
Abstract
Leishmaniasis is one of the infectious diseases caused by protozoa and is considered the second most significant parasitic disease after malaria. In this research, thiosemicarbazone Schiff base derivatives were synthesized via a one-pot, two-step, three-component reaction. Then, the effectiveness of the compounds against the two forms of Leishmania major and Leishmania tropica called amastigote and promastigote, was tested. All synthesized compounds displayed weak to good anti-amastigote and anti-promastigote activities. Notably, compounds 5g and 5p were the most potent compounds against amastigote and promastigote forms, respectively, of Leishmania major, with IC50 = 26.7 µM and 12.77 µM. Analogues 5e and 5g were the most potent compounds against amastigote and promastigote forms of Leishmania tropica, with IC50 = 92.3 µM and 12.77 µM, respectively. The cytotoxicity activity of the compounds was also evaluated using the J774.A1 cell lines. Some of the screened derivatives displayed low cytotoxicity to macrophage infection. Among compounds, 5p and 5e showed the highest SI (95.4 and 34.6) against L. major and L. tropica, respectively. In the next phase, the most effective thiosemicarbazone derivatives were examined for their ability to induce apoptosis in promastigotes. According to the results, these compounds displayed different early and late apoptosis as well as necrotic effects on promastigotes of Leishmania major and Leishmania tropica. Furthermore, the compounds’ drug-likeness and pharmacokinetic parameters were predicted in silico. All compounds showed acceptable drug-likeness and pharmacokinetic profiles. Furthermore, the most likely sites of the compounds metabolized by the major cytochromes were identified. Additionally, the in silico compounds’ cardiotoxicity potential was assessed. This investigation showed compounds 5m-p were cardiotoxic. Lastly, molecular docking and molecular dynamics simulations were performed to explore the potential mechanisms of anti-leishmanial activity in the LmPRT1 active site.
Similar content being viewed by others
Introduction
Infectious diseases have been a fundamental aspect of human health illnesses since ancient times. Bacteria, fungi, viruses, protozoa, parasites, and other pathogens are responsible for these infections1. Some of these pathogens cause life-threatening diseases, whereas others have relatively minor impact on human health. A broad spectrum of pathogens—including protozoa, viruses, bacteria, and helminths—are responsible for the neglected tropical diseases (NTDs), which primarily affect populations in tropical and subtropical regions. With more than one billion people affected worldwide—particularly in underprivileged areas of Africa, Asia, and Latin America—these diseases represent a critical concern for global public health2. Leishmaniasis is one of the infectious diseases caused by protozoa and is considered the second most significant parasitic disease after malaria3. Skin ulcers, skin sores, stuffy nose/lips/gums, and nose bleeding are typical symptoms of this disease4. The impoverished, malnourished people, migrants, and those with weakened immune systems are particularly vulnerable. Environmental factors such as deforestation, climate change, dam construction, and water projects also contribute significantly to the spread of leishmaniasis5. This disease is present in over 98 countries, infecting more than 13 million people and causing an average of 300,000 cases every year. Leishmaniasis threatens around 1.7 billion individuals in endemic regions. Clinical manifestations are categorized into three types: mucocutaneous, cutaneous, and visceral6. Leishmania is transmitted by the bite of an infected female Phlebotomine sandfly, after which the protozoan invades the spleen, liver, and bone marrow via macrophages7. Leishmania’s life cycle is dimorphic, with parasites living as external promastigotes in vectors and intracellular amastigotes in mammalian host macrophages8. The genome of Leishmania major (L. major) was the first to be sequenced, laying the groundwork for subsequent genomic studies in the Leishmania genus. Cutaneous leishmaniasis, most commonly caused by L. major, usually heals on its own in humans but often leads to cosmetically disfiguring scarring9.
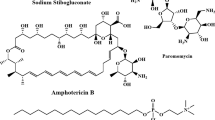
In January 2021, the World Health Organization (WHO) unveiled its 2021–2030 roadmap in Geneva, outlining a comprehensive strategy to combat 20 NTDs. Among its primary objectives is the control and eventual elimination of leishmaniasis10. The current treatment of leishmaniasis relies on a limited selection of drugs, administered either monotherapy or in combination, depending on factors such as the disease type, the causative species, the geographic distribution, and the patient condition9. Drug discovery for NTDs faces numerous challenges. Economically, the low income of affected populations leads to minimal expected return on investment, discouraging pharmaceutical development. Biologically, the intricate and often poorly understood life cycles of the parasites pose significant obstacles. Chemically, new compounds must meet strict criteria, including low cost, ease of handling, chemical stability, low toxicity, and favorable pharmacokinetics—all of which are difficult to predict in the early stages. In addition, resistance development must be avoided. Pharmacologically, therapies must be suitable for diverse patient populations, including individuals with varying ages and comorbidities. These multifaceted challenges contribute to the already high attrition rates in drug development pipelines, and leishmaniasis is no exception to this pattern11. The main target of anti-leishmanial drugs is Leishmania amastigotes that live in macrophage phagolysosomes; however, this is not an easy target because drugs must overwhelm key structural barriers12. Although several treatment options exist for leishmaniasis, therapeutic management remains challenging due to the disease’s clinical diversity and complexity. Current anti-leishmanial therapies are far from optimal, primarily because of their considerable toxicity, limited efficacy, parenteral administration—which reduces patient compliance—high treatment costs, emerging drug resistance, and poor accessibility in endemic regions. Moreover, the effectiveness of certain compounds varies significantly depending on the Leishmania species, the clinical manifestation, and the geographic origin of the infection13. The continued rise in resistance to existing anti-leishmanial agents highlights the need to reassess the widespread reliance on monotherapies. Combination therapy offers a promising strategy to address these challenges. It is designed to delay or prevent the development of resistance, enhance therapeutic efficacy through synergistic mechanisms, allow for lower individual drug dosages, and shorten treatment duration—thereby minimizing overall toxicity. An important consideration in combination regimens is the pharmacokinetic profile of the drugs, particularly their half-lives. The ideal approach involves pairing a highly potent short-acting compound with a longer-acting partner to ensure sustained parasite clearance. Additionally, factors such as tissue distribution, volume of distribution, and macrophage uptake are crucial in determining therapeutic success—especially against intracellular Leishmania amastigotes residing in organs of the reticuloendothelial system13. The FDA-approved drugs for leishmaniasis are illustrated in Fig. 1. Leishmaniasis poses a mammoth task in the field of drug discovery due to the outrageous limitations associated with clinically available anti-leishmanial drugs, namely stibogluconate (Pentostam®), meglumine antimoniate (Glucantime®), ketoconazole, liposomal amphotericin B (AmBisome®), miltefosine, and paromomycin. Long-term use in highly endemic regions has contributed to growing drug resistance. Due to the limited effectiveness of available chemotherapeutics, the absence of vaccines, and the lack of novel agents, there is an urgent need to develop safe, affordable, and short-course treatments with novel mechanisms of action14,15. As a result, there is a crucial need to produce more effective and safer drugs that may function through other pathways than previously described treatments. In this regard, research efforts by scientists around the world resulted in the discovery and development of some new anti-leishmanial drugs, some of which are currently in numerous stages of clinical trials. Anti-leishmanial drugs under clinical development include GSK3186899, DNDI-0690, 18-methoxy coronaridine, meglumine, and pentoxifylline (Fig. 1)5.
Thiosemicarbazones are simply generated through the condensation of a thiosemicarbazide with ketones and/or aldehydes. With the general formula R1R2CH = N-NH-(C = S)-NH2, thiosemicarbazones have captured maximum attention for their multifunctional coordination modes to transition metal ions and a huge spectrum of biological activities16particularly their leishmanicidal action. The medical application of thiosemicarbazones dates back to the 1950s, when their biological activity against tuberculosis and leprosy was first reported. Their therapeutic efficacy is influenced by various factors, such as the electronic configuration of the coordinated metal ions, the nature and position of substituent groups, and the number of available donor atoms involved in coordination16. Methisazone is an FDA-approved drug that inhibits protein synthesis and mRNA, particularly in pox viruses17. Thioacetazone is an oral antibiotic that is used in the treatment of tuberculosis. Also, triapine (3-AP) is a potent inhibitor of ribonucleotide reductase (Fig. 2)18. Amithiozone has been applied in trials studying the treatment of mycobacterium avium-intracellulare infection. Perchlozone is a thiosemicarbazone used for the treatment of multidrug-resistant tuberculosis. Perchlozone is a prodrug that is activated by ethA and inhibits the HadABC complex (Fig. 2). Thiosemicarbazone analogues have demonstrated efficiency against leishmania amastigotes and promastigotes, with likely mechanisms of action elucidated, including mitochondrial depolarization, DNA interaction, parasite death via apoptosis, and parasite protein inhibition15,19. The thiosemicarbazone scaffold was previously identified in the arena of anti-parasitic compounds. Thiosemicarbazones were first developed as parasitic cysteine protease inhibitors, like rhodhesain in T. brucei and cruzain in T. cruzi, two enzymes that play critical roles throughout the parasite’s life cycle. Some members of this group of derivatives were inactive on the enzyme but active on parasites, implying that cruzain was not the main target for some of these derivatives. Thiosemicarbazone metal-complexes or their formation of strong metal chelates recommend that the mechanism of action is mostly because of a dual pathway, including the production of metal complex-DNA interaction or toxic free radicals by thiosemicarbazone bio-reduction20.
Multi-component reactions (MCRs) represent a valuable synthetic strategy in both organic and medicinal chemistry. They have been widely utilized in diverse synthetic applications where conventional methods often require multiple steps and labor-intensive procedures. MCRs offer numerous advantages, including high product yields, atom and step economy, shorter reaction times, and environmental friendliness. Moreover, they serve as powerful tools for rapidly generating libraries of new chemical entities (NCEs), particularly valuable in the drug discovery process. Significant advancements have been made in the development and application of MCRs through extensive research. Among the three main types of one-pot synthetic methodologies—(1) cascade reactions, (2) multicomponent reactions (MCRs), and (3) one-pot stepwise synthesis (OPSS)—the OPSS approach is considered more adaptable and practical. This is largely due to its stepwise execution, which allows for modification of reaction conditions at each stage to optimize the overall synthetic process21. Under basic conditions, MCRs can generate both carbon-heteroatom and carbon-carbon bonds22,23,24,25.
In the present study, a series of derivatives with a thiosemicarbazone scaffold have been synthesized via a one-pot, two-step, three-component approach and evaluated against promastigote and amastigote forms of L. major and L. tropica. Finally, in silico insights regarding compounds for future study and stability were obtained.

(A) FDA approved anti-leishmanial drugs (B) Compound drugs in clinical trials under diverse phases.

