MOFs reported in the literature. However, they fall short of the U.S. Department of Energy’s (DOE) recommended target (6.5 wt% and 50 gH2 L− 1)25 for the operational temperature range of −40–60 °C. To address this limitation, researchers have proposed selecting MOFs based on their physical parameters as an effective strategy6,21,23,24,26. Prior studies have shown that hydrogen storage performance in MOFs depends on the combination of crystallographic features, including single crystal density, gravimetric and volumetric surface areas, pore volume, pore diameter, cavity diameters, and void fractions21. While hydrogen adsorption studies have traditionally focused on gravimetric performance, works by Goldsmith et al.26 and Ahamed et al.24 have emphasized the need to concurrently consider volumetric uptake at 77 K—keeping the fact that the materials like MOF-5 and HKUST-127 meet the gravimetric targets but lacks in sufficient volumetric capacity.
Recognizing the limitations of focusing on a single performance metric, Gómez-Gualdrón et al.28 and Chen et al.29 stressed that both gravimetric and volumetric capacities must be concurrently balanced to satisfy the volume and weight constraints of onboard hydrogen storage. However, achieving this balance remains a challenge at higher temperatures. For instance, Chen et al.29 synthesized hypothetical MOFs based on the NU-1500 framework and identified NU-1501-Al as a relatively balanced structure, but it delivered only 2.5 wt% at 298 K. This performance drop can be attributed to the fact that existing studies have largely relied on maximizing the crystallographic features or altering topologies to enhance hydrogen storage30. However, they did not establish the specific combinations of crystallographic features required for effective storage. Quantitative benchmarks, such as the optimal range of surface area, pore volume and crystal density were also not defined to leverage gravimetric and volumetric hydrogen storage. Without these defined targets, identifying or designing MOFs that can satisfy DOE requirements under real-world conditions remains a challenge.
To overcome the limitations of crystallographic feature-driven studies, researchers have adopted optimization-based approaches to identify the best performing MOFs31. For example, Ghude et al.32 used Bayesian optimization and evolutionary particle swarm optimization to screen over 98,000 MOF structures, aiming to identify optimal candidates for hydrogen storage under temperature-pressure swing conditions (100 bar/77 K to 5 bar/160 K).combinations. Singh et al.33 used optimization algorithms focused on linker functional groups and identified IRMOF-10 as a best candidate (3.9 wt% and 23 gH2 L− 1 at 77 K). However, these studies primarily relied on single-objective optimization approaches (see Supplementary Table 2) and thus, optimal combinations of crystallographic features capable of simultaneously maximizing both gravimetric and volumetric hydrogen storage capacities remain unexplored.
Thus, a multi-objective optimization-based screening framework is proposed in this work to identify the MOFs having the optimal crystallographic features for achieving the desired trade-off between gravimetric and volumetric capacities (Fig. 1). A bootstrap aggregate random forest tree (B-RFT) technique embedded with multi-objective particle swarm optimization (MOPSO)39,40,41 was used to screen the MOFs from nearly a million MOF structures reported in various databases. Among those, 152 feasible combinations of MOF features were identified that represent optimal trade-offs between hydrogen storage densities. Using the Nearest Neighbor Search (NNS) algorithm, MOF-2087 was identified as the top-best performing MOF based on optimized crystallographic feature similarity. Hereafter the term top-best performing candidate is referred as ‘global best MOF’ for ease of reference. GCMC simulations confirmed that MOF-2087 demonstrates the desired trade-off between gravimetric and volumetric hydrogen storage capacities, surpassing the classical benchmark MOFs at 298 K. Furthermore, molecular dynamics (MD) simulations indicate that C_2, C_R, and Zn atoms in MOF-2087 are the critical sites for hydrogen adsorption and are responsible for achieving a better tradeoff between gravimetric and volumetric storage capacities. Although feature optimization was performed at 77 K due to the scarcity of reliable adsorption data at high temperatures, the optimum features are structural and remains stable up to 298 K. GCMC simulations across 77–298 K showed consistent hydrogen storage performance for MOF-2087, and MD simulations confirmed its structural stability. This supports that optimization at 77 K can reliably identify MOFs performing well at room temperature. Therefore, our results propose MOF-2087 as the new benchmark material for hydrogen storage applications.

Steps involved in the B-RFT-MOPSO-based deliverable gravimetric and volumetric hydrogen storage optimization of MOFs. (1) Before all else, the realm of 925,229 MOF structures was collected from HyMARC. (2) Formation and parameter tuning of B-RFT framework to predict the hydrogen storage densities in MOFs, later the multi-objective framework has been designed by embedding the B-RFT framework with MOPSO. (3) The NNS was applied to match the obtained Pareto-optimal solutions with the existing structures. Further, TOPSIS identifies the global best one. (4) The hydrogen storage performance and features of the global best MOF will be demonstrated by using GCMC studies. (5) The hydrogen storage mechanism of the global best MOF will be evaluated by using MD simulations.
Results
To implement the proposed optimization-based screening strategy, the workflow began with collecting 733,792 open-source MOFs from the HyMARC data hub (Fig. 1). This resource complies crystallographic information from 19 databases worldwide (see Methodology Sect. "Data mining and screening"). This collection is hereafter referred to as ‘global database’. Using this dataset, the B-RFT—model was trained to predict gravimetric and volumetric hydrogen storage capacities based on crystallographic features. Following hyperparameter tuning and validation, the model was integrated into MOPSO framework to identify optimal design features. The global best MOF was further evaluated through GCMC and MD simulations. The subsequent sections detail the model development, optimization process, and performance evaluation.
Hyperparameter tuning of B-RFT framework
The bootstrap-random forest tree (B-RFT) model combines bootstrap bagging with decision-tree ensembles, where each tree is trained on a random subset of the data. Bagging inherently provides an unbiased internal cross-validation via Out-Of-Bag (OOB) samples, allowing direct estimation of prediction error without a separate hold-out set. An OOB error-based sensitivity was performed to optimize the number of trees (nTrees) and number of leaves per tree (nLeaves) on a representative training subset. Although OOB cross-validation sufficed, a separate validation using 20% of the dataset was maintained throughout to confirm the model’s predictive performance. The nTrees were varied from 1 to 500 to balance computational efficiency with prediction accuracy (Supplementary Fig. 1). For gravimetric capacity; the OOB mean square error stabilized at 0.037 after 30 trees, whereas for volumetric capacity, the minimum error of 1.97 was observed at 48 trees, leading to the choice of nTrees as 50. The higher OOB error in volumetric capacity prediction is likely due to the marginal contributions of pore volume, large cavity and pore limiting diameters of MOFs (Supplementary Fig. 2). With nTrees fixed at 50, varying the nLeaves between 1 and 100 showed that the OOB error reached a minimum of 0.033 for gravimetric and 1.97 for volumetric capacities with five leaves. Increasing the nLeaves beyond five added model complexity and increased computation time without significantly improving the prediction accuracy42,43. Therefore, the final configuration of nTrees = 50 and nLeaves = 5 was selected considering the balance between computational efficiency and prediction accuracy.
Dataset size variation
Beyond nLeaves and nTrees, the quality and quantity of the training dataset significantly affect the prediction accuracy of the B-RFT model21,44. However, determining the exact amount of data required to train a B-RFT model for hydrogen storage prediction with a coefficient of determination (R2) > 0.95 remains unclear. To address this, a sensitivity analysis was performed by varying the dataset size from 5 to 80% (up to 78,956 MOFs), with the model tested on 9870 MOFs. Figure 2A and B illustrates the prediction performance of the B-RFT framework for gravimetric hydrogen storage. As shown in Fig. 2C and D, when less than 45% of the dataset was used, the performance metric R2 was substantially lower, while the average unsigned error (AUE), root mean square error (RMSE), and median average error (MAE) were notably higher, especially for volumetric capacity predictions. This decrease in accuracy is likely due to crystallographic inaccuracies and the inclusion of MOFs with zero surface area in the training dataset21 (Supplementary Fig. 2). Increasing the size of the training dataset beyond 45% consistently improved the AUE and R2 metrics. The best balance between computational cost and prediction accuracy was achieved with 75% of the dataset (74,022 MOFs) and was selected for training the optimal B-RFT model. This optimized B-RFT model was subsequently used as the objective function in the MOPSO framework (Fig. 1). Table 2 summarizes the predictive performance of the optimized B-RFT model.

The performance assessment of the B-RFT framework concerning the training dataset size is tested by using 9870 MOFs uniformly for all training dataset combinations. (A) and (C) variation of R2 (navy blue) and AUE (green) concerning the training dataset size for both the UG and UV uptakes. (B) and (D) RMSE (orange) and MAE analysis (navy blue) of the trained B-RFT framework concerning the training dataset size utilized for UG and UV uptakes.
Trends and correlation between variables
Figure 3 presents an analysis of the crystallographic feature trends for predicted hydrogen storage capacities across MOFs in the global database, using the best-performing B-RFT model. The analysis reveals that an increase in gravimetric surface area, pore volume, void fraction, pore limiting diameter, and large cavity diameter enhances volumetric and gravimetric hydrogen uptakes. In contrast, single crystal density and volumetric surface area correlate negatively with both capacities, suggesting that minimizing these features leads to higher hydrogen storage densities. These trends provide a base for defining bounds for crystallographic features utilized in the B-RFT-MOPSO optimization process.
Constraints for hydrogen storage optimization in MOFs
The global MOF database is composed predominantly of hypothetical MOFS (hMOFs) − 97.3% of 733,792 - characterized by low single crystal densities (< 0.3 g cm− 3), high surface areas (> 5300 m2 g− 1), and pore volume exceeding 3.3 cm3 g− 1. In contrast, experimental MOFs (eMOFs) tend to have void fractions below 0.87, with only a few exceeding this threshold (refer to Fig. 3). When considering feature combinations such as density < 0.3 g cm− 3, surface area > 5300 m2 g− 1, pore volume > 3.3 cm3 g− 1, and void fraction ≥ 0.87, no MOFs in the global database satisfy all simultaneously. Although Fig. 3G, H suggests that the optimal void fraction range lies between 0.87 and 0.92, only MOFs within the narrower range of 0.89–0.91 meet all three other criteria concurrently. This observation is consistent with recent computational and experimental studies29,31. Therefore, the void fraction range in this work has been refined to 0.89–0.91 for effective screening.
To eliminate non-viable solutions for MOF crystallographic features, specific parametric constraints were incorporated into the B-RFT-MOPSO framework (Supplementary Note 3). Based on the feature trend patterns, it is evident that MOFs with low densities and high surface areas are particularly conducive to achieving better gravimetric and volumetric densities, especially under pressure swing conditions (100 bar − 5 bar). Considering the availability of MOFs and feature correlations, maximum and minimum bounds for B-RFT-MOPSO have been established, as detailed in Supplementary Table 3. Supplementary Fig. 3 provides a two-dimensional visualization of the search domain designed to identify optimal MOFs with enhanced hydrogen storage capacities.

Correlation and trends of seven crystallographic features of MOFs concerning UG and UV.
Variation of deliverable capacities of global database concerning the single crystal density, surface areas, void fraction, accessible pore volume, cavity diameters, and pore limiting diameters of MOFs assuming the pressure swing conditions (100-5 bar) at 77 K. (A, C, E, G, I, K, M) represents the UGs of MOFs and (B, D, F, H, J, L, N) represents the UVs of MOFs.
Performance evaluation of B-RFT-MOPSO
A series of performance assessment indicators were utilized to evaluate the diversity and convergence of the B-RFT-MOPSO optimization framework (Supplementary Method 3). As the true Pareto front of the multi-objective hydrogen uptake problem is unknown, a reference Pareto front was constructed using 152 feasible points obtained from 15 independent runs of B-RFT-MOPSO, serving as a baseline for evaluation (Supplementary Fig. 4, red markers). Generally, the size of the obtained Pareto set reflects the ability of the B-RFT-MOPSO to identify feasible non-dominated solutions. On average, approximately 47 feasible non-dominated solutions were generated per run, consistent with optimization models reported in the literature45 (Supplementary Fig. 5A).
Diversity in the obtained Pareto-optimal solutions was evaluated using the standard spacing metric (SSM) indicator, where a lower SSM value signifies better diversity in the predicted non-dominated solutions. The mean SSM value of 0.0002 indicates that the B-RFT-MOPSO consistently achieves well-diverged non-dominated solutions across independent runs (Supplementary Fig. 5B). The range of SSM values observed aligns with the existing studies46.
The quality of the Pareto points generated by the B-RFT-MOPSO was assessed by their proximity to the reference or true solution sets, measured by the standard generational distance (SGD) indicator (Supplementary Method 3.3). A smaller SGD value indicates that the non-dominated solutions obtained in each run are closer to the reference Pareto points. The mean SGD value of 0.0235 for the B-RFT-MOPSO framework demonstrates its ability to effectively identify MOF features that enhance hydrogen storage (refer to Supplementary Fig. 5C).
Additionally, the search domain of the B-RFT-MOPSO in the global search space is significantly influenced by the convergence rate of the framework. Forcing a multi-objective model to achieve rapid convergence can limit the search space exploration. Therefore, the total number of framework evaluations required to reach the desired Pareto points was used to assess the convergence rate of the designed multi-objective framework47. On average, 109,437 simulations were performed in each independent B-RFT-MOPSO run to obtain feasible non-dominated solutions.
Feasible crystallographic features of MOFs and their usable hydrogen storage capacities
Gravimetric hydrogen storage in MOFs
The B-RFT-MOPSO optimization framework identified 152 non-dominated crystallographic features of MOFs (Supplementary Fig. 4–navy blue) that exhibit a feasible tradeoff between hydrogen storage capacities under pressure swing conditions at 77 K. The resulting non-dominated solution set indicates that reducing single-crystal density (0.24 g cm− 3 to 0.10 g cm− 3) and volumetric surface area (1800 m2 cm− 3 to 500 m2 cm− 3) enhances gravimetric hydrogen uptake in MOFs (Supplementary Fig. 6A and B). In contrast, maximizing gravimetric surface area (~ 7200 m2 g− 1) results in improved hydrogen storage, aligning with Chahine’s rule29 (Supplementary Fig. 6C). Void fractions from 0.905 to 0.91 also favor gravimetric hydrogen storage. Additionally, an increase in accessible pore volume correlates with enhanced gravimetric uptake. The highest gravimetric hydrogen storage was observed in MOFs with large cavity diameters between 16 and 18 Å and pore-limiting diameters between 14 and 18 Å (Supplementary Fig. 6F and G).
Volumetric hydrogen storage in MOFs
In contrast to gravimetric hydrogen storage, an increase in single-crystal density enhances volumetric hydrogen storage (Supplementary Fig. 6A). A similar trend is observed for MOFs’ volumetric and gravimetric surface areas. Void fractions in the range of 0.905 to 0.91 result in higher volumetric hydrogen storage, with a maximum of approximately 38 gH2 L− 1 (as the dataset is capped below 40 gH2 L− 1)21 for large cavity and pore-limiting diameters between 16 and 18 Å, and 14 Å and 18 Å, respectively.
The identified non-dominated solutions represent feasible combinations of crucial crystallographic features in MOFs and serve as theoretical benchmarks for optimizing hydrogen storage tradeoffs. However, it is challenging to computationally construct or reconstruct new MOF structures based on these crystallographic features. To overcome this, existing MOFs from the global database were matched with the optimal features using the nearest neighbor search (NNS) algorithm. If a MOF’s features closely align with the optimal solution, it is identified as a locally best MOF and stored in a local repository.
Local best MOFs
Applying the nearest neighbor search (NNS)48,49 to the global database of 733,792 MOFs, the crystallographic features of each MOF were compared to the query points (optimal solutions found from B-RFT-MOPSO) to identify the local best MOFs. The degree of similarity between the features of each MOF and query points was determined using a customized 7-dimensional Euclidean distance (ΔEx) metric. MOFs with the smallest ΔEx for a specific query point in the global repository were identified as the local best MOF and transferred to the local-best repository. In total, 43 out of 733,792 MOFs were identified as the closest match to the 152 Pareto optimal solutions. These identified local best MOFs are illustrated in Supplementary Fig. 4. Notably, all the screened local best MOFs were hypothetical.
Among the various MOF databases, the TobaCCo and UO databases demonstrated the most feasible tradeoffs between hydrogen storage densities under pressure swing conditions at 77 K. However, 10 MOFs were identified as significantly distant from their respective query points. To refine the selection, a local domain was implemented to narrow down further MOFs that were sufficiently close to the optimal solutions (Supplementary Fig. 4). Figure 4 presents the shortlisted local best MOFs and the corresponding Pareto optimal solutions obtained through the B-RFT-MOPSO framework.