Structure of some thiosemicarbazide shiff base-containing approved drugs.
Materials and methods
Synthesis
All chemicals were acquired from Sigma-Aldrich Chemical Company and were used without further purification. Melting points were measured using an Electrothermal 9100 apparatus and were uncorrected. FT-IR spectra were recorded using a Shimadzu 8400 S spectrophotometer. The NMR spectra were obtained on a Bruker AVANCE DMX400 spectrometer, operating at 100 MHz13C-NMR) and 400 MHz1H-NMR), with DMSO as the solvent and TMS as an internal standard.
General process for the synthesis of 2-aryl-N-alkylhydrazine-1-carbothioamide (5a-p)
The mixture of hydrazine (1 mmol) and substituted isothiocyanate (1 mmol) was heated in ethanol (20 mL) in the presence of a Et3N as catalyst. After precipitate formation, aromatic aldehyde (1 mmol), sodium sulfate, and acetic acid (1 drop) were added to the mixture, which was then refluxed. The progress of the reaction was monitored by TLC. Upon completion, the mixture was allowed to cool to room temperature and then placed in an ice bath. The synthesized solid was filtered, washed with water, and re-crystallized from water/ethanol (1:1) to obtain the desired pure products.
2-(3,4-Dimethoxybenzylidene)-N-methylhydrazine-1-carbothioamide (5a)26
Yellow solid; Yield 80%; m.p. 194–195 ºC; Rf = 0.52 (n-hexane/ethyl acetate 2:1.2); FT-IR (KBr, υ) cm− 1: 3353, 3152, 3005, 2896, 1603, 1549, 1402, 1271, 1237, 1085, 1018, 639;1H-NMR (400 MHz, DMSO-d6) δ/ppm: 11.40 (1 H, s, NH), 8.43 (1 H, d, J = 4.8 Hz, CH), 7.98 (1 H, s, Ph), 7.46 (1 H, d, J = 2.0 Hz, NH), 7.20 (1 H, dd, J1 = 1.6 Hz, J2 = 2.0 Hz, Ph), 6.98 (1 H, d, J = 8.4 Hz, Ph), 3.83 (3 H, s, OCH3), 3.79 (3 H, s, OCH3), 3.04 (3 H, d, J = 4.8 Hz, CH3);13C-NMR (100 MHz, DMSO-d6) δ/ppm: 177.5, 150.2, 148.7, 141.9, 126.6, 121.5, 111.3, 108.6, 55.7, 55.6, 30.4; MS (m/z, %): 253.10 (M), 223.20 (67), 179.11 (52), 164.21 (100); Anal. Calcd for C11H15N3O2S: C, 52.15; H, 5.97; N, 16.59; S, 12.66%. Found: C, 51.76; H, 5.76; N, 16.76; S, 12.51%.
2-(4-Hydroxy-3-methoxybenzylidene)-N-methylhydrazine-1-carbothioamide (5b)27
Yellowish solid; Yield 83%; m.p. 145–146 ºC; Rf = 0.62 (n-hexane/ethyl acetate 2:1.2); FT-IR (KBr, υ) cm− 1: 3358, 3142, 3002, 1617, 1560, 1400, 1283, 1217, 1158, 1086, 617;1H-NMR (400 MHz, DMSO-d6) δ/ppm: 11.32 (1 H, s, NH), 8.57 (1 H, s, CH), 8.38 (1 H, d, J = 4.4 Hz, Ph), 7.94 (1 H, s, NH), 7.43 (1 H, dd, J1 = 2.0 Hz, J2 = 2.0 Hz, Ph), 7.10 (1 H, dd, J1 = 1.6 Hz, J2 = 2.0 Hz, Ph), 6.79 (1 H, d, J = 8.0 Hz, OH), 3.83 (3 H, s, OCH3), 3.04 (3 H, s, CH3);13C-NMR (100 MHz, DMSO-d6) δ/ppm: 177.5, 148.7, 147.6, 143.1, 125.5, 121.5, 114.9, 109.8, 55.2, 30.4; MS (m/z, %): 239.09 (M), 224.10 (48), 196.10 (35), 151.20 (100), 137.2 (64); Anal. Calcd for C10H13N3O2S: C, 50.19; H, 5.48; N, 17.56; S, 13.40%. Found: C, 50.53; H, 5.33; N, 17.37; S, 13.65%.
N-Methyl-2-(4-methylbenzylidene)hydrazine-1-carbothioamide (5c)28,29
White solid; Yield 82%; m.p. 161–162 ºC; Rf = 0.79 (n-hexane/ethyl acetate 2:1.2); FT-IR (KBr, υ) cm− 1: 3325, 3156, 3009, 1613, 1545, 1401, 1258, 1170, 1089, 1032, 630;1H-NMR (400 MHz, DMSO-d6) δ/ppm: 11.43 (1 H, s, NH), 8.48 (1 H, d, J = 4.8 Hz, CH), 8.02 (1 H, s, NH), 7.69 (2 H, d, J = 8.0 Hz, Ph), 7.23 (2 H, d, J = 8.0 Hz, Ph), 3.02 (3 H, d, J = 4.4 Hz, CH3), 2.33 (3 H, s, CH3);13C-NMR (100 MHz, DMSO-d6) δ/ppm: 177.9, 141.6, 139.1, 131.3, 128.9, 126.7, 30.5, 20.8; MS (m/z, %): 209.15 (M), 194.15 (100), 179.12 (44), 119.01 (48), 91.11 (80); Anal. Calcd for C10H13N3S: C, 57.94; H, 6.32; N, 20.27; S, 15.47%. Found: C, 57.57; H, 6.08; N, 20.46; S, 15.73%.
2-(4-Fluorobenzylidene)-N-methylhydrazine-1-carbothioamide (5d)28
White solid; Yield 92%; m.p. 184–186 ºC; Rf = 0.80 (n-hexane/ethyl acetate 2:1.2); FT-IR (KBr, υ) cm− 1: 3318, 3156, 3007, 1601, 1543, 1401, 1233, 1256, 1086, 1033, 628;1H-NMR (400 MHz, DMSO-d6) δ/ppm: 11.50 (1 H, s, NH), 8.55 (1 H, d, J = 4.4 Hz, CH), 8.03 (1 H, s, NH), 7.87 (2 H, dd, J1 = 5.6 Hz, J2 = 5.6 Hz, Ph), 7.25 (2 H, t, J = 8.8 Hz, Ph), 3.02 (3 H, s, CH3);13C-NMR (100 MHz, DMSO-d6) δ/ppm: 177.9, 163.8, 161.5, 140.3, 129.4, 115.5, 31.8; MS (m/z, %): 211.15 (M), 196.15 (32), 180.20 (100), 139.20 (30); Anal. Calcd for C9H10FN3S: C, 51.17; H, 4.77; N, 19.89; S, 15.18%. Found: C, 51.55; H, 6.08; N, 20.46; S, 15.73%.
2-(4-Bromobenzylidene)-N-methylhydrazine-1-carbothioamide (5e)30
White solid.; Yield 81%; m.p. 210–211 ºC; Rf = 0.88 (n-hexane/ethyl acetate 2:1.2); FT-IR (KBr, υ) cm− 1: 3341, 3154, 3006, 2882, 1588, 1542, 1482, 1397, 1266, 1161, 1087, 1032, 622;1H-NMR (400 MHz, DMSO-d6) δ/ppm: 11.55 (1 H, s, NH), 8.58 (1 H, d, J = 4.4 Hz, CH), 8.01 (1 H, s, NH), 7.77 (2 H, d, J = 8.4 Hz, Ph), 7.61 (2 H, d, J = 8.4 Hz, Ph), 3.02 (3 H, d, J = 4.4 Hz, CH3);13C-NMR (100 MHz, DMSO-d6) δ/ppm: 177.5, 140.2, 133.6, 131.5, 128.8, 122.4, 30.7; MS (m/z, %): 273 [M + 2], 271 [M], 199 (35), 120 (40), 88 (100); Anal. Calcd for C9H10BrN3S: C, 39.72; H, 3.70; N, 15.44; S, 11.78%. Found: C, 40.03; H, 3.99; N, 15.23; S, 11.60%.
2-(4-Nitrobenzylidene)-N-methylhydrazine-1-carbothioamide (5f)31,32
Yellow solid.; Yield 77%; m.p. 247–249 ºC; Rf = 0.80 (n-hexane/ethyl acetate 2:1.2); FT-IR (KBr, υ) cm− 1: 3374, 3144, 3005, 1608, 1511, 1400, 1340, 1252, 1096, 1043, 624;1H-NMR (400 MHz, DMSO-d6) δ/ppm: 11.79 (1 H, s, NH), 8.77 (1 H, d, J = 4.4 Hz, CH), 8.25 (2 H, d, J = 9.2 Hz, Ph), 8.13 (1 H, s, NH), 8.09 (2 H, d, J = 8.8 Hz, Ph), 3.04 (3 H, d, J = 4.8 Hz, CH3);13C-NMR (100 MHz, DMSO-d6) δ/ppm: 176.8, 151.1, 142.3, 128.2, 121.4, 111.3, 30.5; MS (m/z, %): 238.20 [M], 207.21 (41), 152.20 (100), 120.25 (72), 91.20 (40); Anal. Calcd for C9H10N4O2S: C, 45.37; H, 4.23; N, 23.51; S, 13.46%. Found: C, 45.04; H, 4.35; N, 22.99; S, 13.63%.
N-Methyl-2-(4-methoxybenzylidene)hydrazine-1-carbothioamide (5g)28
Yellow solid.; Yield 83%; m.p. 192–193 ºC; Rf = 0.75 (n-hexane/ethyl acetate 2:1.2); FT-IR (KBr, υ) cm− 1: 3320, 3158, 3010, 2987, 1602, 1509, 1401, 1254, 1166, 1083, 1033, 632;1H-NMR (400 MHz, DMSO-d6) δ/ppm: 11.50 (1 H, s, NH), 8.55 (1 H, s, CH), 8.04 (1 H, d, J = 2.8 Hz, NH), 7.89–7.87 (2 H, m, Ph), 7.29–7.24 (2 H, m, Ph), 3.84 (3 H, s, OCH3), 3.02 (3 H, d, J = 2.8 Hz, CH3);13C-NMR (100 MHz, DMSO-d6) δ/ppm: 177.8, 147.1, 140.8, 138.9, 127.7, 123.5, 53.9, 30.5; MS (m/z, %): 239.20 [M], 208.10 (45), 196.10 (67), 91.20 (100); Anal. Calcd for C9H13N3OS: C, 53.79; H, 5.87; N, 18.82; S, 14.36%. Found: C, 53.45; H, 6.15; N, 18.71; S, 14.55%.
N-Methyl-2-(4-N, N-dimethylbenzylidene)hydrazine-1-carbothioamide (5h)33
Green solid.; Yield 70%; m.p. 240–241 ºC; Rf = 0.78 (n-hexane/ethyl acetate 2:1.2); FT-IR (KBr, υ) cm− 1: 3328, 3147, 2996, 2871, 1594, 1521, 1408, 1266, 1172, 1080, 1036, 618;1H-NMR (400 MHz, DMSO-d6) δ/ppm: 11.22 (1 H, s, NH), 8.30 (1 H, d, J = 4.4 Hz, CH), 7.93 (1 H, s, NH), 7.59 (2 H, d, J = 8.8 Hz, Ph), 6.71 (2 H, d, J = 9.2 Hz, Ph), 3.01 (3 H, d, J = 4.8 Hz, CH3), 2.97 (6 H, s, 2 × CH3);13C-NMR (100 MHz, DMSO-d6) δ/ppm: 177.2, 151.3, 142.5, 128.5, 121.1, 110.9, 42.6, 30.4; MS (m/z, %): 224.20 [M], 193.10 (48), 180.20 (36), 120.05 (30), 91.15 (100), 60.10 (40); Anal. Calcd for C11H16N4S: C, 55.90; H, 6.82; N, 23.71; S, 13.57%. Found: C, 55.59; H, 6.66; N, 23.99; S, 13.86%.
2-(4-Fluorobenzylidene)-N-nitrohydrazine-1-carbothioamide (5i)
White solid.; Yield 80%; m.p. 158–159 ºC; Rf = 0.25 (n-hexane/ethyl acetate 2:0.5); FT-IR (KBr, υ) cm− 1: 3318, 3157, 3003, 2984, 1600, 1506, 1403, 1234, 1066, 633;1H-NMR (400 MHz, DMSO-d6) δ/ppm: 11.55 (1 H, s, NH), 8.60 (1 H, d, J = 4.4 Hz, CH), 8.09 (1 H, s, NH), 7.92 (2 H, dd, J1 = 5.6 Hz, J2 = 5.6 Hz, Ph), 7.32 (2 H, t, J = 8.8 Hz, Ph);13C-NMR (100 MHz, DMSO-d6) δ/ppm: 175.3, 143.4, 136.7, 134.5, 131.8, 125.4; MS (m/z, %): 242.10 [M], 196.03 (59), 182.10 (62), 137.16 (100), 119.04 (46); Anal. Calcd for C8H7FN4O2S: C, 39.67; H, 2.91; N, 23.13; S, 13.24%. Found: C, 40.04; H, 3.05; N, 22.88; S, 13.14%.
2-(4-Bromobenzylidene)-N-nitrohydrazine-1-carbothioamide (5j)
White solid.; Yield 82%; m.p. 200–201 ºC; Rf = 0.25 (n-hexane/ethyl acetate 2:0.6); FT-IR (KBr, υ) cm− 1: 3341, 3154, 3005, 2968, 1585, 1542, 1481, 1266, 1162, 1064, 1032, 622;1H-NMR (400 MHz, DMSO-d6) δ/ppm: 11.60 (1 H, s, NH), 8.63 (1 H, d, J = 4.4 Hz, CH), 8.06 (1 H, s, NH), 7.82 (2 H, d, J = 8.4 Hz, Ph), 7.66 (2 H, d, J = 8.4 Hz, Ph);13C-NMR (100 MHz, DMSO-d6) δ/ppm: 177.8, 140.2, 133.1, 131.5, 128.6, 122.4; MS (m/z, %): 305.15 [M + 2], 303.07 [M], 271.10 (95), 199.01 (35), 120.15 (48), 88.05 (100); Anal. Calcd for C8H7BrN4O2S: C, 31.70; H, 2.33; N, 18.48; S, 10.58%. Found: C, 32.04; H, 2.50; N, 18.22; S, 10.45%.
2-(3,4-Dimethoxybenzylidene)-N-nitrohydrazine-1-carbothioamide (5k)
White solid; Yield 91%; m.p. 198–199 ºC; Rf = 0.37 (n-hexane/ethyl acetate 2:1.5); FT-IR (KBr, υ) cm− 1: 3353, 3151, 3004, 2971, 1602, 1549, 1401, 1270, 1237, 1085, 1018, 639;1H-NMR (400 MHz, DMSO-d6) δ/ppm: 11.40 (1 H, s, NH), 8.43 (1 H, J = 4.4 Hz, CH), 7.98 (1 H, s, NH), 7.46 (1 H, d, J = 1.6 Hz, Ph), 7.20 (1 H, dd, J1 = 2.0 Hz, J2 = 1.6 Hz, Ph), 6.98 (1 H, d, J = 8.0 Hz, Ph), 3.83 (3 H, s, OCH3), 3.79 (3 H, s, OCH3);13C-NMR (100 MHz, DMSO-d6) δ/ppm: 177.4, 150.3, 148.8, 141.6, 126.5, 121.4, 111.1, 108.3, 55.3, 55.2; MS (m/z, %): 284.15 [M], 255.17 (100), 221.10 (37), 179.18 (65), 119.15 (45); Anal. Calcd for C10H12N4O4S: C, 42.25; H, 4.25; N, 19.71; S, 11.28%. Found: C, 42.62; H, 4.12; N, 19.96; S, 11.16%.
2-(4-Methylbenzylidene)-N-nitrohydrazine-1-carbothioamide (5l)
White solid.; Yield 88%; m.p. 188–189 ºC; Rf = 0.35 (n-hexane/ethyl acetate 2:1.5); FT-IR (KBr, υ) cm− 1: 3325, 3157, 3006, 2987, 1605, 1545, 1402, 1256, 1174, 1088, 1032, 629;1H-NMR (400 MHz, DMSO-d6) δ/ppm: 11.44 (1 H, s, NH), 8.48 (1 H, d, J = 4.8 Hz, CH), 8.02 (1 H, s, NH), 7.69 (2 H, d, J = 8.0 Hz, Ph), 7.23 (2 H, d, J = 8.0 Hz, Ph), 3.02 (3 H, d, J = 4.4 Hz, CH3);13C-NMR (100 MHz, DMSO-d6) δ/ppm: 177.8, 142.6, 138.5, 132.7, 128.2, 126.5, 30.7; MS (m/z, %): 338.10 [M], 192.10 (54), 133.12 (100), 119.15 (67); Anal. Calcd for C9H10N4O2S: C, 45.37; H, 4.23; N, 23.51; S, 13.46%. Found: C, 45.05; H, 4.13; N, 23.23; S, 11.72%.
2-(4-Methoxybenzylidene)-N-(pyridin-3-yl)hydrazine-1-carbothioamide (5m)
White solid.; Yield 85%; m.p. 154–155 ºC; Rf = 0.62 (n-hexane/ethyl acetate 1:1.5); FT-IR (KBr, υ) cm− 1: 3395, 3159, 3008, 2975, 1588, 1533, 1401, 1215, 1182, 1048, 1029, 630;1H-NMR (400 MHz, DMSO-d6) δ/ppm: 11.98 (1 H, s, NH), 10.09 (1 H, s, br, CH), 8.74 (1 H, s, NH), 8.40 (2 H, d, J = 4.0 Hz, Ph), 8.05 (3 H, brs, Ph), 7.46 (3 H, dd, J1 = 4.8 Hz, J2 = 4.8 Hz, Ph), 3.42 (3 H, brs, OCH3);13C-NMR (100 MHz, DMSO-d6) δ/ppm: 177.8, 146.5, 144.7, 141.8, 139.7, 137.6, 128.1, 123.4, 115.2, 111.3, 111.1, 48.2; MS (m/z, %): 285.05 [M] (100), 271.15 (38), 185.15 (68), 153.05 (71), 89.14 (15); Anal. Calcd for C14H14N4OS: C, 58.72; H, 4.93; N, 19.57; S, 11.20%. Found: C, 58.42; H, 5.02; N, 19.36; S, 11.37%.
2-(4-Bromobenzylidene)-N-(pyridin-3-yl)hydrazine-1-carbothioamide (5n)
White solid.; Yield 80%; m.p. 157–158 ºC; Rf = 0.75 (n-hexane/ethyl acetate 1:0.25); FT-IR (KBr, υ) cm− 1: 3416, 3134, 3005, 2991, 1633, 1502, 1400, 1212, 1180, 1083, 1025, 630;1H-NMR (400 MHz, DMSO-d6) δ/ppm: 11.86 (1 H, s, NH), 9.85 (1 H, s, CH), 8.28 (1 H, d, J = 2.4 Hz, NH), 8.21 (1 H, dd, J1 = 1.2 Hz, J2 = 1.2 Hz, Ph), 7.77 (1 H, s, Ph), 7.62–7.56 (3 H, m, Ph), 7.03 (1 H, dd, J1 = 4.8 Hz, J2 = 4.8 Hz, Ph), 6.87 (2 H, t, J = 8.8 Hz, Ph);13C-NMR (100 MHz, DMSO-d6) δ/ppm: 192.2, 168.9, 141.8, 141.5, 138.1, 135.5, 132.1, 131.4, 130.5, 123.2, 122.6; MS (m/z, %): 337.05 [M + 2], 335.17 [M], 273.10 (98), 271.10 (95), 199.05 (45), 120.10 (52), 88.18 (100); Anal. Calcd for C13H11BrN4S: C, 46.58; H, 3.31; N, 16.71; S, 9.57%. Found: C, 46.22; H, 3.19; N, 16.45; S, 9.38%.
2-(3,4-Dimethoxybenzylidene)-N-(pyridin-3-yl)hydrazine-1-carbothioamide (5o)
Light yellow solid.; Yield 91%; m.p. 175–176 ºC; Rf = 0.25 (n-hexane/ethyl acetate 1:2); FT-IR (KBr, υ) cm− 1: 3337, 3136, 2996, 1600, 1510, 1400, 1222, 1143, 1024, 625;1H-NMR (400 MHz, DMSO-d6) δ/ppm: 11.87 (1 H, s, NH), 10.07 (1 H, s, CH), 8.61–8.63 (1 H, m, NH), 8.33–8.34 (1 H, m, Ph), 8.04–8.11 (1 H, m, Ph), 7.92–7.94 (1 H, m, Ph), 7.49 (1 H, d, J = 2.0 Hz, Ph), 7.28–7.37 (1 H, m, Ph), 7.23 (1 H, dd, J1 = 1.6, J2 = 1.6 Hz, Ph), 6.93 (1 H, d, J = 8.0 Hz, Ph), 3.76 (3 H, s, OCH3), 3.73 (3 H, s, OCH3);13C-NMR (100 MHz, DMSO-d6) δ/ppm: 176.2, 150.5, 149.1, 147.3, 145.8, 143.5, 135.6, 133.1, 125.7, 122.6, 121.8, 111.2, 108.6, 54.9, 54.7; MS (m/z, %): 316.10 [M], 288.15 (81), 223.10 (24), 183.16 (100), 152.05 (67); Anal. Calcd for C15H16N4O2S: C, 56.94; H, 5.10; N, 17.71; S, 10.14%. Found: C, 57.28; H, 4.95; N, 17.45; S, 9.94%.
2-(4-Fluorobenzylidene)-N-(pyridin-3-yl)hydrazine-1-carbothioamide (5p)
Yellow solid.; Yield 82%; m.p. 200–201 ºC; Rf = 0.75 (n-hexane/ethyl acetate 1:0.25); FT-IR (KBr, υ) cm− 1: 3268, 3149, 3000, 2984, 1601, 1505, 1400, 1230, 1153, 1083, 1042, 627;1H-NMR (400 MHz, DMSO-d6) δ/ppm: 12.04 (1 H, s, NH), 10.27 (1 H, s, CH), 8.68 (1 H, d, J = 2.4 Hz, NH), 8.40 (1 H, dd, J1 = 1.2 Hz, J2 = 1.2 Hz, Ph), 8.17 (1 H, s, Ph), 8.02–7.96 (3 H, m, Ph), 7.43 (1 H, dd, J1 = 4.8 Hz, J2 = 4.8 Hz, Ph), 7.29 (2 H, t, J = 8.8 Hz, Ph);13C-NMR (100 MHz, DMSO-d6) δ/ppm: 176.6, 161.9, 147.3, 141.8, 135.5, 133.4, 130.5, 129.7, 129.6, 122.8, 115.2; MS (m/z, %): 274.12 [M], 182.13 (100), 152.17 (49), 137.05 (30); Anal. Calcd for C13H11FN4S: C, 56.92; H, 4.04; N, 20.42; S, 11.69%. Found: C, 56.61; H, 4.13; N, 20.17; S, 11.93%.
In vitro anti-leishmanial assay
Reagents
Novy-MacNeal-Nicolle (NNN) (Brain-Heart Blood Agar, Luria Broth, Merck, Germany) and Roswell Park Memorial Institute (RPMI 1640) mediums, fetal bovine serum (FBS), and Pen-Strep (penicillin 100 U/mL)/streptomycin 100 µg/mL) (Gibco, Germany), DMSO, tetrazolium salt (MTT, Sigma-Aldrich), and FITC Annexin V Apoptosis Detection Kit (Mab Tag, Germany) were procured and stored at the recommended temperatures until testing. All synthesized derivatives (5a-p) were initially dissolved/dispersed in DMSO (Sigma-Aldrich) to prepare a stock solution of 5mg/mL. The final concentration of DMSO was maintained below 1%, which does not exhibit harmful effects on the parasites34,35. Derivative dilutions were prepared in PBS to evaluate the impact of serial concentrations of 3.12–1000 µM on the macrophage, amastigote, and promastigote cells36,37,38. Amphotericin B 50 mg, Cipla (AmpB), and meglumine antimoniate (Glucantime®, Sanofi, 1.5 5g/mL ampules) (GLU) were used as positive control drugs in promastigote and amastigote forms, respectively.
Cultivation of Leishmania spp.
The standard Iranian strains of L. tropica (MRHO/IR/02/Mash10) and L. major (MRHO/IR/75/ER) were kindly provided by Dr. Ghaffarifar, Department of Parasitology, Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran. Promastigotes were cultured in NNN medium and subsequently sub-cultured in RPMI 1640 enriched with 20% heat-inactivated FBS and 1% Pen-Strep, including streptomycin (100 µg/mL) and penicillin (200 IU/mL), at 18–24 °C36,37.
J774 A.1 macrophage cell culture
J774A.1, the murine macrophage cell line, was bought from the Pasteur Institute, Iran. The cells were cultured in RPMI 1640 supplemented with 1% penicillin-streptomycin and 12% FBS in a humidified atmosphere at 37 °C and 5% CO2. Upon reaching confluency, fresh media was added to the flasks36,37,38.
Anti-leishmanial activity
Anti-promastigote effects
Parasite counting method
The number of viable promastigotes of L. major and L. tropica was counted using the microscopic counting method to evaluate the effects of different concentrations of various compounds36,38,39,40. To do this, 100 µL of RPMI 1640 medium with 1 × 106 promastigotes/mL during the growth phase was placed into 96-well culture plates, and then different amounts (3.12–1000 µM) of each compound were added for 24, 48, and 72 h at 24 °C. Further, the number of fixed parasites was directly counted in a Neubauer chamber at 400x magnification under the light microscope for three consecutive days. Each experiment was performed in triplicate. The positive control, AmB, 0.93 µM, was produced in sterile PBS according to the manufacturer’s instructions. A negative control, CTRL (-), was defined as three wells in each plate that did not contain any compounds.
MTT assay for promastigote viability
The MTT reduction method (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was applied to investigate cellular viability. In this assay, 100 µL of RPMI 1640 containing 1 × 106/mL promastigotes at the logarithmic phase was cultivated in each of the 96-well ELISA plates. Then, the parasites were exposed to different concentrations of compounds (3.12 to 1000 µM). The remaining experimental procedures followed the previously described protocols35,36. The percentage of promastigote viability was calculated using the following formula: 100 × (absorbance of treated cells-absorbance of the blank/absorbance of control cells-absorbance of the blank), while the alive promastigotes were postponed in PBS without any drug, and the environment without promastigote was applied as a negative control, CTRL-(-), and blank, respectively. The 50% inhibition concentration (IC50) of all compounds was evaluated as the 50% decrease in cell viability of treated cells compared to untreated cells36,37.
Anti-intracellular amastigote activity
For evaluation of the anti-leishmanicidal activity against L. major and L. tropica intracellular amastigotes, 200 µL of 105 J774A.1 macrophage was cultured in each well of 12-well plates that had small round glass coverslips at the bottom and was incubated for 24 h at 37 °C, 5% CO2. After incubation, the non-adhered cells were removed by washing with sterile PBS. Then, 200 µL of metacyclic promastigotes (1 × 106) at the stationary phase (10:1) were added to the adherent macrophages to infect macrophage cells. After 24 h, the infected cells were incubated with different concentrations of compounds (1.32–1000 µM) for 72 h at 37 °C, 5% CO2, and 95% relative humidity. Following incubation, cells were washed with PBS, fixed with methanol, and Giemsa stained. The number of infected macrophages and intracellular amastigotes (Leishman bodies) were counted under an optical microscope. The number of amastigotes in 100 macrophage cells was used to calculate the IC50. Each experiment was performed in triplicate. GLU 14.61 µM was used as the positive control36,37.
Maintenance macrophages or macrophage cytotoxicity assay
The primary and major host cells for Leishmania parasites are macrophages. The J774A.1 murine macrophage cell line was applied to evaluate the toxicity of compounds through the MTT colorimetric assay. Macrophage cells were cultured in a 96-well plate at a density of 1 × 106 cells/well and incubated at 37 °C in a 5% CO2 atmosphere. After 24 h, the supernatant containing non-adhered cells was removed, and then in the presence of numerous concentrations of the derivatives (1.32–1000 µM), the cells were incubated for another 72 h. The optical density of solubilized formazan crystals was evaluated using an ELISA reader (Bio-Rad, USA) at a wavelength of 570 nm. Each experiment was performed in biological triplicates. The 50% cytotoxic concentration (CC50) of various compounds was determined as the 50% decrease in macrophage cell viability of treated cells compared to untreated cells. The selectivity index (SI) is the ratio of the CC50 of macrophage cells to the IC50 of amastigotes36,37.
Flow cytometry assay
The Annexin V Apoptosis Detection Kit (Mab Tag, Germany) was used to assess apoptosis induction in promastigotes exposed to IC50 concentrations of some effective compounds. Briefly, 1 × 106 promastigotes at the logarithmic stage, treated with IC50 concentrations of the compounds, were harvested, washed with a cold sterile PBS solution, and centrifuged for 15min at 1000 g. Subsequently, 5 µL each of Annexin-V and propidium iodide (PI), along with 400 µL of binding buffer, were added to cell pellets. The promastigotes were incubated at 25 °C in a dark place for 15min. The test was read using CyFlow Space flow cytometry (Sysmex-Partec, USA), and data were analyzed by FlowJo TM software (Vancouver, BC, version 10.5.3). All tests were accomplished in triplicate, and the percentage of necrosis and apoptosis was investigated for each tested compound36,40.
Statistical analysis
The mean and standard deviation (SD) were calculated for all triplicate experiments. Based on the mean percentage viability of promastigotes, amastigotes, and macrophage cells, the IC50 values for promastigotes and amastigotes, and CC50 values for J774A.1 macrophages, were determined. GraphPad Prism software was used to evaluate the results and analyze them. Repeated measures ANOVA was employed for the parasite counting method results analysis. Additionally, the one-way ANOVA test followed by Tukey’s post hoc test was applied to compare the IC50 and CC50 values of derivatives. The ratio of CC50 and IC50 of amastigotes was shown by the SI.
Computational studies
In silico pharmacokinetic prediction and physicochemical properties
The ADMETLab3.0 web server (https://admetlab3.scbdd.com) was used to evaluate the compounds’ drug-likeness and ADMET properties. ADMETlab 3.0 provides a comprehensive and efficient platform for assessing physicochemical properties and ADMET-related parameters in the early stages of drug discovery.
In silico metabolism of compounds in the presence of cytochrome P450
The SMARTCyp 3.0 online server (https://smartcyp.sund.ku.dk/mol_to_som) was used to predict the sites of metabolism of the derivatives by the cytochrome isoforms CYP3A4, CYP2D6, and CYP2C9.
In silico cardiotoxicity prediction
The Pred-hERG web server (http://www.labmol.com.br) was used to predict the cardiotoxicity of the tested derivatives. This method estimates the potential for hERG channel inhibition, which is associated with cardiotoxic effects, with an accuracy, sensitivity, and specificity of approximately 89–90%.
Molecular docking study
The molecular docking study of the derivatives into the L. major Pteridine reductase 1 (LmPRT1) active site was performed using Autodock 4.2 software41,42. The crystal structure of LmPRT1 (PDB ID: 5L42) was retrieved from the PDB. The docking protocol followed the procedure described in a previous report43. Before docking, crystallized water molecules and bound ligands were removed from the protein structure. Kollman charges and polar hydrogen atoms were then added. The structures of derivatives were prepared and energetically minimized in HyperChem 8.0 software by the MM+ method. Then, the Gasteiger charges were added to compounds. Lastly, the pdbqt files of the enzyme and compounds, which were applied in docking, were provided with AutoDock tools.
Molecular dynamics simulations study
The GROMACS 2022.6 computational software performed molecular dynamics (MD) simulations. The method was done by the MD technique from the previous work44. The Acpype tool was used to generate the topology files and other force field parameters for the selected compounds. Before energy minimization, water molecules were represented using a tip3p model and approximately 11,864 water molecules were present in the system. Appropriate amounts of counter-ions were added by replacing water molecules to ensure the overall charge neutrality of the simulated system that was comprised of LmPTR1, ligand, and water molecules. Amber99sb force field was used for the determination of all bonding and nonbonding interactions. Steepest descent followed by conjugate gradient algorithms was used for energy minimization of the protein-ligand complex. After the energy minimization process, the position restraint procedure was performed in association with NVT and NPT ensembles. An NVT ensemble was adopted at a constant temperature of 300 K with a coupling constant of 0.1 ps and a time duration of 500 ps. After temperature stabilization, an NPT ensemble was performed. In this phase, a constant pressure of 1 bar was employed with a coupling constant of 5.0 ps and a time duration of 1000 ps. The pressure was maintained at 1 bar with the Parrinello-Rahman barostat. The Particle Mesh Ewald, Lincs algorithm, and Lennard-Jones potential were used to treat the long-range electrostatic, covalent bond constraints, and van der Waals interactions, respectively. Finally, a 100 ns MD simulation was performed with monitoring of equilibration by examining the stability of the energy, temperature, and density of the system as well as the root-mean-squared deviations (RMSDs) of the backbone atoms.
Results and discussions
Rationale and design
Chemically, thiosemicarbazones are intermediates in the synthesis of 2-amino-1,3,4-thiadiazoles, and they reside in solution at an equilibrium between the ring-closed and open-chain forms (ring-chain tautomerism)45,46. The thiosemicarbazone analogues were previously recognized in the anti-parasitic compounds. Thiosemicarbazones were initially discovered as inhibitors of parasitic cysteine proteases, such as rodhesain in T. brucei and cruzain in T. cruzi. Some members of this group of derivatives were inactive on the enzyme but active on parasites47,48. Several studies have explored the anti-leishmanial activity of thiosemicarbazones. For instance, Eldehna et al.. reported the synthesis of arylnicotinic acids conjugated with arylthiosemicarbazides and evaluated their anti-leishmanial properties. Their results showed promising activity, particularly for thiosemicarbazide derivatives, which outperformed the semicarbazide counterparts. This enhanced activity may be attributed to the greater lipophilicity of thiosemicarbazides, facilitating their penetration through parasitic membranes and improving target accessibility. This hypothesis is further supported by the higher CLogP values observed for thiosemicarbazide derivatives (Compound 1, Fig. 3)49. Fonseca et al. developed sugar analogues of thiosemicarbazones that inhibited parasite cysteine proteases. Structural modifications, such as the removal or substitution of the thiosemicarbazone moiety with semicarbazone, led to loss of activity, while alterations to the sugar component also reduced potency. These compounds showed efficacy against S. mansoni, T. brucei, and T. cruzi (Compound 2, Fig. 3)50. Moreira et al. described a series of aryl thiosemicarbazones against cruzain and anti-T. cruzi inhibitors. According to findings, thiosemicarbazones were introduced as selective and potent anti-T. cruzi agents. Anti-T. cruzi activity was shown to be reliant on the nature of the substituent used, whereas the substituent position in the phenyl ring had less significance for activity (Compound 3, Fig. 3)51. Linciano et al. identified several thiosemicarbazone derivatives with activity against intracellular amastigotes of T. cruzi, L. infantum, and T. brucei. Their findings supported the use of thiosemicarbazones as a viable scaffold for anti-parasitic drug development (Compound 4, Fig. 3)48. Manzano et al.. recognized some thiosemicarbazones with good activity against L. donovani amastigotes, with the most active compound showing an EC50 of 0.8 µM and minimal toxicity on two diverse mammalian cell lines (Compound 5, Fig. 3)52. Tenório et al. examined thiosemicarbazones substituted with nitro groups at the arylhydrazone moiety for their anti-Toxoplasma gondii activity. These compounds demonstrated efficacy against T. gondii without inducing morphological changes in host cells (Compound 6, Fig. 3)53. Thiosemicarbazones, on the other hand, are well-known for their wide range of biological activities, among which anti-leishmanial potential stands out (Compounds 7–14, Fig. 3)53,54,55,56,57,58,59,60.

Design plan of thiosemicarbazone analogues as leishmanial agents.
According to previous studies, the structure-activity relationships (SAR) of thiosemicarbazone derivatives against leishmania can be summarized as follows: Halogen substituents, such as chlorine and bromine were well tolerated at the meta-position on the phenyl ring. Furthermore, modification of the methyl substituent at the ortho-position had only a minor effect. The presence of sulfur and methine bonds was found to be critical for biological activity. Moreover, the incorporation of bulky substituents on the side chain was generally associated with a reduction in efficacy24,61. The introduction of fluorine atoms on the phenyl or benzyl group attached to the N-iminothiourea moiety had a positive influence on the anti-leishmanial activity. This effect is likely attributed to the increased lipophilicity of the C-F bond compared to the C-H bond, despite their similar atomic sizes60. Based on the results, the hydrophobic character of the phenyl moiety plays a significant role in optimizing anti-leishmanial activity57. Furthermore, the SAR findings have shown that the activity of thiosemicarbazone derivatives largely depends on the electronic nature of the substituents attached to the NH group of the thiosemicarbazone core and the phenyl ring55. The present research has synthesized thiosemicarbazone analogues with electron donor substituents, electron-withdrawing substituents, and heteroaromatic rings at the NH of the thiosemicarbazone moiety and different groups at the para- and/or meta-phenyl rings. These analogues are currently being evaluated against Leishmania parasites, marking the first report of such compounds in this context. Additionally, in-silico studies were conducted to support the structure-based design. Previous research served as the foundation for the current study’s design rationale (Fig. 3).
Synthesis
We have previously reported the synthesis as well as the antioxidant, anti-cancer, and anti-bacterial activities of a series of substituted 3-benzylidene-2-imino-4-phenyl-3H-thiazole62. In this study, we reported a one-pot, two-step, three-component protocol for the synthesis of thiosemicarbazone analogues, starting from hydrazine, substituted benzaldehyde, and various isothiocyanates. The reaction sequence begins with the nucleophilic attack of hydrazine on the substituted isothiocyanate, forming a thiosemicarbazone intermediate. This is followed by a condensation reaction between the resulting intermediate and an aromatic aldehyde, leading to the formation of the final thiosemicarbazone analogues. Pyridine-3-isothiocyanate, methyl isothiocyanate, and nitro-isothiocyanate were used as starting materials in the synthesis. The preparation of the thiosemicarbazone analogues (5a-p) is illustrated in Scheme 1, and their chemical structures are presented in Fig. 4.