Pareto optimal solutions obtained from B-RFT-MOPSO. The orange-coloured points indicate the shortlisted local best MOFs and their respective closeness to the Pareto solutions. The greenish point represents the global best MOF feature identified by the Technique for Order of Preference by Similarity to the Ideal Solution (TOPSIS).
Global best MOF
Each crystallographic feature combination derived from the B-RFT-MOPSO framework represents a unique tradeoff between gravimetric and volumetric hydrogen uptake. Thus, identifying the single best solution that demonstrates a feasible trade-off between hydrogen storage capacities in MOFs is essential. To determine the global best solution, the TOPSIS (Technique for Order of Preference by Similarity to Ideal Solution) multi-criteria decision-making method was employed50 (Supplementary Method 3.4). Supplementary Table 4 presents the top 5 hMOFs ranked by the closeness index. The feature ranked first exhibits the most feasible tradeoff between hydrogen storage capacities, making it an optimal solution suitable for inclusion in the decision-maker’s repository.
Using the identified global best solution as a benchmark for the best-performing MOF, the NNS identified MOF-2087 from the ToBaCCo database22 as the optimal MOF with the minimal ΔEx value of 17.8, surpassing the other MOFs in the global database. Table 3 provides the crystallographic features of MOF-2087, along with the single-objective dominant solutions and their respective hydrogen storage capacities. The MOF from the UO database20 exhibits high volumetric storage of 37.85 gH2 L− 1 (Str_m3_o7_o19_f0_nbo.sym.22.out, a volumetric dominant solution) with a moderate gravimetric capacity of 12.2 wt% under pressure conditions at 77 K. Conversely, MOF-12358 from the ToBaCCo database was identified as the gravimetric storage-dominant solution, achieving 21.86 wt% and 31.84 gH2 L− 1 among the local best MOFs. As listed in Table 3, the crystallographic features of MOF-2087 align with the theoretical benchmark features.
Hydrogen storage performance of global best MOF: MOF-2087
Structural descriptions of MOF-2087
MOF-2087 framework consists of a zinc (II) metal cluster bonded to the nitrogen atom in the pyridine ring of the organic ligand (3-ethynyl-7-(pyridine-4yl) dibenzofuran), forming a 3D structure with a dia net topology (4/6/c1; sqc6), as shown in Fig. 5. The ethynyl terminals of the four ligand units under cyclo-addition, resulting in the formation of an 8-membered C-cluster (bicycle-[2,2,2]-octane) in a tetrahedral arrangement (Fig. 5C). Pore size distribution analysis of MOF-2087 reveals two adjacent peaks at 23.5 Å and 23.9 Å, indicating the mesoporous nature of the material (Supplementary Fig. 7). Furthermore, the crystallographic information of MOF-2087 can be found in the Supplementary document.

The pictorial representation of MOF-2087. (A) 2 × 2 × 2 supercell of MOF-2087 facing towards the hexagonal pore geometry. (B) Organicligand. (C) 8 membered C-cluster. (D) hexagonal pore geometry. C-grey, H-white, N-blue, O-red, and Zn-violet.
Hydrogen storage mechanism
Preferential sites of hydrogen interaction
The hydrogen density distribution and radial distribution functions (RDF) at 77 K and 5 bars were calculated to determine the preferential hydrogen adsorption sites in MOF-2087 (Fig. 6). The strongest hydrogen interactions were observed with C atoms at 3.8 Å, followed by O at 4.0 Å, and H at 4.04 Å. Additionally, N–H2 interactions peaked at 4.8 and Zn–H2 interactions were observed at 5.31 Å. These results suggest that organic elements such as C, O, and H exhibit stronger interactions with H2 molecules than the Zn metal center. The observed results were consistent with the results from literature51,52.
The supercell (2 × 2 × 2) of MOF-2087 was meshed into 500 × 500 × 500 grids to compute Gaussian and normalized H2 density distributions, visualized using ParaView®. Figure 7A, B and C represents the face and axillary projections of MOF-2087 before adsorption, while Fig. 7D, E and F illustrates MOF-2087 after hydrogen adsorption at 50 bar and 77 K. Correlating the RDF with the mass center probability, significant hydrogen interactions were observed around the 8-membered C-clusters with tetrahedrally coordinated dibenzofuran rings, forming an irregular, diagonally stretched square-shaped adsorption site (Fig. 6A, labeled as Site I). Additional interactions were identified at the Zn sites (Fig. 6B and C, labeled as Site II) and around the dibenzofuran rings (labeled as Site III). Hydrogen density mapping reveals comprehensive H2 molecule distribution within the hexagonal pore, spanning the (011) and (110) crystallographic planes (Fig. 7E and F, and Supplementary Fig. 8). This distribution highlights trapping of hydrogen within the pores surrounded by dibenzofuran rings, Zn sites, and 8-membered C-clusters, contributing to a high hydrogen uptake of 22.3 wt% and ~ 38 gH2 L− 1 at 77 K and 100 bar. These findings indicate that hydrogen adsorption sites in MOF-2087 are primarily located in the organic ligands (Site I and III), rather than the metal site (Site II)51,52.
Simulated annealing
To further understand the hydrogen storage mechanism of MOF-2087 and validate the GCMC findings, classical MD simulations were conducted. C atoms in the ligand were classified as C_2 (from Site I and III), C_3 (from Site I), and C_R (from Site III), based on their geometry and force field classifications53 (Fig. 8). Preliminary GCMC simulations revealed that a MOF-2087 supercell can adsorb approximately 188 H2 molecules at 77 K and 1 bar; therefore, 188 H2 molecules were introduced into the simulation cell for the MD simulations54. Figure 9 displays the annealed structure of the MOF-2087-H2 system within the NVT ensemble. The thermal motion and equilibrium zones of H2 were characterized by measuring the peak RDF distances between host atoms and H2 (Table 4). These distances indicate that, as the system cools, H2 molecules tend to move closer to specific atoms, suggesting favorable adsorption interactions. This behavior is likely due to reduced thermal motion at lower temperatures, allowing H2 molecules to settle into lower-energy adsorption sites within the framework55.
Table 4 shows a significant decrease in the average distance between C_2 and H2 from 5.9 Å at 300 K to 3.6 Å at 14 K, indicating that these atoms become favorable adsorption sites, likely due to π-π interactions resulting from resonance in the rings. Similarly, the distance between C_R and H2 decreases from 6.1 to 3.6 Å, reflecting stronger π-π stacking interactions. As the temperature increases, the distance between Zn and H2 increases. However, as the system cools, this distance shortens from 8.3 to 5.0 Å. This suggests that while Zn centers are not the main adsorption sites, they may still participate in adsorption at lower temperature or under high pressure, possibly due to polarization effects. O atoms also exhibit strong interactions, with the distance between O and H2 decreasing from 6.2 to 4.2 Å, indicating an affinity for H2 molecules at lower temperatures. Additionally, the distance between host H atoms and H2 decreases from 6.8 to 3.9 Å, indicating preferential adsorption. These H atoms belonging to the pyridine and furan moieties54are accounted for alongside the C_R atoms throughout the study. Overall, the annealed structure demonstrates that C_R and C_2 carbons are the most favorable adsorption sites for H2, primarily due to the electronegativity of the sp2 hybridized carbon atoms, followed by O atoms. These findings support the preliminary adsorption sites identified in the GCMC simulations (Fig. 6).

Mass center probability of hydrogen distribution in MOF-2087 (red-x axis, green-y axis, and blue-z axis) (A, B, and C) respectively. (D) Represents the radial distribution function of gaseous hydrogen concerning the radial distances of the elements presented in MOF-2087, evaluated at 77 K and 5 bar.

Pictorial representation of the empty MOF-2087 supercell (A, B, and C) and hydrogen density distribution of the MOF-2087 under 77 K and 5 bar (D, E, and F) seen through the respective crystallographic planes (red-x axis, green-y axis and blue-z axis).

Pictorial representation of classified carbon atoms according to the DERIDING force field.

Annealed structure of MOF-2087 at different projections (A and B). The H2 molecules found in the first interaction zone (g(r)) are mapped in orange, magenta, green, yellow, and red for O, C_2, C_R, N, and Zn, respectively. For visualization purposes, the H2 molecules are scaled larger than their actual size.
H2 diffusion in MOF-2087
Mean square displacement (MSD) statistics were collected from equilibrated conditions throughout 2000 ps. The calculated MSD for all temperatures considered (77 K, 160 K, 200 K, and 298 K) exhibits a linear trend, making it suitable for computing H2 diffusivities within the MOF, as shown in Fig. 10. The diffusion characteristics of H2 inside MOF-2087 demonstrate a clear trend of increasing diffusion rates with rising temperature, from 3.78 × 10− 8 m2 s− 1 at 77 K to 2.92 × 10− 7 m2 s− 1 at 298 K. This trend indicates enhanced H2 mobility with increasing thermal energy, enabling H2 molecules to overcome potential energy barriers within the MOF and explore the structure more freely56.
This behavior aligns with the simulated annealing results, where reduced thermal energy at lower temperatures leads to minimal diffusion rates, allowing H2 molecules to access and occupy favorable adsorption sites. As illustrated in Fig. 10B, the diffusion coefficients of MOF-2087 exhibit Arrhenius behavior (R2 = 0.98) across the studied temperature range. The calculated activation energy of 0.017 eV is lower than that of MOFs without open metal sites, such as Al-soc-MOF-1d (0.034 eV)56IRMOF-1 (0.026 eV), IRMOF-8 (0.0222 eV), and IRMOF-18 (0.032 eV)57and slightly higher than that of MOF-5 (0.014 eV)56. This moderate activation energy, combined with the lower self-diffusion coefficient of MOF-2087, suggests that the strong interaction between H2 and host adsorption sites tends to localize H2 molecules around organic nodes, particularly carbon clusters, resulting in reduced diffusivity.

MSD of H2 at various temperatures (A) and Arrhenius plot of MOF-2087 (R2 = 0.98) (B).
H2 displacement in MOF-2087
To gain a deeper understanding of H2 displacement within MOF-2087, radial distribution functions of H2 molecules relative to host atoms were computed across different temperature zones (Figs. 11 and 12). A sharp peak at 3.65 Å for C_2 and Zn indicates that many H2 molecules preferentially settle at these sites. At ambient temperatures, the H2 molecules are more dispersed and accumulate around 4.65 Å and 8.46 Å near Zn, likely due to increased thermal motion at higher temperatures. Significant H2 interactions were observed at 4.4 Å for C_3 at 77 K, reflecting the low-temperature effects noted during simulated annealing. However, this high-intensity peak for C_3 becomes saturated and shifts to 4.53 Å and 6.53 Å at 298 K, indicating that, similar to Zn, C_3 effectively participates in H2 adsorption at lower temperatures.
The O–H2 interaction peaks were observed at 3.71 Å at 77 K. While a shoulder peak is observed at 2.90 Å, inferring O becomes a more favorable adsorption site at 298 K. The number of H2 molecules distributed at 77 K and 298 K provides insights into the nature of hydrogen interactions with specific host atoms at varying distances (Table 5). The average number of H2 molecules around C_2 and C_R atoms is significantly higher at 77 K, especially within the 5 Å to 6 Å range. Although strong interactions persist at ambient temperature, the number of H2 molecules decreases due to the increased thermal motion. This suggests that C_2 and C_R are more favorable adsorption sites than others.
As a metal site, Zn exhibits moderate interaction with hydrogen at 77 K and 298 K, indicating that while Zn is not the primary adsorption site, it likely plays a supportive role in stabilizing hydrogen molecules. At 77 K, the H2 distribution number around O atoms is 0.48 at a 6 Å distance, underscoring O’s significant role in H2 adsorption. However, this value decreases at 298 K, showing that higher temperatures reduce the likelihood of sustained H2 adsorption at O sites. It is important to note that comparing these numbers with the enhanced H2 uptake of MOF-2087 might be misleading, as the number of atoms in the supercell is relatively high; for instance, MOF-2087 contains 820 C_2 atoms within its supercell. Therefore, the collective contribution of individual host atoms results in substantial H2 adsorption, exceeding that of any single adsorption site. Correlating the MD results with GCMC findings confirms that C_2 and C_R (from sites I and III) are the primary adsorption sites in MOF-2087. While O and Zn atoms also contribute to adsorption, their effectiveness is more temperature-dependent.

Computed radial distribution function of host-H2 interaction at 77 K. (A) Classified carbon atoms. (B) Host atoms from site II and III.

Computed radial distribution function of host-H2 interaction at 298 K. (A) Classified carbon atoms. (B) Host atoms from site II and III.
In addition to the structural parameters and preferential adsorption sites, the relative binding characteristics of MOFs contribute to hydrogen storage capacities. The estimated isosteric heat of hydrogen (Qst) of MOF-2087 is 3.1 kJ mol− 1, indicating physisorption based hydrogen storage rather than chemisorption (Supplementary Fig. 9). It is also noteworthy that the changes in pressure or hydrogen loading has shown minimal variations in Qst, with a maximum observed value of 3.18 kJ mol− 1 at 100 bar (298 K). This lower Qst suggests that MOF exhibits modest guest-host interactions. While the substantial H2 storage capacities observed in MOF 2087 is driven by sites I and II and the framework’s considerable porosity29.
Hydrogen storage performance of MOF 2087
The hydrogen sorption characteristics of MOF-2087 were calculated using the RASPA code58. Figure 13 illustrates the estimated total gravimetric and volumetric H2 uptake of MOF-2087 across varying pressures (0-100 bar) and temperatures (77 K to 298 K). The hydrogen adsorption isotherm at 77 K (Fig. 13) shows a steep increase in H2 uptake at low-pressure regimes (< 20 bar), indicating a physisorption-based hydrogen storage mechanism59. The uptake reaches a maximum of approximately 38 g H2 L− 1 and 22.3 wt% at 100 bar. Despite MOF-2087 exhibiting a relatively low isosteric heat of adsorption, which influences low-pressure hydrogen storage, its substantial free volume, surface area, and adsorption sites contribute to enhanced H2 uptake at medium and high-pressure regimes, achieving an optimal trade-off between hydrogen storage capacities6.
The H2 adsorption isotherm of MOF-2087 demonstrates a monotonic increase in uptake with increasing pressure and does not saturate at 100 bar, indicating structural stability under high-pressure conditions. Additionally, hydrogen density maps at low, medium, and high pressures show active H2 interactions primarily at the site I, with contributions from sites II and III during higher loadings (Supplementary Fig. 8). The observed linearity in the isotherm at elevated temperatures (160 K to 298 K) is attributed to the dilution of the adsorbed H2 layer on the surface of the MOF, a behavior similar to that reported by Assoulaye et al.38.
The hydrogen storage capacities of MOF-2087 were evaluated at operating temperatures ranging from 77 K to 298 K, as shown in Fig. 13C. Under pressure swing operations, MOF-2087 achieves a maximum hydrogen storage capacity of 32.4 gH2 L− 1 and 18.3 wt% at 77 K, in alignment with the B-RFT-MOPSO predictions (Table 3; Fig. 13C). To the best of our knowledge, MOF-2087 demonstrates a more trade-off hydrogen storage density compared to classical MOFs available within the specified search domain. As expected, MOF-2087 retained this trade-off at elevated temperatures, as shown in Fig. 13C. A reduction in less than 10% in both gravimetric and volumetric capacities was observed between 200 and 298 K, indicating the MOF’s stability in maintaining enhanced hydrogen uptake at DOE-recommended operating temperatures. Specifically, at ambient temperature, MOF-2087 achieves a maximum uptake of 5.3 wt% and 7.4 gH2 L− 1, indicating an optimized gravimetric and volumetric hydrogen storage performance.

Hydrogen adsorption characteristics. (A) Total gravimetric and (B) volumetric hydrogen adsorption of MOF-2087. (C) UG and UV of MOF 2087 under pressure swing conditions (100 bar−5 bar) concerning the temperature.
Discussion
The primary objective of the present study is to identify a MOF having a superior tradeoff between gravimetric and volumetric hydrogen capacities under pressure swing conditions at 77 K. By coupling the prediction accuracy of the B-RFT model with the search efficiency of MOPSO, the optimization-based screening approach effectively optimized the crystallographic features of MOFs and identified a better solution from the vast global database. The hyperparameter tuning and optimized training dataset size for the B-RFT model significantly improved prediction accuracy (Supplementary Table 1). Additionally, single-crystal density was found as a key feature, negatively correlating with both hydrogen storage capacities, unlike other features21,23. Specific sweet spots were observed for volumetric hydrogen storage in terms of single crystal density (~ 0.5 g cm− 3)23, the void fraction (~ 0.90), pore volume (~ 5 cm3 g− 1), large cavity diameter (~ 15 Å), and pore limiting diameter (~ 8 Å) of MOFs (Refer to Fig. 3). The B-RFT-MOPSO framework successfully maintained these sweet spots (Supplementary Fig. 6F and G), underscoring the accuracy and search efficiency of the approach. While these features exhibit their respective sweet spots, achieving the optimal combination remains hypothetical; hence, a 7–9 Å difference between the rank one feature and cavity and pore limiting diameters of MOF-2087 was observed.