Synthesis of thiosemicarbazone analogues (5a-p).

Structural synthesized thiosemicarbazone analogues (5a-p).
Spectroscopic techniques were employed to identify and confirm the structures of all synthesized compounds (FT-IR1, H-NMR13, C-NMR, as well as mass and elemental analysis).
The FT-IR spectra of compounds (5a-p) showed NH stretching bonds in the range of 3416 –3318 cm− 1 and 3159 − 3134 cm− 1. The -C = N stretching vibrations appeared between 1633 –1585 cm− 1, while aromatic C = C bonds were observed in the ranges of 1560 –1503 cm− 1 and 1481 − 1400 cm− 1. The C = S stretching was identified between 1089 –1048 cm− 1, whereas C-N and C-S stretching bonds were determined at 1237 –1156 cm− 1 and 639 − 617 cm− 1, respectively. In the ¹H-NMR spectra, NH protons of the carbothioamide group appeared as distinct signals between 12.04 and 11.22 and 8.68–7.93 ppm. The imine proton resonated in the range of 10.27–8.30 ppm, and aromatic hydrogen was observed between 6.7 and 8.4 ppm. Peaks belonging to methoxy and methyl proton bonds were detected at 3.84–3.42 and 3.04–2.33 ppm, respectively. The ¹³C-NMR spectra exhibited thiourea carbon (C = S) resonances between 177.8 and 175.3 ppm, and the imine carbon appeared between 140.2 and 148.8 ppm. Aromatic carbon atoms resonated within the range of 150.3-125.6 ppm. Peaks belonging to methoxy and methyl carbons were observed in the range of 20.8-115.5 ppm.
The plausible mechanism for the one-pot, two-step, and three-component synthesis of thiosemicarbazone Schiff base derivatives (5a-p) is illustrated in Scheme 2. In step I of the reaction, hydrazine reacts with a substituted isothiocyanate to form a thiosemicarbazone intermediate 3. Subsequently, in step II, this intermediate undergoes condensation with a substituted benzaldehyde to initially generate a carbinolamine intermediate. Under acidic conditions, the carbinolamine undergoes dehydration, eliminating a water molecule to afford the final thiosemicarbazone Schiff base analogues.