Sorting the features in the HyMARC dataset based on the design space constraints reveals a volumetric ceiling of 40 gH2 L− 1, indicating that no MOF in the dataset meets the DOE’s volumetric targets (Supplementary Fig. 10). For example, the H74A database contains MOFs with the gravimetric surface areas between 3226 and 3704 m2 g− 1, below the desired range of 5300–7200 m2 g− 1. Meanwhile, databases like UO, SNW, and ToBaCCo show promising numbers of MOFs (5169, 346, and 794, respectively) with favorable gravimetric-volumetric tradeoffs. However, some entries in CSM-2018-I have densities (0.33–3.22 g cm− 3) and void fractions in MTV (beyond 0.89–0.91) that fall outside the design range. Analyzing the non-dominated crystallographic features of MOFs (Supplementary Fig. 6), a reduction in single crystal density and volumetric surface area (refer to Sect. 2.3) enhances gravimetric hydrogen uptake. This should be prioritized in designing MOFs for hydrogen storage applications, where maximizing energy density per unit weight is crucial, especially for automotive applications. In contrast, high-density MOFs with void fractions between 0.905 and 0.91, large cavity diameters (16–18 Å), and pore-limiting diameters (14–18 Å) provide an optimal trade-off for achieving storage capacities approaching DOE targets.
The B-RFT-MOPSO framework was restricted to a defined search domain, but additional best-performing MOFs may exist outside this space.
One of the significant challenges to consider here is synthesizing these hypothetical MOFs and achieving the desired levels of porosity and surface area21,23,24. Despite this, observations using B-RFT-MOPSO combined with NNS and TOPSIS identified the Zn-based MOF-2087 as the global best-performing MOF in terms of optimal crystallographic feature combinations. Previous studies demonstrated the classical benchmark MOFs such as MOF-5, IRMOF-20, and PCN-610, are capable of having better tradeoffs at 77 K21,23,26. However, these MOFs perform poorly at ambient temperatures, achieving volumetric density of 8.8–9.42 gH2 L− 1 but exhibiting poor gravimetric uptake (0.8–2.66 wt%)38. Consequently, achieving DOE targets at 77 K does not guarantee performance at DOE-recommended temperatures. In contrast, MOF-2087 exceeds the gravimetric storage of MOF-5, IRMOF-20, and PCN-61021 by 621.2%, 342.75%, and 186.8%, respectively, under pressure swing conditions at 298 K, showcasing exceptional; gravimetric hydrogen storage at ambient temperature. These performance differences stem from MOF-2087’s enhanced porosity and surface areas. For instance, the modest surface area (2200–2500 m2 g− 1), limited pore volume (0.4–1.3 cm3 g− 1), and simple Zn4O secondary building unit of MOF-5result in rapid drop in hydrogen uptake at elevated temperatures. In contrast, the larger unit cell (39 Å x 39 Å x 39 Å), higher pore volume (~ 5 cm3 g− 1) and presence of aromatic carbon clusters in MOF-2087 provide an efficient network for better hydrogen adsorption. Although MOF-2087’s volumetric capacity reduces by 21.02%, 26.22%, and 24.2% compared with MOF-5, IRMOF-20, and PCN-610, there is still room to enhance volumetric uptake through strategies such as alkali metal decoration, monolith formation, or alternative operating scenarios like temperature-pressure swing conditions23.

As expected, the gravimetric hydrogen storage of MOF-2087 (4.97 wt%) is particularly noteworthy when compared to MOFs with dominant volumetric capacity (other than benchmarked MOFs, dotted red lines in Fig. 14), which often exhibit low gravimetric uptake. For example, Mg-MOF-74 and Zn-MOF-74 offer exceptional volumetric capacities of 12.3 gH2 L− 1 and 11.96 gH2 L− 1, respectively, but their gravimetric capacities are considerably lower at 0.91 wt% and 0.67 wt%, highlighting the tradeoff where enhanced volumetric capacity comes at the expense of gravimetric uptake (refer to Fig. 14). Although MOF-2087 has a modest volumetric uptake of 6.95 gH2 L− 1, it achieves a superior gravimetric uptake, suggesting a favorable structure that delivers significant hydrogen uptake without compromising volumetric density.
Further comparison with NU-1501-Al, which has a near-similar topology and crystallographic features, reveals that MOF-2087, with its higher number of carbon atoms and 8-membered C-cluster-like porous aromatic frameworks61exhibits greater hydrogen uptake29. While both MOFs have similar volumetric capacities, the gravimetric capacity of MOF-2087 is double that of NU-1501-Al (Fig. 14). The metal centers and oxygen atoms in MOF-2087 contribute to hydrogen adsorption due to significant electronegativity differences, while the C-cluster traps hydrogen via van der Waals interactions, as indicated by the C_2 and C_R carbon interaction results from MD simulations. The presence of diverse, active sites, including the metal center, oxygen atoms, and C-cluster, as revealed by the mass center probability distribution (Fig. 6), likely accounts for the higher gravimetric capacity. At the same time, the slight deviation of crystallographic features of NU-1501-Al from the ideal solution, especially with void fraction and volumetric surface area, could have also been attributed to the reduced gravimetric uptake. As the temperature increases from 77 to 298 K, a significant drop in hydrogen uptake is observed, similar to NU-1501-Al, which is attributed to the increased kinetic energy of hydrogen molecules.
Nonetheless, the uptake of MOF-2087 at 298 K and 100 bar (5.3 wt% and 7.4 gH2 L− 1) remains notable, demonstrating one of the best deliverable capacities with an optimal tradeoff under ambient conditions. The observed performance at 298 K can be attributed to the structural features identified during optimization at 77 K, which remains stable across this temperature range. This was confirmed through simulated annealing, where the structure of MOF-2087 was relaxed at each temperature point, including 300 K. The framework remained stable throughout the range of 77–298 K, supporting the use of 77 K-optimized features for performance evaluation at elevated temperatures. Additionally, previous studies have shown that MOFs such as SNU-77 and Cd-doped variants retain their structural features from cryogenic to above ambient temperatures (373 K), further supporting this assumption62,63. Therefore, MOF-2087 is expected to maintain the optimum trade-off in hydrogen storage performance even at ambient conditions.
Regarding comparative methods analysis, the B-RFT-MOPSO hybrid framework offers a significantly faster alternative to the brute-force high throughput GCMC screening process. This study’s B-RFT framework required approximately 6-120 CPU seconds to calculate hydrogen storage densities for the global MOF features. The MOPSO algorithm then took around 168 CPU hours to conduct an average of 109,437 framework evaluations to identify the local best solutions and less than 10 CPU minutes to determine the global best features from the local repository. Although the B-RFT-MOPSO framework necessitates pre-training and validation datasets from experimental GCMC simulations, it is scalable to larger search domains encompassing a wide range of features. Once evaluated and validated, the framework enables computationally efficient evaluation of new feature combinations, potentially running on a desktop workstation rather than requiring HPC servers, as the current study employed an Intel Xeon 24-core machine. In contrast, brute-force GCMC screening is computationally expensive and intensive, often dependent on the number of equilibration and production cycles the user sets. For instance, Snurr et al. reported that screening a dataset of 130,000 hMOFs would require approximately 500,000 CPU hours, impractical for low-configuration machines64. Employing the optimization-based screening approach centered on the statistical ideal solution; the same task could be accomplished in less than 200 CPU hours for a single run, effectively isolating feasible MOF feature combinations.
Hence, in a nutshell, the hydrogen sorption of MOF-2087 is undoubtedly superior and can fit into the leaderboard of benchmarked MOFs. This peculiar nature of MOF-2087 also highlights the reliability, search ability, and tendency to reach optimality of our optimization-based screening approach (B-RFT-MOPSO). In summary, these observations show our optimization-based searching route as a reliable and the most promising route for material selection from a realm of available materials.
Conclusions
The present work demonstrates the B-RFT-MOPSO hybrid computational framework as a powerful approach for optimizing and screening MOFs for hydrogen storage. MOF-2087 was identified as the best-performing framework from a global repository of 733,792 MOFs, achieving an optimal tradeoff between gravimetric and volumetric hydrogen storage capacities, 38 g H2 L−1, and 22.3 wt% at 77 K, 100 bar, and 5.3 wt% and 7.4 gH2 L−1 at 298 K, 100 bar, outperforming classical benchmarked MOFs like MOF-5, IRMOF-20, PCN-610.