Plausible mechanism for the formation of thiosemicarbazone Schiff base 5a-p.
The efficiency and yield of the synthesis of these compounds were primarily influenced by several key factors: electronic effects of substituent groups (electron-withdrawing or electron-donating), steric effects, hydrogen bonding ability, and nature of the heterocyclic ring substituent. Based on these factors, compounds having methoxy at the para-phenyl ring and especially two methoxy at the para- and meta-phenyl ring showed the highest increases in yield. This enhancement is attributed to the strong resonance electron-donating nature of methoxy groups, which stabilizes intermediates and facilitates the condensation reaction. Similarly, compounds with methyl at the para-phenyl ring also provide high yields but less than methoxy substituent because the methyl group is an inductive electron-donating substituent. In contrast, compounds bearing electron-withdrawing substituents such as fluorine or bromine at the para-phenyl ring tend to lower yields, especially when strong resonance electron-donating substituents are absent. Beyond aromatic substitution, the nucleophilicity of the hydrazine derivative significantly influenced reaction efficiency. N-methylhydrazine generally exhibits good nucleophilicity with higher yields, while N-nitrohydrazine has weaker nucleophilicity but strong electron-donating substituents on the aromatic ring (like dimethoxy at the para- and meta-phenyl ring) can compensate and maintain high yields. N-(pyridin-3-yl), due to its heterocyclic and electron-withdrawing nature, showed lower nucleophilicity, yet still provided moderate yields when combined with potent resonance donors. Steric effects also played a crucial role. Bulkier substituents, such as bromine atom, introduced steric hindrance that modestly decreased yield. In contrast, smaller substituents like fluorine and methyl cause less steric hindrance. Overall, the highest yields were achieved when a favorable balance of electronic effects (resonance and inductive), nucleophilicity, and steric hindrance was attained. In general, compounds with strong resonance donors and synthesized from highly nucleophilic hydrazines demonstrated the greatest synthetic efficiency (Table 1).
Anti-leishmanial activity
Anti-promastigote effects
The average number of L. major and L.tropica promastigotes at prearranged concentrations was counted in the presence and absence of all analogues and AmB for 24, 48, and 72 h at 24 °C (Table 2). Among the tested derivatives, compounds 5g, 5b, 5a, 5e, and 5f demonstrated a significant reduction in promastigote count relative to both the untreated control and less active derivatives such as 5p, 5i, and 5l (p < 0.001). However, these active compounds did not exhibit statistically significant differences in comparison with AmB as a positive control (p > 0.05), suggesting a comparable anti-promastigote effect. The results also revealed that the proliferation profile followed a dose-dependent response. The impact was more intense at higher concentrations (p < 0.05). On the other hand, 1000, 800, 400, and 6.12 µM concentrations showed the highest and lowest efficacies, respectively, in inhibiting the growth of L. major and L. tropica promastigotes. Notably, compounds 5c and 5h showed only mild and/or no effect, respectively, on both L. major and L. tropica promastigotes after 72 h. Similarly, compound 5j showed a mild effect, while 5k-m, 5n, and 5o demonstrated limited activity, with no statistically significant difference compared to the negative and positive controls (p > 0.05).
The metabolic activity of promastigotes and cytotoxicity of compounds were evaluated by MTT assay for 72 h of normal incubation. This colorimetric method was employed to validate the findings obtained through routine microscopic examination. Among the tested compounds, compounds 5a, 5b, 5e, 5f, and 5g exhibited notably higher activity, significantly reducing promastigote viability in both L. major and L. tropica compared to other derivatives (p < 0.001). The IC50 values in the L. major promastigotes treated with more efficient compounds, including compounds 5g, 5b, 5a, 5e, and 5f, were 26.7, 38.3, 54.3, 159.7, and 159.7 µM after 72 h, respectively. These results in L. tropica promastigotes were 101.5, 199.8, 258.0, 92.3, and 240.0 µM for compounds 5g, 5b, 5a, 5e, and 5f, respectively. The IC50 values of compounds 5p, 5i, and 5l were in the range of 31.9, 88.8, and 346.0 µM, respectively, against L. major and 313.9, 185.9, and 331.1 µM, against L. tropica promastigotes. Notably, none of the tested compounds showed a statistically significant difference in activity when compared to AmB as a reference drug (p > 0.05). This indicates that although several compounds were promising but not superior to the standard treatment.
Anti-amastigote effects
Moreover, it is well known that the clinical stage of the Leishmania genus is related to the amastigote parasite, which is the intracellular proliferation form in mammalian cells5. Therefore, the synthesized compounds at various concentrations were tested against L. tropica and L. major amastigotes. As revealed in Table 2, most of the screened compounds were notably toxic to macrophage infection and decreased the overall number of intracellular amastigotes (compounds 5g, 5b, 5a, 5d, 5e, and 5f); one possesses less anti-leishmanial activity (compound 5h), whereas compound 5c had moderate activity on reducing macrophage infection and amastigotes of both L. tropica and L. major toward the untreated control groups (P < 0.001). Among these, compounds 5g, 5b, 5a, 5e, and 5f also showed statistically significant differences in activity compared to GLU, as standard drug used in the amastigote assay (P < 0.05). The best IC50 value was observed first in 5g (18.8 µM), then 5e (31.8 µM), and 22.7 µM and 24.5 µM against L. major and L. tropica amastigotes. These results were significantly superior to GLU (P < 0.001), suggesting the strong potential of these compounds as anti-leishmanial agents (Table 2).
Among the synthesized derivatives, compounds 5p, 5i, and 5j, even at the lowest concentration (6.12 µM), reduced significantly macrophage infections and decreased the number of intracellular amastigotes compared to the positive control groups (P < 0.001). Additionally, derivatives 5o, 5n, and 5k-m remarkably reduced the number of amastigotes by up to 20–25% toward the untreated negative control (p < 0.05) (Table 1). Regarding potency, compound 5p exhibited the most effective activity against L. major amastigotes, with an IC₅₀ value of 31.2 µM, showing significant superiority over GLU (P < 0.001). In contrast, compound 5i showed the least effect on L. tropica amastigotes with an IC50 value of 185.9 µM (Table 2).
These findings also highlight a notable distinction between the parasite’s two life stages: the IC₅₀ values for intracellular amastigotes were consistently higher than those observed for promastigote forms, suggesting greater resistance of amastigotes to treatment (P < 0.001). This variation underscores the importance of targeting the clinically relevant amastigote stage in anti-leishmanial drug development.
Macrophage cytotoxicity assay
To further evaluate the safety profile of the synthesized compounds, a cytotoxicity MTT assay was conducted on uninfected J774A.1 macrophage cells, which were incubated with varying concentrations of the derivatives over 72 h (Table 2). Compounds cytotoxicity for J774A.1 macrophages and amastigotes IC50 was compared using the selectivity index (SI), the ratio between the CC50 and IC50 amastigote values. An SI ≥ 10 was considered indicative of a non-toxic and selective compound. As shown in Table 2, among the derivatives showing notable anti-leishmanial efficacy, compounds 5e-g, 5i, and 5p demonstrated low cytotoxicity toward macrophages and maintained favorable SI values, thereby indicating good selectivity toward Leishmania amastigotes. In contrast, several other derivatives exhibited either lower SI values or higher cytotoxicity. Notably, the selectivity of these active compounds was significantly better than GLU, the reference drug used in the assay (P < 0.001).
Apoptosis study
To investigate the mechanism underlying the anti-leishmanial activity of the most effective thiosemicarbazone derivatives (5a, 5b, 5e, 5f, 5g, 5i, and 5p), apoptosis induction was assessed in L. major and L. tropica promastigotes treated with their respective IC₅₀ concentrations. The results showed that these compounds displayed different early and late apoptosis as well as necrotic activities against L. major and L. tropica promastigotes. The apoptosis percent at IC50 concentrations for compounds 5a, 5b, 5e, 5f, 5g, 5i, 5p, and GLU were 36.2%, 41.5%, 37.7%, 16.19%, 52.9%, 22.14%, 42.2%, and 30.14%, respectively. All tested compounds induced more apoptotic effects than GLU (P < 0.05) except compounds 5f and 5i (P > 0.05). Compounds 5g and 5p showed higher apoptotic effects than other analogues and GLU (p < 0.001) (Fig. 5).