Simulated annealing and MD simulations revealed that in MOF-2087, C2 and C_R atoms are the most favorable adsorption sites for hydrogen due to their π-π interactions and the electronegativity of sp2 hybridized carbons. The radial distribution function showed a significant decrease in the distance between C2 and H2 from 5.9 Å at 300 K to 3.6 Å at 14 K, indicating enhanced adsorption at lower temperatures. Similarly, C_R–H2 distances decreased, highlighting strong π-π stacking interactions. O atoms also demonstrated strong adsorption potential, with distances reducing from 6.2 to 4.2 Å, while Zn showed moderate interaction, likely due to polarization effects. H2 diffusion analysis indicated increased mobility with temperature, but lower diffusion rates at reduced temperatures facilitated access to favorable adsorption sites. These findings, consistent with GCMC simulations, underscore the role of C2 and C_R atoms as primary adsorption sites, contributing to the enhanced hydrogen uptake in MOF-2087.
In future, the proposed machine learning and multi-objective optimization framework can be extended to optimize functional group combinations and metal ion selections, enabling the design of new MOFs with enhanced tradeoffs at 298 K. Furthermore, the proposed computational framework offers a scalable and versatile approach for identifying high-promising MOFs specifically tailored for onboard hydrogen storage in fuel cell vehicles. Beyond this primary focus, the method can also be adapted to other real-world gas storage applications such as methane by retraining the B-RFT model with domain-specific datasets.
Methods
Data mining and screening
A global repository of 925,229 MOFs was mined from the HyMARC (Hydrogen Materials Advanced Research Consortium) data hub, which complies with data from 19 known databases and was deposited by Ahmed et al.21. Initial screening classified 191,437 MOFs as non-open-source structures (i.e., their crystallographic information is not publicly available) and 733,792 MOFs as open-source structures. These open-source MOFs were shortlisted and stored in the labelled repository, the global database. Supplementary Fig. 11 illustrates the distribution of MOFs across the global database based on their respective sources. The seven key crystallographic features, crystal density, gravimetric and volumetric surface areas, pore volume, pore diameter, cavity diameters, and void fractions, were identified using HyMARC data hub21,23,65. A subset of the global database, consisting of MOFs with deliverable hydrogen storage capacities measured at 100-5 bar pressure swing at 77 K21,23was extracted to train the B-RFT framework (Supplementary Table 5). This training dataset includes hydrogen storage capacities of 12,764 experimental MOFs (eMOFs) and 85,930 hypothetical MOFs (hMOFs). The distribution of usable gravimetric and volumetric capacities within the subset is depicted in Supplementary Fig. 12.
Including both eMOFs and hMOFs in the training dataset significantly enhances the performance of the B-RFT in predicting hydrogen storage densities across the global database. The dataset was shuffled before training and testing to prevent the B-RFT from memorizing trends in the training data. Correlation analysis of the training data revealed that gravimetric capacity is predominantly influenced by pore volume and gravimetric surface area. In contrast, volumetric capacity is mainly governed by a void fraction of MOFs (Supplementary Fig. 2). For simplicity, we refer to the repository of 733,792 open-source MOFs as the global database. Detailed steps involved in the present study are provided in Supplementary Fig. 13.
B-RFT framework and performance evaluation
The B-RFT (Bootstrap Aggregated Random Forest Tree) framework couples randomization with decision trees, enabling efficient prediction for larger datasets66. By implementing parallel decision trees trained on random subsets of data, the framework reduces the chances of overfitting and improves prediction accuracy for complex datasets. This adaptive bootstrapping strategy enhances scalability and efficiency in large-scale onboard applications. Supplementary Fig. 14 outlines the hydrogen storage density prediction steps using the B-RFT framework, with additional details in Supplementary Method 1.
The B-RFT model aims to predict the usable gravimetric and volumetric hydrogen storage densities in MOFs under pressure swing conditions at 77 K. Separate B-RFT models were designed for each density to reduce the complexities, evaluated using performance metrics such as R2RMSE, AUE, and MEA21. Model sensitivity analysis was performed on training dataset sizes ranging from 4935 to 78,965 MOFs, with 9870 MOFs used for validation and testing21,67. The B-RFTs best-performing model was used as the fitness function for subsequent multi-objective particle swarm optimization (MOPSO) due to the lack of an explicit fitness function for optimizing hydrogen storage tradeoffs.
MOPSO
The MOPSO68,69 algorithm was employed to identify MOFs with optimal tradeoffs between hydrogen storage capacities. In multi-objective optimization, a local repository was created to store non-dominated solutions, updated during iterations. To ensure diversity of search, repository size was constrained, and the algorithm continued until convergence criteria were met. The B-RFT-MOPSO framework was configured to maximize MOF’s hydrogen storage densities, with design parameters such as population size, learning coefficients, and damping ratios tailored from previous optimization studies39,70,71. For additional details refer to Supplementary Fig. 15 and Supplementary Methodology 2. Performance metrics including the standard spacing metric (SSM), standard generational distance (SGD), and solution set size were used to evaluate the framework’s effectiveness. Further details of MOPSO, performance indicators, nearest neighbor search, and multi-criteria decision-making technique are provided in Supplementary Methodologies 2 and 3.
GCMC and MD approach
The hydrogen adsorption properties of potential MOFs identified through B-RFT-MOPSO were simulated using the GCMC method via the RASPA computational package58. Adsorption isotherms for each pressure point were calculated over 10,000 cycles (5000 for equilibration and 5000 for data collection). In GCMC, each cycle corresponds to ‘m’ Monte Carlo moves, where ‘m’ is either 20 or the number of adsorbate molecules in the supercell, whichever is higher. Each simulation move includes translation, rotation, insertion, or deletion of guest molecules with equal probabilities. The intermolecular interactions between the host framework and adsorbate molecules were modeled using Lennard-Jones (LJ) potentials (Eq. 1). Long-range corrections were applied with a cutoff radius of 12 Å.
Here,\(\:\:{\epsilon\:}_{ij}\) represents the depth of potential, \(\:{r}_{ij}\) is the distance between atoms i and j, and \(\:{\sigma\:}_{ij}\) corresponds to the distance at which the intermolecular potential between two particles equals zero. Cross-interactions between host and adsorbate molecules were calculated using the Lorentz–Berthelot missing rule:
Hydrogen molecules were modeled using a single van der Waals (vdW) site, as Fischer et al.72 proposed. The adsorbate-adsorbate columbic interactions were computed using Ewald summation.
The adsorbate-host framework and adsorbate-adsorbate interactions were corrected using pseudo-Feynman-Hibbs correction24,73 to account for the quantum effects in low-temperature regimes, which adjusts the LJ potential based on temperatures:
Where \(\:{k}_{B}\) is the Boltzmann constant, \(\:\mu\:\) the reduced mass, h is the Planck constant, and T the temperature. Framework molecules were modeled using the universal force field74 and DREIDING53 for the metal and non-metal framework atoms respectively75. The supercell structure was tailored for each MOF to ensure it was sufficiently large relative to twice the cutoff radius, with all MOFs treated as rigid.
The probability density distribution of hydrogen and radial distribution functions (RDFs) were calculated to identify preferred adsorption sites within the MOFs. The RDF between hydrogen and host atoms was calculated as follows51:
Where V is the system volume, \(\:{N}_{i}\) and \(\:{\:N}_{j}\) are the number of host and guest atomic pairs, and r is the distance between the centers of mass of the pairs. The isosteric heat of adsorption(Qst), representing the interaction energy between host and guest molecules, was calculated based on potential energy (v) and the number of adsorbate molecules (N) as shown in Eq. 5:
Considering the ensemble fluctuations, Qst can also be expressed as76,77,
The GCMC approach was validated by calculating the hydrogen storage capacities of 10 randomly selected MOFs from the training dataset21. Supplementary Fig. 15 shows a close agreement between the training dataset and present GCMC computed capacities, confirming the model’s reliability.