The necrotic and apoptotic profiles of the L. major and L. tropica promastigotes at IC50 concentrations of various syntheses compared to Glucantime as a general drug in Leishmaniasis treatment.
Structure-activity relationship studies
Anti-leishmanial activity against L. major
The thiosemicarbazone analogues (5a-p) were investigated against the amastigote and promastigote L. major. The IC50 values of the synthesized derivatives were determined and compared to the GLU and AmB drugs (Table 2).
Within the 2-(substituted benzylidene)-N-methylhydrazine-1-carbothioamide series (5a-h), electron-donating groups (EDGs) or electron-withdrawing groups (EWGs) at the para-position of benzylidene ring maintained the effect on amastigotes and promastigotes. Anti-promastigote and anti-amastigote activities were further decreased for substitution with moderately-sized EDG (-CH3 and N(CH3)2) (compounds 5c and 5h). In contrast, within the 2-(substituted benzylidene)-N-nitrohydrazine-1-carbothioamide (5i-l) and the 2-(substituted benzylidene)-N-(pyridine-3-yl)hydrazine-1-carbothioamide (5m-p), incorporation of a small, lipophilic EWG such as fluorine (compounds 5i and 5p) enhanced the anti-amastigote and anti-promastigote activities compared to other substitutions. Conversely, the introduction of EDGs (e.g., -CH3 and OCH3; compounds 5l and 5m) and/or moderately-sized EWGs like bromine (compound 5j) further reduced anti-amastigote and anti-promastigote activities.
Within the 2-(substituted benzylidene)-N-methylhydrazine-1-carbothioamide (5a-h) series, replacing OCH3 substitution in compound 5g with bromine in compound 5e decreased anti-promastigote and anti-amastigote activities. Changing moderately EWG (bromine, compound 5e) with strongly EWG (NO2 and flourine, compounds 5d and 5f) dramatically reduced activity against amastigote and promastigote forms. Notably, the presence of fluorine at the para-position of the benzylidene ring decreased activity compared to nitro at the same position, suggesting that increased volume, electron-withdrawing strength, and hydrophilicity may negatively impact leishmanicidal activity, contrary to earlier hypotheses suggesting these factors would enhance efficacy56. In contrast, a comparison of three compounds, 5c, 5g, and 5h, with EDGs at the para-position of benzylidene ring showed that compound 5g with OCH3 substitution improved anti-leishmanial activity compared to 5c (CH3) and 5h (N(CH3)2). This observation contradicts the previously assumed trend, where increased volume and lipophilicity were expected to correlate with decreased activity against L. major. Such discrepancies may be explained by the complex interplay between the electronic nature (EDG vs. EWG) of substituents and other physicochemical properties influencing the compounds’ behavior against both promastigote and amastigote forms. Two-substituted benzylidene in compound 5a showed lower anti-leishmanial activity than mono-substituted benzylidene in compound 5g. This can be due to the existence of two ortho OCH3 substitutions relative to each other, which changes the conformation of the compound compared to mono-substituted benzylidene. A comparative analysis of compounds 5a and 5b with two substituents on the benzylidene displayed that replacing the OCH3 group with OH at the para-position of benzylidene increased anti-promastigote activity while it decreased anti-amastigote activity. These findings suggest that the presence of EDG and lipophilic at the para-position benzylidene changed activity. Moreover, the potential formation of intramolecular hydrogen bonds between hydroxyl and methoxy groups may further impact the molecular conformation, thereby altering biological activity. Thus, subtle structural modifications, especially in the substitution pattern and electronic properties of the benzylidene ring, play a critical role in modulating compound efficacy against Leishmania species.
Within the 2-(substituted benzylidene)-N-nitrohydrazine-1-carbothioamide (5i-l) series, compound 5i, with fluorine at the para-position of the benzylidene ring, revealed the highest activity on promastigote and amastigote forms. A comparative analysis of three compounds, 5i (fluorine), 5j (bromine), and 5l (CH3), exhibited that an increase in EWG strength and lipophilicity positively influenced biological activity. However, two-substituted benzylidene (5k, meta- and para-OCH3) showed the lowest activity. According to the results, two-substituted benzylidene exhibited lower anti-leishmanial activity compared to mono-substituted benzylidene, which could be due to the change in the conformation of the compound.
Within the 2-(substituted benzylidene)-N-(pyridine-3-yl)hydrazine-1-carbothioamide (5m-p) series, compound 5p with fluorine at the para-position benzylidene ring revealed significantly higher anti-promastigote and anti-amastigote activities than compounds 5m (OCH3) and 5n (bromine). These results suggest that small-sized and strong EWGs increased anti-leishmanial activity. Furthermore, consistent with trends observed in other structural series, this set of compounds also demonstrated that two-substitution on the benzylidene ring markedly reduces biological activity. This reduction is presumably due to conformational changes and an increase in molecular volume, which may hinder proper interaction with the biological target or reduce membrane permeability.
Generally, two series of the 2-(substituted benzylidene)-N-nitrohydrazine-1-carbothioamide (5i-l) and the 2-(substituted benzylidene)-N-(pyridin-3-yl)hydrazine-1-carbothioamide (5m-p) showed lower anti-leishmanial activity than the 2-(substituted benzylidene)-N-methylhydrazine-1-carbothioamide (5a-h) series. This suggests that small-sized, EDG, and lipophilic substituents (e.g., CH3) at the carbothioamide moiety are more favorable for activity against both amastigote and promastigote forms of L. major than bulkier, EWG, and more hydrophilic groups (e.g., NO2 and/or pyridine).
Anti-leishmanial activity against L. tropica
The thiosemicarbazone analogues (5a-p) were investigated on amastigotes and promastigotes of L. tropica. The IC50 values of the synthesized derivatives were determined and compared to the GLU and AmB drugs (Table 2).
Within the 2-(substituted benzylidene)-N-methylhydrazine-1-carbothioamide series (5a-h), compound 5e, having bromine at the para-position of the benzylidene ring, showed the highest anti-leishmanial activity. Replacing bromine with OCH3, compound 5g, was the most potent compound after compound 5e. The existence of OCH3 somewhat reduced activity on the promastigote form while it was maintained against the amastigote form. Placing NO2, fluorine, CH3, and N(CH3)2 at the para-position of benzylidene in compounds 5f, 5d, 5c, and 5h, respectively, significantly decreased anti-promastigote and anti-amastigote activities of L. tropica. Based on findings, the presence of EWG and moderate size displayed a positive effect on anti-leishmanial effect. Besides, a comparison of two compounds, 5a and 5b, having two-substituted benzylidene showed that compound 5b with an OH group at the para-position was more potent than compound 5a with an OCH3 group at the same position. Similarly, the presence of EDG and bulk groups decreased anti-leishmanial activity.
Within the 2-(substituted benzylidene)-N-nitrohydrazine-1-carbothioamide (5i-l) series, compound 5i (fluorine) was significantly more potent than compounds 5j (bromine) and 5l (CH3). The results indicate that increases in lipophilicity, EDG, and bulkier substituents negatively impacted both anti-promastigote and anti-amastigote activities. On the other hand, the presence of two-substituents on the benzylidene ring in compound 5k decreased anti-leishmanial activity compared to mono-substituted (compounds 5i and 5l). This decrease may be attributed to the presence of the OCH3 group at the ortho-position, which likely alters the molecule’s conformation.
Within the 2-(substituted benzylidene)-N-(pyridine-3-yl)hydrazine-1-carbothioamide (5m-p) series, compound 5p (fluorine) showed higher anti-leishmanial activity than compounds 5m (OCH3) and 5n (bromine). It seems that the presence of small size, moderate lipophilicity, and strong EWG substitution at the para-position of benzylidene increased anti-leishmanial activity. Additionally, consistent with observations in the previous series, two-substituted the benzylidene ring decreased anti-leishmanial activity compared to mono-substituted (compound 5o vs. compounds 5m, 5n, and 5p).
Similar to anti-leishmanial activity observed against L. major, two series of the 2-(substituted benzylidene)-N-nitrohydrazine-1-carbothioamide (5i-l) and the 2-(substituted benzylidene)-N-(pyridine-3-yl)hydrazine-1-carbothioamide (5m-p) showed lower anti-leishmanial activity than the 2-(substituted benzylidene)-N-methylhydrazine-1-carbothioamide (5a-h) series.
Generally, all three series of derivatives exhibited higher activity against L. major than L. tropica, while the overall tendency of anti-leishmania activity against promastigote and amastigote forms was similar in both L. major and L. tropica species. Among two-substituted compounds (5a, 5k, and 5o), compound 5a with a CH3 at the hydrazinecarbothioamide moiety was significantly more potent than 5k (NO2) and 5o (pyridine). While 5k and 5o showed almost similar activity against L. major. Moreover, compound 5a showed remarkably higher activity compared to compounds 5k and 5o. Besides, compound 5k was strangely more potent than 5o against L. tropica. These findings show that the presence of CH3 (EDG) at the hydrazinecarbothioamide moiety remarkably increased anti-leishmanial activity compared to nitro and pyridine (EWGs) against both L. major and L. tropica. On the other hand, the presence of nitro and pyridine exhibited almost similar activity against L. major, while nitro showed stronger activity against L. tropica. It seems that the presence of bulk and EWG had a negative effect on L. tropica, but the bulk feature was not important for L. major.
In the group of fluorine-substituted para-phenyl compounds (5d, 5i, and 5p), compound 5p, which incorporates a pyridine ring, exhibited the highest potency overall. Among the others, 5d was more active than 5i against L. major, while 5i showed greater activity against L. tropica compared to both 5d and 5p. Additionally, compound 5d was slightly less active compared to 5p against the amastigote form, though their overall activities were comparable against L. tropica. These observations suggest that increased lipophilicity at the hydrazinecarbothioamide moiety enhances anti-leishmanial activity, particularly against L. major. Among mono-substituted compounds with bromine in the para-phenyl ring (5e, 5j, and 5n), compound 5e showed remarkably higher activity compared to compounds 5j and 5n. Compound 5j was slightly more potent than compound 5n against L. major. A similar trend was observed against L. tropica, with the exception that compound 5n showed considerably higher activity than 5j. According to findings, in these compounds, EDG and small-size groups at the hydrazinecarbothioamide moiety showed higher anti-leishmanial activity against L. major and L. tropica. However, increasing lipophilicity and size is not critical against L. major, but it is effective against L. tropica. Among mono-substituted compounds with OCH3 in the para-phenyl ring (5g and 5m), compound 5g displayed superior activity than compound 5m against both L. major and L. tropica. It looks like, in these compounds, the presence of EDG and a small size group at the hydrazinecarbothioamide moiety showed higher anti-leishmanial activity. In general, all evaluated derivatives exhibited greater potency against L. major than L. tropica.
Overall, the EDG or EWG substitution at the para-position of the phenyl ring retains the anti-promastigote and anti-amastigote activities56,63. Notably, small and lipophilic EWGs, such as fluorine, improved the anti-leishmanial activity56,57,59. Moreover, moderately sized and lipophilic substituents, including the EDG (-OCH3) and EWG (-Br), also contributed to improved activity. Substitution with EWGs like nitro and pyridine at the hydrazinecarbothioamide moiety revealed less activity than the CH3 group56,59. All derivatives exhibited higher anti-promastigote activity against L. major than GLU, except for compounds 5c, 5h, 5k, 5m, 5n, and 5o. Moreover, several compounds were more potent than GLU against the L. major amastigote form. Regarding L. tropica, all compounds exhibited higher anti-promastigote activity than GLU except compounds 5b, 5h, 5j, and 5o. While almost all compounds were more potent than GLU against the L. tropica amastigote form. Besides, all compounds displayed lower anti-leishmanial activity compared to AmB against both L. major and L. tropica. In general, the findings of current research are congruent with those of prior investigations57,58,59,63,64.
Furthermore, the cytotoxicity of compounds on the J774A.1 cell line was assessed. Among different parasite stages, amastigote inhibition is mostly more relevant therapeutically. Among the tested compounds, compounds 5c, 5h, and 5n showed the lowest cytotoxicity toward macrophages. However, compounds 5e and 5g exhibited the highest cytotoxicity (Table 1). The SI defined as the ratio of CC₅₀ to IC₅₀, was used to assess compound safety. For a molecule to be considered non-toxic, the SI should be at least 1020. Compound 5p with anti-leishmanial activity against L. major (IC50 = 31.2 µM on promastigote form and 12.77 µM on amastigote form; CC50 = 1218 µM) showed a SI of 95.4, showing a safe profile with admiration to macrophages. Following that, compounds 5a, 5e, and 5g showed the lowest toxicity for macrophages (CC50 = 1654, 847, and 733.2 µM) and showed promising anti-leishmanial activity (SI = 27.5, 26.6, and 39, respectively), indicating a safe profile for macrophages. In contrast, 5h (IC50 > 4450 on promastigote form and 2400 µM on amastigote form; CC50 = 3392 µM; SI = 1.4), 5l (IC50 = 346 µM on promastigote form and 680 µM on amastigote form; CC50 = 1062 µM; SI = 1.5), 5m (IC50 = 1000 µM on promastigote form and 891.6 µM on amastigote form; CC50 = 1332 µM; SI = 1.5), and 5o (IC50 = 1079 µM on promastigote form and 886 µM on amastigote form; CC50 = 1806 µM; SI = 2) because of the low anti-L. major activity, showed low selectivity.
Compounds 5e and 5g with anti-leishmanial activity against L. tropica (IC50 = 92.3 µM on promastigote form and 24.5 µM on amastigote form; CC50 = 847 µM) and (IC50 = 101.5 µM on promastigote form and 22.7 µM on amastigote form; CC50 = 733.2 µM) showed SIs of 34.6 and 32.2, resulting in a safe profile with admiration for macrophages. These results indicate a favorable safety profile, particularly with limited toxicity toward macrophages. In comparison, GLU showed SI equal to 10 and 8.09 against L. major and L. tropica, respectively, highlighting the superior selectivity of compounds 5e and 5g. Therefore, the most promising anti-leishmanial derivatives have a suitable scaffold to be investigated for further enhanced anti-parasitic drugs since they have an appropriate in-vitro early toxicity profile and selectivity on macrophages, particularly when it comes to compounds active against L. major and L. tropica.
Computational studies
Predicting in silico metabolic procedure in the existence of cytochrome P450
Drug-drug interactions may occur when foreign compounds, particularly drugs and their metabolites, influence the bioavailability of other drugs. Such interactions can lead to adverse effects, either due to the co-administration of multiple drugs or through drug–metabolite interactions65. Notably, more than half of all currently used drugs are metabolized by the three most significant enzymes in drug metabolism: CYP3A4, CYP2D6, and CYP2C9. Tables 3, 4 and 5 provide the top three atoms in compounds that have the highest metabolic priority.
According to the findings, the metabolic reactivity values of the derivatives are appropriate and comparable, providing a reliable basis for understanding their interactions with cytochrome P450 isoforms. Based on the SMARTCyp 3.0 analysis, the three atomic sites with the lowest scores in each compound were considered the most likely sites of metabolic transformation. For instance, the site with the highest score in derivatives 5a, 5c, 5d, 5e, and 5m is atomic site C11, and in compounds 5i, 5j, 5k, 5l, 5n, and 5p is atomic site S1 for isoform 2C9 (Table 3). For isoform 3A4, S1 is the main adduct formation site in derivatives 5i, 5j, 5k, 5l, 5m, 5n, 5o, and 5p, while in second rank, C11 is the main adduct formation site in derivatives 5a, 5c, 5d, and 5e (Table 4). Moreover, in compounds 5a, 5c, 5d, 5e, 5m, and 5o and compounds 5i, 5j, 5k, 5l, and 5p, atomic positions C11 and S1, respectively, are the key adduct formation sites for isoform 2D6 (Table 5).
In silico estimates of cyto-safe and cardiotoxicity
Cyto-safe and cardiotoxicity predictions are crucial for evaluating a sufficient level of safety earlier administration into the body. According to pred-hERG analysis, all derivatives—except for compounds 5m through 5p—were predicted to be non-hERG blockers, suggesting a low risk of cardiotoxicity (Table 6).
In silico estimate of pharmacokinetic and physicochemical features
Lipinski’s rule of five criteria includes molecular weight (MW) ≤500, hydrogen bond acceptors ≤10, hydrogen bond donors ≤5, and LogP value ≤5. Violations of these criteria may impair oral bioavailability. As shown in Table 7, all synthesized analogues complied with the MW and LogP requirements, having values below 500 and LogP < 5, respectively. Further, the compounds exhibited total polar surface areas (TPSA) ranging from 36.42 to 98.02 Ų and 4 to 7 rotatable bonds, which fall within the accepted thresholds for good oral bioavailability (TPSA ≤ 140 Ų; rotatable bonds ≤ 10). Additionally, all derivatives had an average of 3–8 hydrogen bond acceptors (nHA) and 2–3 hydrogen bond donors (nHD), satisfying Lipinski’s criteria and reinforcing their potential as orally active drug candidates.
P-glycoprotein (P-gp) is a transmembrane efflux transporter belonging to the ATP-binding cassette superfamily. It is widely expressed in biological barrier tissues, including the small intestine, liver, kidneys, and blood-brain barrier (BBB). P-Gp influences drug intestinal absorption since it can remove them from the cell66. According to the predictions, none of the synthesized analogues were identified as P-gp substrates, indicating a reduced risk of P-gp-mediated drug efflux and potentially improved oral bioavailability.
The ability of drugs to traverse the BBB is an important pharmacokinetic indication. Only drugs that have traversed the BBB can alter the physiological processes occurring in the brain67. The results showed that none of the produced analogues could pass through the BBB, suggesting limited central nervous system (CNS) exposure (Table 8).
Some CYP450 isoforms play a main role in drug metabolism. Derivatives were tested as inhibitors of Cyp3A4, Cyp2D6, and Cyp2C9. None of the analogues inhibited Cyp2D6. Analogues were not CYP3A4 inhibitors, except derivatives 5a, 5b, 5k, and 5m-p. Additionally, compounds 5j, 5k, 5m, and 5n were Cyp2C9 inhibitors, and compounds 5c, 5g, 5l, 5o, and 5p can possibly be Cyp2C9 inhibitors, whereas other analogues were not Cyp2C9 inhibitors (Table 8).
Carcinogenicity is one of the most critical adverse effects in the development of new drugs. Since carcinogenesis typically involves a long latency period before clinical manifestation, comprehensive long-term (often lifetime) animal studies are necessary to assess the carcinogenic potential of novel compounds. According to the predictions, all tested compounds were identified as potentially carcinogenic. Moreover, compounds 5a-c, 5e, 5f, and 5j-l did not display drug-induced nephrotoxicity, indicating a favorable renal safety profile for these analogues. Additionally, compounds 5f and 5i-l were not drug-induced neurotoxic, suggesting low neurotoxic risk (Table 8).
Molecular docking study
Computational approaches, such as molecular docking, are valuable tools for investigating enzyme-molecule interactions and aid in identifying the pharmacophoric features for the design of potential bioactive molecules with therapeutic uses68. This section aims to rationalize the observed in vitro anti-leishmanial activity by targeting a specific enzyme of the parasite. The enzyme of L. major pteridine reductase 1 (LmPTR1) was studied as a potential target. PTR1 is a short-chain NADPH-dependent oxidoreductase enzyme that plays a central role in the pterin salvage pathway69. Because the Leishmania parasite has a distinct ability to salvage pterins from its host, which absences PTR1 activity and instead synthesizes pterin analogues from GTP, the PTR1 gene becomes a critical target for therapeutic progression70. Many biochemical studies have confirmed that PTR1 is NADPH-dependent and functions in its tetrameric form71. This enzyme converts biopterin to H2 and H4 biopterin while also degrading other folate forms such as 7,8-dihydrofolate, and tetrahydrofolate. The removal of the PTR1 gene causes the insect phase promastigotes’ death; though, this result can be mitigated by giving lower pterins but not folates, implying that H4-biopterin serves an important function. Recent findings indicated that H4-biopterin is involved in regulating parasite differentiation. Mutants lacking PTR1 displayed decreased H4-biopterin levels, causing parasites in the sand fly vector to mature into the highly infectious metacyclic promastigote phase72.
The catalytic site of PTR1 contains a triad/tetrad composed of the Tyr, Asn, Ser, and Lys residues. It is identified that the Asp (e.g., Asp181) is more stable, comparable to a Ser residue. The pyrophosphate of the co-factor interacts with Arg17, stabilizing the enzyme-cofactor complex. Additionally, Phe113 plays a critical role in ligand stabilization within the active site, engaging in hydrophobic interactions that help anchor the substrate or inhibitor37,73. The inhibitor or substrate must bind to PTR1 in the presence of the co-factor NADPH74. The nicotinamide ring of NADPH engages in hydrophobic contact with Phe113, a key interaction that is essential for substrate recognition and catalytic activity. The catalytic residues Tyr191, Asp181, Lys198, and Tyr194 are highly conserved, suggesting that the catalytic mechanism of PTR1 is preserved across different Leishmania species75.
The docking poses were selected based on their optimal binding interactions and the top-scored conformations, as determined by the docking search algorithm and scoring functions. The binding affinities to PTR1’s active site were estimated based on high-scoring functions, the hydrogen bonds formed with key amino acid residues, and the spatial alignment of the docked compounds relative to the co-crystallized ligand. The docking methodology was validated by re-docking the co-crystallized ligand into the PTR1 binding site, which duplicated the initial pose determined from the PDB with an RMSD of 1.89 Å.
All of the analogues 5a-p were docked into the LmPTR1 active site to identify the optimum conformational position. Among them, compounds 5g and 5p showed the highest activity and the lowest free binding energy, correlating well with their observed in vitro activity. As illustrated in Fig. 6, the NHs of the thiosemicarbazides formed a hydrogen bond with Gly225 and two hydrogen bonds with co-factor NADPH in compound 5g. The methoxyphenyl moiety engaged in multiple hydrophobic interactions with Asp181, Pro187, Leu188, Phe113, Tyr194, Tyr191, Thr195, and Thr184. Furthermore, the thiosemicarbazone moiety formed hydrophobic interactions with Val230, Ser227, Leu229, Leu226, and Met183, contributing further to the stability of the binding pose (Fig. 6).