Classical molecular dynamics simulations were performed in LAMMPS package, to understand the global best MOF’s hydrogen storage mechanism. Similar force fields and hydrogen model used in GCMC were applied. MD simulated annealing was carried out to identify the minimum potential energy sites in the MOF. The number of hydrogen molecules in the simulation cell was based on GCMC, adsorption data. Hydrogen molecules were randomly introduced into the cell, equilibrated at 300 K for 10,000 steps using NVT ensemble, and annealed from 300 K to 1 K over 10.5 ps. A longer MD simulation lasting 2000 ps, was performed at various temperatures (77 K, 160 K, 200 K, and 298 K) to compute the mean square displacement (MSD)54:
Where R(t) is the center of mass of hydrogen molecule at time t, and N is the number of hydrogen molecules in the system. The diffusion coefficients (DH) were then calculated using Einstein’s relation78.
Where \(\:{\text{n}}_{\text{d}\text{i}\text{m}}\) is the diffusion dimensionality (3 for present system). The number of hydrogen molecules around the host atoms at increased radial distances was computed by using RDF g(r).
Where p is the average hydrogen density around the host atoms.
Data availability
All the relevant information and necessary datasets were available in the Supplementary information file. The Supplementary Excel file showed the Pareto optimal solutions and local best MOFs. Further details or data will be available from the corresponding author upon reasonable request. The CIF information of MOF-2087 can be found in the Supplementary information file. The pretrained B-RFT models can be found in figshare public repository: https://doi.org/10.6084/m9.figshare.28925930.
References
Martin, R. L. & Haranczyk, M. Exploring frontiers of high surface area metal–organic frameworks. Chem. Sci. 4, 1781–1785 (2013).
Chae, H. K. et al. A route to high surface area, porosity and inclusion of large molecules in crystals. Nature 427, 523–527 (2004).
Li, L. et al. Selective gas adsorption and separation in flexible metal-organic frameworks. Huagong Jinzhan/Chem. Ind. Eng. Prog. 35, 1794–1803 (2016).
Cohen, S. M. Postsynthetic methods for the functionalization of metal-organic frameworks. Chem. Rev. 112, 970–1000 (2012).
Yaghi, O. M. et al. Reticular synthesis and the design of new materials. Nature 423, 705–714 (2003).
Frost, H., Düren, T. & Snurr, R. Q. Effects of surface area, free volume, and heat of adsorption on hydrogen uptake in metal- organic frameworks. J. Phys. Chem. B. 110, 9565–9570 (2006).
Archana, K. et al. Enhanced isosteric heat of adsorption and gravimetric storage density of hydrogen in GNP incorporated Cu based core-shell metal-organic framework. Int. J. Hydrogen Energy. 45, 33818–33831 (2020).
KV, S. S., Seth, A., Mohapatra, D., Ramachandran, S. & Sujith, R. Iron decorated defective phosphorene as a viable hydrogen storage medium–a DFT study. Int. J. Hydrogen Energy. 47, 34976–34993 (2022).
Gangadhar, J., Tirumuruhan, B. & Sujith, R. Applications and future trends in mesoporous materials. Adv. Funct. Porous Mater. Macro Nano Scale Lengths. https://doi.org/10.1007/978-3-030-85397-6_8 (2022).
Wang, Z. et al. Accelerated discovery of superoxide-dismutase nanozymes via high-throughput computational screening. Nat. Commun. 12, 6866 (2021).
Kwon, O. et al. Computer-aided discovery of connected metal-organic frameworks. Nat. Commun. 10, 3620 (2019).
Rosen, A. S. et al. High-throughput predictions of metal–organic framework electronic properties: Theoretical challenges, graph neural networks, and data exploration. Npj Comput. Mater. 8, 112 (2022).
Schneider, C., Bodesheim, D., Keupp, J., Schmid, R. & Kieslich, G. Retrofitting metal-organic frameworks. Nat. Commun. 10, 4921 (2019).
Wilmer, C. E. et al. Large-scale screening of hypothetical metal-organic frameworks. Nat. Chem. 4, 83–89 (2012).
Lin, L. C. et al. In Silico screening of carbon-capture materials. Nat. Mater. 11, 633–641 (2012).
Comlek, Y., Pham, T. D., Snurr, R. Q. & Chen, W. Rapid design of top-performing metal-organic frameworks with qualitative representations of building blocks. Npj Comput. Mater. 9, 170 (2023).
Mohamed, S. A., Zhao, D. & Jiang, J. Integrating stability metrics with high-throughput computational screening of metal–organic frameworks for CO2 capture. Commun. Mater. 4, 79 (2023).
Borboudakis, G. et al. Chemically intuited, large-scale screening of MOFs by machine learning techniques. Npj Comput. Mater. 3, 40 (2017).
Cui, J. et al. Direct prediction of gas adsorption via spatial atom interaction learning. Nat. Commun. 14, 7043 (2023).
Boyd, P. G. et al. Data-driven design of metal–organic frameworks for wet flue gas CO2 capture. Nature 576, 253–256 (2019).
Ahmed, A. & Siegel, D. J. Predicting hydrogen storage in MOFs via machine learning. Patterns 2, 2 (2021).
Anderson, R. & Gómez-Gualdrón, D. A. Increasing topological diversity during computational synthesis of porous crystals: How and why. CrystEngComm 21, 1653–1665 (2019).
Ahmed, A. et al. Exceptional hydrogen storage achieved by screening nearly half a million metal-organic frameworks. Nat. Commun. 10, 10 (2019).
Ahmed, A. et al. Balancing gravimetric and volumetric hydrogen density in MOFs. Energy Environ. Sci. 10, 2459–2471 (2017).
DoE, U. Target explanation document: Onboard hydrogen storage for light-duty fuel cell vehicles. US Drive. 1, 1–29 (2017).
Goldsmith, J., Wong-Foy, A. G., Cafarella, M. J. & Siegel, D. J. Theoretical limits of hydrogen storage in metal-organic frameworks: opportunities and trade-offs. Chem. Mater. 25, 3373–3382 (2013).
Vaishnavi, A. L. et al. Enhanced hydrogen uptake of high surface area copper carboxylate frameworks/graphene nanoplatelets nanocomposite. J. Energy Storage. 104, 114629 (2024).
Gómez-Gualdrón, D. A. et al. Understanding volumetric and gravimetric hydrogen adsorption trade-off in metal–organic frameworks. ACS Appl. Mater. Interfaces. 9, 33419–33428 (2017).
Chen, Z. et al. Balancing volumetric and gravimetric uptake in highly porous materials for clean energy. Science. 368, 297–303 (2020).
Liu, K. et al. Exploring the chemical space of metal–organic frameworks with Rht topology for high capacity hydrogen storage. J. Phys. Chem. C. 128, 7435–7446 (2024).
Wang, L. et al. Artificial intelligence and High-Throughput computational workflows empowering the fast screening of metal–organic frameworks for hydrogen storage. ACS Appl. Mater. Interfaces. 16, 36444–36452 (2024).
Ghude, S. & Chowdhury, C. Exploring hydrogen storage capacity in Metal-Organic frameworks: A bayesian optimization approach. Chem. Eur. J. 29, e202301840 (2023).
Singh, S. K., Sose, A. T., Wang, F., Bejagam, K. K. & Deshmukh, S. A. Data driven discovery of MOFs for hydrogen gas adsorption. J. Chem. Theory Comput. 19, 6686–6703 (2023).
Farha, O. K. et al. De Novo synthesis of a metal–organic framework material featuring ultrahigh surface area and gas storage capacities. Nat. Chem. 2, 944–948 (2010).
Suresh, K. et al. Optimizing hydrogen storage in MOFs through engineering of crystal morphology and control of crystal size. J. Am. Chem. Soc. 143, 10727–10734 (2021).
Li, H., Eddaoudi, M., O’Keeffe, M. & Yaghi, O. M. Design and synthesis of an exceptionally stable and highly porous metal- organic framework. Nature 402, 276–279 (1999).
Kapelewski, M. T. et al. Record high hydrogen storage capacity in the metal–organic framework Ni2 (m-dobdc) at near-ambient temperatures. Chem. Mater. 30, 8179–8189 (2018).
Assoualaye, G. & Djongyang, N. Influence of pore size and isosteric heat of adsorption of some metal–organic frameworks on the volumetric and gravimetric adsorption capacities of hydrogen at room temperature. Polym. Bull. 78, 4987–5001 (2021).
Hojjati, A., Monadi, M., Faridhosseini, A. & Mohammadi, M. Application and comparison of NSGA-II and MOPSO in multi-objective optimization of water resources systems. J. Hydrol. Hydromech. 66, 323–329 (2018).