(A) 3D and (B) 2D interactions of compound 5g with LmPRT1 residues active site.
The phenyl ring formed a π-alkyl stacking interaction with the co-factor NADPH and a π-π stacking interaction with Phe113 in compound 5p. Moreover, the fluorine atom at the para-phenyl ring in compound 5p made a hydrogen bond with Ser111. The pyridine ring of compound 5p showed hydrophobic interactions with Pro187, Leu188, Thr184, Asp181, Tyr194, Thr195, and Tyr191. Furthermore, the sulfur atom of the thiosemicarbazone moiety made hydrophobic interactions with Tyr283, and the phenyl ring formed this interaction with Met183 and Gly225 (Fig. 7).

(A) 3D and (B) 2D interactions of compound 5p with PRT1 residues active site.
A comparative analysis of the binding modes of three compounds, 5a, 5k, and 5o, within the LmPRT1 active site showed distinct interaction profiles. Compound 5a formed four hydrogen bonds, including three hydrogen bonds between thiourea moiety NHs and Gly225 as well as co-factor NADPH. Moreover, the methoxy group at the para-phenyl ring formed a hydrogen bond with Leu188, while the phenyl ring of the compound showed a π-alkyl interaction with Met183. In contrast, the thiourea moiety NH in compound 5k exhibited a hydrogen bond with Asp181. Meanwhile, compound 5o formed three hydrogen bonds: the thiourea moiety NH, the methoxy group at the para-phenyl ring, and the sulfur atom with Tyr194, Arg17, and Gly225, respectively. The Phe113 made a π-π stacking interaction with the phenyl ring in both compounds 5k and 5o (Fig. 8). Based on findings, thiourea moiety NHs play an important role in binding to the active site through hydrogen bonds. Gly225 and co-factor NADPH were also key for this bond. In addition, probably due to the more appropriate binding mode of compound 5a to the strong affinity at the LmPTR1 active site compared to the two compounds 5k and 5o, it showed stronger anti-leishmanial activity.
The docking results of compounds 5a and 5b showed that Asp181 made two hydrogen bonds with thiourea moiety NHs, while Arg17 and co-factor NADPH formed hydrogen bonds with the methoxy group at the para-phenyl ring in compound 5b (Fig. 8). Consistent with their observed anti-leishmanial activities, Leu188, Gly225, Asn181, and co-factor NADPH were key to hydrogen bond formation with the thiourea moiety NH and methoxy group of the para position.
Among compounds 5d, 5i, and 5p, the observed order of activity against L. major was 5p > 5i > 5d. The docking findings showed that para-fluorophenyl in 5p made a hydrogen bond with Ser111, while the other two compounds did not form any hydrogen bonds. Thus, the formation of hydrogen bond with active site residues was essential for anti-leishmanial activity. Besides, compound 5p made a π-π stacking interaction with Phe113 and a π-alkyl interaction with co-factor NADPH via a phenyl ring. Whereas, in compound 5d we observe none of these interactions, which may explain its lower activity. Meanwhile, the phenyl ring of compound 5i formed two π-alkyl interactions with Phe113 and co-factor NADPH. It seems that hydrophobic interactions of the compound with Phe113 and co-factor NADPH in order to π-π stacking interaction > π-alkyl interaction play a main role against L. major (Figs. 7 and 9).

2D binding modes of compounds (A) 5a, (B) 5k, (C) 5o, and (D) 5b with LmPRT1 residues active site.

2D binding modes of (A) compound 5d and (B) compound 5i with LmPRT1 residues active site.
Compound 5e showed more anti-leishmanial activity than compounds 5j and 5n. According to binding modes in the PRT1 active site, compound 5e formed three hydrogen bonds between Gly225 and the co-factor NADPH and thiourea moiety NHs. In contrast, compound 5j showed two hydrogen bonds: the nitro group of Met183 and thiourea moiety NH with Asp181. Similarly, compound 5n also made two hydrogen bonds: the thiourea moiety NHs with Gly225 and Asp181. On the other hand, the phenyl ring of two compounds, 5e and 5n, exhibited π-π stacking and π-alkyl interactions with Phe113 and co-factor NADPH, respectively. These interactions are consistent with the binding patterns observed in the most active compounds. Collectively, the results highlight that Gly225 and NADPH play a critical role in hydrogen bond formation, exerting a greater influence on binding affinity than other residues in the active site. Similar to the best compounds results showed that Gly225 and co-factor NADPH compared to other residues were important to form hydrogen bonds. Moreover, similar results were observed in compounds 5g and 5m.
Two compounds, 5c and 5l, showed weak anti-leishmanial activity, which correlates with their docking results. Neither compound formed hydrogen bonds with Gly225 or the co-factor NADPH, both of which were identified as critical interaction sites in more active analogues. Instead, the thiourea moiety NHs of compound 5c formed two hydrogen bonds with Asp181, while the thiourea moiety NH in compound 5l made a hydrogen bond with Asp181. Although both compounds showed π-π stacking and π-alkyl interactions with Phe113 and co-factor NADPH, respectively, these interactions alone were insufficient to confer strong activity (Fig. 10). These findings support the broader trend observed in the study: hydrogen bonding with Gly225 and NADPH is essential for potent anti-leishmanial activity, and its absence corresponds with reduced efficacy.

2D binding modes of (A) compound 5c and (B) compound 5l with PRT1 residues active site.
According to findings, favorable hydrogen bonding and hydrophobic interactions were observed with Asp181, Phe113, Leu188, Gly225, Arg17, Tyr194, and Ser111 residues and co-factor NADPH. These findings support the 5g and 5p’s great capacity to inhibit the catalytic activity of LmPTR176. In terms of spatial disposition, the inhibitor is identical to the pterin head group of the prototypic antifolate medication methotrexate, and it binds to the PTR1 active site using sandwiched hydrophobic stacking. The planar aromatic ring of the inhibitor also forms a π-π stacking interaction with Phe113 aromatic rings, a key feature in stabilizing the ligand within the catalytic pocket75.
It can be observed that Gly225, Asp181, and co-factor NADPH are the main amino acids in the hydrogen bond interactions. Hydrophobic interactions, on the other hand, require Met183, Leu229, Tyr191, Phe113, Thr195, and Leu188 residues. Furthermore, the Phe113 residue was important for π-π stacking interaction, which is essential for stabilizing the ligand within the enzyme’s catalytic pocket37,77,78,79.
The thiourea moiety NHs as hydrogen bond acceptors, π-π stacking and π-alkyl interactions, and hydrophobic aromatic characteristics are pharmacophore properties that have been recognized as critical to complexes’ interaction (Fig. 11). As a result, a suitable molecule for the enzyme includes hydrogen bond acceptors, a hydrophobic group, and an aromatic ring system.