Jalili, S., Khani, R., Maheri, A. & Hosseinzadeh, Y. Performance assessment of meta-heuristics for composite layup optimisation. Neural Comput. Appl. 34, 2031–2054 (2022).
Slowik, A. Particle swarm optimization. In The Industrial Electronics Handbook - Five Volume Set 1942–19484. https://doi.org/10.1007/978-3-319-46173-1_2 (2011).
Genuer, R., Poggi, J. M. & Tuleau-Malot, C. Villa-Vialaneix, N. Random forests for big data. Big Data Res. 9, 28–46 (2017).
Guo, J. et al. Optimized random forest method for 3D evaluation of coalbed methane content using geophysical logging data. ACS Omega. 9, 35769–35788 (2024).
Butler, K. T., Davies, D. W., Cartwright, H., Isayev, O. & Walsh, A. Machine learning for molecular and materials science. Nature 559, 547–555 (2018).
Pellegrino, E. et al. Machine learning random forest for predicting oncosomatic variant NGS analysis. Sci. Rep. 11, 21820 (2021).
Bhowmik, A. R. & Chakraborty, A. K. Solution of optimal power flow using Non dominated sorting multi objective opposition based gravitational search algorithm. Int. J. Electr. Power Energy Syst. 64, 1237–1250 (2015).
Si, B. et al. Multi-objective optimization design of a complex building based on an artificial neural network and performance evaluation of algorithms. Adv. Eng. Inf. 40, 93–109 (2019).
Kollenda, D., Reher, A. S. & de Haas, B. Individual gaze predicts individual scene descriptions. Sci. Rep. 15, 9443 (2025).
Aumüller, M. & Ceccarello, M. The role of local dimensionality measures in benchmarking nearest neighbor search. Inf. Syst. 101, 101807 (2021).
Kim, Y., Chung, E. S., Jun, S. M. & Kim, S. U. Prioritizing the best sites for treated wastewater instream use in an urban watershed using fuzzy TOPSIS. Resour. Conserv. Recycl. 73, 23–32 (2013).
Peng, L., Zhu, Q., Wu, P., Wu, X. & Cai, W. High-throughput computational screening of metal–organic frameworks with topological diversity for hexane isomer separations. Phys. Chem. Chem. Phys. 21, 8508–8516 (2019).
Lu, X., Xie, Z., Wu, X., Li, M. & Cai, W. Hydrogen storage metal-organic framework classification models based on crystal graph convolutional neural networks. Chem. Eng. Sci. 259, 117813 (2022).
Mayo, S. L., Olafson, B. D. & Goddard, W. A. DREIDING: A generic force field for molecular simulations. J. Phys. Chem. 94, 8897–8909 (1990).
Koizumi, K., Nobusada, K. & Boero, M. Hydrogen storage mechanism and diffusion in metal–organic frameworks. Phys. Chem. Chem. Phys. 21, 7756–7764 (2019).
Nguyen-Thuy, T. et al. Hydrogen adsorption mechanism of MOF-74 metal–organic frameworks: An insight from first principles calculations. RSC Adv. 10, 43940–43949 (2020).
Liu, S. et al. Machine learning potential for modelling H2 adsorption/diffusion in MOFs with open metal sites. Chem. Sci. 15, 5294–5302 (2024).
Yang, Q. & Zhong, C. Molecular simulation of adsorption and diffusion of hydrogen in metal- organic frameworks. J. Phys. Chem. B. 109, 11862–11864 (2005).
Dubbeldam, D., Calero, S., Ellis, D. E. & Snurr, R. Q. RASPA: Molecular simulation software for adsorption and diffusion in flexible nanoporous materials. Mol. Simul. 42, 81–101 (2016).
Ding, L. & Yazaydin, A. O. Hydrogen and methane storage in ultrahigh surface area metal–organic frameworks. Microporous Mesoporous Mater. 182, 185–190 (2013).
Xuan Huynh, N. T., Chihaia, V. & Son, D. N. Hydrogen storage in MIL-88 series. J. Mater. Sci. 54, 3994–4010 (2019).
Kim, D. W. et al. High hydrogen storage in trigonal prismatic monomer-based highly porous aromatic frameworks. Adv. Mater. 36, 2401739 (2024).
Park, H. J., Lim, D. W., Yang, W. S., Oh, T. R. & Suh, M. P. A highly porous metal–organic framework: structural transformations of a guest-free MOF depending on activation method and temperature. Chem. Eur. J. 17, 7251–7260 (2011).
Tahier, T. & Oliver, C. L. A cd mixed-ligand MOF showing ligand-disorder induced breathing behaviour at high temperature and stepwise, selective carbon dioxide adsorption at low temperature. CrystEngComm 19, 3607–3618 (2017).
Bucior, B. J. et al. Energy-based descriptors to rapidly predict hydrogen storage in metal–organic frameworks. Mol. Syst. Des. Eng. 4, 162–174 (2019).
Ahmed.A, S. D. Computational Prediction of Hydrogen Storage Capacitites in MOFs-PS_Usable_Hydrogen_storage_Capacity_ML-HyMARC Data Hub (2019).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 (2001).
Fernandez, M., Woo, T. K., Wilmer, C. E. & Snurr, R. Q. Large-scale quantitative structure-property relationship (QSPR) analysis of methane storage in metal-organic frameworks. J. Phys. Chem. C. 117, 7681–7689 (2013).
Reyes-Sierra, M., Coello, C. A. C. et al. Multi-objective particle swarm optimizers: A survey of the state-of-the-art. Int. J. Comput. Intell. Res. 2, 287–308 (2006).
Lin, Q., Li, J., Du, Z., Chen, J. & Ming, Z. A novel multi-objective particle swarm optimization with multiple search strategies. Eur. J. Oper. Res. 247, 732–744 (2015).
Reddy, M. J. & Kumar, D. N. Performance evaluation of elitist-mutated multi-objective particle swarm optimization for integrated water resources management. J. Hydroinformat. 11, 79–88 (2009).
Zhao, S. Z., Iruthayarajan, M. W., Baskar, S. & Suganthan, P. N. Multi-objective robust PID controller tuning using two lbests multi-objective particle swarm optimization. Inf. Sci. (Ny). 181, 3323–3335 (2011).
Fischer, M., Hoffmann, F. & Fröba, M. Preferred hydrogen adsorption sites in various MOFs–a comparative computational study. ChemPhysChem 10, 2647–2657 (2009).
Liu, J. et al. Experimental and theoretical studies of gas adsorption in Cu3(BTC)2: An effective activation procedure. J. Phys. Chem. C. 111, 9305–9313 (2007).
Rappé, A. K., Casewit, C. J., Colwell, K. S., Goddard, W. A. & Skiff, W. M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 114, 10024–10035 (1992).
Chen, H. & Snurr, R. Q. Computational screening of metal-catecholate-functionalized metal–organic frameworks for room-temperature hydrogen storage. J. Phys. Chem. C. 125, 21701–21708 (2021).
Snurr, R. Q., Bell, A. T. & Theodorou, D. N. Prediction of adsorption of aromatic hydrocarbons in silicalite from grand canonical Monte Carlo simulations with biased insertions. J. Phys. Chem. 97, 13742–13752 (1993).
Brand, S. K., Colón, Y. J., Getman, R. B. & Snurr, R. Q. Design strategies for metal alkoxide functionalized metal–organic frameworks for ambient temperature hydrogen storage. Microporous Mesoporous Mater. 171, 103–109 (2013).
Keffer, D. J. & Adhangale, P. The composition dependence of self and transport diffusivities from molecular dynamics simulations. Chem. Eng. J. 100, 51–69 (2004).
Acknowledgements
The authors thank the financial support provided by the Science and Engineering Research Board, Department of Science and Technology, India, grant no. CRG/2020/001066. Also, we thank the high-performance computational resource Sharanga, provided by Birla Institute of Technology and Science Pilani, Hyderabad Campus.
Author information
Authors and Affiliations
Contributions
A.P: Data curation, formal analysis, validation, visualization, writing-original draft. R.D: data curation, formal analysis, visualization, writing-original draft. S.R.: funding acquisition, conceptualization, analysis, supervision. A.A.: supervision, funding acquisition, data analysis R.S.: conceptualization, supervision, funding acquisition, analysis, editing. All authors contributed to manuscript review and editing and have approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Anbumani, P., Duvvuri, R., Radhika, S. et al. A multi-objective optimization-driven screening approach for maximizing hydrogen storage capacities in MOFs. Sci Rep 16, 242 (2026). https://doi.org/10.1038/s41598-025-09654-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-09654-z