Pharmacophore characteristics that have been recognized as essential factors in the complexes’ interaction in LmPRT1 active site.
The observed in vitro and in silico results provide several SAR insights for this series of compounds: (I) aromatic substitution: the para-position of substituents on the aromatic ring plays a critical role in enhancing biological activity. Substitution at this position significantly improves the anti-leishmanial potential; (II) thiosemicarbazone core: the presence of the thiosemicarbazone moiety is essential for activity. This group contributes to key hydrogen bonding interactions within the enzyme’s active site; (III) substitution on the thiosemicarbazone NH: introducing a methyl group on the NH position of the thiosemicarbazone moiety significantly improves activity compared to nitro or pyridine substitutions. So small electron-donating groups enhance anti-leishmanial effects; (IV) pyridine and fluorine substituents: notably, the combination of a pyridine ring with a fluorine atom enhances biological activity, likely due to favorable hydrophobic and electronic interactions within the enzyme’s active site.
These findings provide valuable structural guidelines for the rational design of more potent anti-leishmanial agents in future research.
Molecular dynamics simulations study
Molecular dynamics (MD) simulations are used to investigate the flexibility and stability of docking complexes at different time intervals in a hydrated media: optimizing docking, considering the flexibility of both the ligand and protein and evaluating ligand binding affinities. MD simulations validated the docking study’s findings, demonstrating the role of residues in hydrophobic interactions and hydrogen bonds. Compounds 5e-, 5g-, and 5p-LmPTR1, as well as LmPTR1 only (apo protein), were selected for MD simulations. To accomplish this, MD simulations of 150 ns periods was performed. In this study, we analyzed multiple parameters on the MD trajectories, including Root Mean Square Deviation (RMSD), Root Mean Square Fluctuations (RMSF), Hydrogen Bonds (H-Bond), Radius of Gyration (Rg), and Solvent Accessible Surface Areas (SASA).
System stability and flexibility (root mean square deviation (RMSD))
Backbone RMSDs were investigated over a 150 ns simulation period to establish the ligands’ active site stability based on their predicted affinity for the LmPRT1 active site. After the MD simulation, the RMSD was used to determine the stability and the conformation change of the complex structure from its original. Figure 12 shows that the apo form of LmPRT1 remained stable during the 150 ns simulation run, with an average RMSD of 2.38 Å. The three candidate compounds remained stable in the protein backbone. The 5g-LmPRT1 complex exhibited the lowest average RMSD of 1.74 Å, whereas the 5e- and 5p-LmPRT1 complexes showed average RMSD of 3.06 Å and 2.93 Å, respectively. As a result, the 5g-enzyme showed lower deviations than the apo protein and others, suggesting higher conformational stability. Smaller fluctuations show that the 5g-LmPRT1 complex is more stable. A range of 1–3 Å for fluctuations is measured as acceptable in the case of a small globular protein. However, fluctuations more than this range display major changes in the conformation of protein80. Candidate complexes revealed good stability based on the average RMSD values, indicating that the structure of the enzyme kept on being consistent throughout simulations. According to the results, the candidates were stable within the active site, with no appreciable diffusion or dissociation from the initial binding site.
The RMSD plot for the 5g-LmPRT1 complex, shown in Fig. 12, shows that in the early 15 ns, the complex reached stability. In contrast, the RMSD plot of the 5p-LmPRT1 complex is presented in Fig. 12; it reached stability after 16 ns. The complex displayed a small rise from 106 to 150 ns simulation time. The detected deviation is most likely due to conformational changes in the compound’s rotatable bonds, as demonstrated by their presence in the two-dimensional depiction of the compound-enzyme interactions and oscillations in the ligand’s torsion angles81. Furthermore, numerous studies show that functional areas of an enzyme are more flexible, resulting in larger RMSD. Protein flexibility is commonly required for their capability to adapt to various biological activities81. Notably, the behavior of the enzyme did not alter significantly in the presence of the selected analogues. Moreover, the RMSD value of 5p was lower than those of the apo protein, while the RMSD value of 5e was slightly higher than apo protein. Compound 5g and apo protein exhibited comparable RMSD values. Based on RMSD compound plots, compounds 5g and 5p showed lower RMSD values than compound 5e. All three compounds display a stable binding with LmPRT1 (Fig. 12). This observation correlates with assay results. Lower RMSD values for compounds could indicate good compound accommodation in the active site. Thus, the RMSD values achieved from the MD showed that the complexes were stable during simulation.

RMSD plot of LmPRT1 with molecules 5e (Green), 5g (Brown), 5p (Blue), and apo protein (Purple) throughout the simulation time.
Likewise, the RMSD of the compounds is also performed separately (Fig. 13). The 5e, 5g, and 5p in the LmPRT1 displayed a stable RMSD figure. Analogues showed stability after 5 ns. It was revealed that analogues remained in the active site of LmPTR1, where they mostly interacted with the co-factor and residues. Three compounds exhibited low RMSD values; however, lower fluctuation (Fig. 13).

Superimposed RMSD of the ligands 5e (Green), 5g (Brown), and 5p (Blue) in complex with LmPRT1 enzyme.
Fluctuations in the residual components (root mean square fluctuations (RMSF))
Root mean square fluctuation (RMSF) analysis is a technique for recognizing rigid and flexible areas in an enzyme and better understanding the stability of complexes. It provides information about how the atom moves in the flexible enzyme areas throughout compound binding82. This was monitored using an analysis of the RMSF values, which define the divergence in the orientation of the molecule relative to its original posture and the amplitude of the enzyme residues’ motions83. RMSF for the complex was computed using C-α atoms. The fluctuation intensity for all molecules was below 3.0 Å, with the exception of those residues that indicate a loop or turn in the enzyme84. There is greater RMSF in areas like turns, loops, and coils in comparison to sheet and helical structures85. The larger the RMSF value, the more flexible and unstable the binding82.

RMSF of the residues of apo LmPRT1 ((Purple) and LmPRT1 in complex with 5e (Green), 5g (Brown), and 5p (Blue), during overall 150 ns MD simulation.
Figure 14 depicts 5e-, 5g-, and 5p-LmPRT1 and apo protein RMSF plots. Throughout the MD simulations, the complexes were stable. All the complexes showed a similar fluctuation. The average RMSF for all complexes was < 1.5 Å. In some regions, there was high fluctuation, whereas other regions had minimal fluctuations. The residues implicated in the active site area had an RMSF value of < 0.4 nm, indicating that the area is very dense, rigid, and stable in a dynamic media86. These findings demonstrated that all of the complexes had an active interaction with the enzyme and kept on stable. The bound complexes and apo protein showed high fluctuations around amino acids 60 to 70, 100 to 120, 200 to 230, and 245 to 264. The loop areas of the LmPTR1 display protuberant fluctuations. At residues 60 to 70 and 100 to 120, fluctuations were higher for the 5e complex than the apo form. Moreover, fluctuations were similar for 5g and 5p complexes with the apo protein. At residues 200 to 230, fluctuations were lower in all complexes than in the apo protein. At residues 245 to 250, compound 5p showed the lowest fluctuations. However, at 250 to 264 residues, this compound showed the highest fluctuations. This proposes that the molecule-binding pretentious the amino acids’ motions. These regions, which undergo significant fluctuations, comprise critical active site amino acids (Gly225, Asp181, Leu188, Tyr191, Leu229, Met183, Phe113) that display low RMSF (< 2.8 Å) and similar fluctuation performance to all complexes. These results show that the complexes (5g and 5p) bind well and undergo negligible conformational changes in comparison to the apo protein form (Fig. 14). This demonstrates that protein in its apo form was more flexible than those in its bound states. In this investigation, residues in these similar areas were discovered to fluctuate for all the derivatives, indicating that they are important for binding. The findings of RMSF were in agreement with previous studies, including the crystal structure of LmPRT1 and LmPRT1-compounds37,78,87,88.
Assessment of structural compactness (radius of gyration (Rg))
For the measure of a system’s compactness, the radius of gyration (Rg) was used. This work examines the compactness, conformation, and folding patterns of complexes during the simulation. Higher Rg values, indicating less compactness (more unfolded), contrast with lower Rg values, which signify greater compactness and increased structural rigidity (more folded). A low Rg indicates stiff packing, while a high Rg suggests less flexibility. Therefore, Rg was calculated to measure the compactness of the system over time, as illustrated in Fig. 15. The complexes display gyration scores ranging from 1.8 to 1.9 nm. Moreover, the average Rg values of 5e-, 5g-, and 5p-LmPRT1 and apo protein were found to be 1.869, 1.856, 1.867, and 1.851 nm, respectively. Rg values do not exhibit fluctuations or changes significantly during MD. The Rg of compounds almost remained stable during simulation (Fig. 15). It indicates that the ligand-enzyme complexes and apo protein were stable. There was no considerable fluctuation in the 5e-, 5g-, and 5p-enzyme Rg in comparison to the apo structure.

The protein backbone Rg during the MD, in the presence of 5e (Green), 5g (Brown), apo (Purple), and 5p (Blue).
For compound 5g, a small Rg was determinated, meaning a relatively compact ligand, confirming its stability during the binding occurrence with the LmPRT1 protein. However, about 115 ns until the end of the simulation, a slight increase in Rg was observed. In comparison to the apo protein, it showed protein compressed until 40 ns, while from 40 ns until the end of the simulation was similar to the apo protein. Compound 5e showed constant Rg until about 70 ns. Then, Rg decreased from 70 ns until the end of the simulation. It exhibited a small fluctuation between 117 and 127 ns. This compound showed a higher Rg value compared to the apo protein during MD simulation. Compound 5p showed almost constant Rg until 107 ns, then it decreased until the end of the simulation (Fig. 15). The Rg results follow a trend similar to the RMSD results, suggesting that the molecules did not induce considerable changes in the enzyme’s compactness. This modest fluctuation in the Rg value demonstrated the stability of complexes. These results show that compounds bind strongly to the active site, as evidenced by the fact that all of the investigated complexes achieved a relatively stable folded shape over the final 40 ns of the MD trajectory. Thus, all complexes tolerate a strong Rg value, which shows the highly folded enzyme-compound structure. Hence, it can be observed that complexes increase the rigidity/compactness of the enzyme structure, leading to more stability.
All complexes’ Rg was not significantly different from the apo protein (Fig. 15). This shows that compound interaction did not alter the total LmPTR1 fold. These findings are congruent with assay results given by Hardy89. The rigidity of the active site prevents LmPTR1 from undergoing a significant conformational change upon ligand binding. The Rg and RMSF findings display that the LmPTR1 active site is rigid, and molecule interaction has no remarkable effect on fluctuations of active site amino acids90.
Solvent-accessible surface area (SASA) analysis
Solvent-accessible surface area (SASA) investigated the impact of solvent accessibility caused by moveable packing on the complexes’ stability. It determines the amount of the interaction between solvents and complexes. This gives us a better understanding of how the complex interacts with solvent molecules. Furthermore, SASA data can be utilized to determine the magnitude of the conformational changes that happen in the binding procedure. Examples include the binding of a foreign material, such as a drug, which increases the solubility of hydrophobic amino acids. Overall, the study’s average SASA and Rg values revealed that the enzyme connected to pharmaceutical compounds underwent minor conformational changes, most probably due to the compounds occupying its active site91. The solvent molecule forms a strong binding with the ligand and can thus be utilized to predict the flexibility and major conformational changes that occur during the compound-enzyme interaction92. A higher SASA shape showed increased enzyme surface area, whereas a lower SASA shape exhibited complex truncation or a decrease85. Figure 16 shows the plot of the fluctuations in SASA values for all the complexes. The average SASA score for the compounds 5e, 5g, and 5p in complex with LmPRT1 and apo protein was found to be 136.97 nm2132.69 nm2and 135.81 nm2 as well as 133.58 nm2respectively (Fig. 16). The findings showed that all the complexes had significantly similar SASA values to the apo protein. Complexes with high and reasonably stable SASA values display that the compound is obtainable for solvent interaction while the enzyme structure remains unaltered81.

The protein backbone SASA during the MD, in the presence of 5e (Green), 5g (Brown), apo (Purple), and 5p (Blue).
During the MD simulation, the complexes and the apo protein followed the same SASA values. The SASA for the apo protein reduced from 139 nm2 to 129 nm2 in the first 30 ns and thereafter kept on steady. A similar trajectory was seen for 5e-, 5g-, and 5p-LmPRT1, where stability happens after 10 ns. However, the 5e-LmPRT1 complex showed a small increase of SASA from 20 to 110 ns, and then it showed a decline until the end of the simulation.
Summary, low SASA values indicate reduced solvent surface charges, causing structural limitations of complexes and a more stable configuration and compact93. As result, using the average of the computed SASA values for the numerous complexes, the order of stability is accordingly apo protein > 5g > 5p > 5e. The low SASA data observed for all the complexes imply their stability during simulation.
Hydrogen bond analysis of complexes
The stability of the compound-enzyme complex is preserved through different interactions such as hydrophobic interactions, hydrogen bonds, and electrostatic interactions. Among the numerous forms of interactions, hydrogen bonding is important in preserving the structural stability of complex since it facilitates different interactions85. The most vital interaction in drug discovery is intermolecular hydrogen bonding. Hydrogen bonds are primarily responsible for ligand binding stability. The amount of hydrogen bonds in intermolecular hydrogen bond interactions determines the rigidity and specificity of the protein-ligand interaction87. The more hydrogen bonds, the stronger the affinity for the enzyme82.
During the 150 ns MD, the hydrogen bonds of each complex were considered. Some hydrogen bonds were made or remade. Figure 17 shows the number of hydrogen bonds formed in complexes. These unsteady hydrogen bonds established as a result of enzyme and compound mobility may account for a tiny portion of the compound’s affinity for LmPRT1 (Fig. 17). In comparison to the 5e-LmPRT1 complex, 5g- and 5p-LmPRT1 exhibited the most hydrogen bonding of any simulated complex. Thus, the total data shows that four complexes form hydrogen bonds with LmPRT1 to keep stability.
5e, 5g, and 5p complexes formed the average hydrogen bonds equal to 1.86, 0.80, and 1.81, respectively. However, the hydrogen bonds in the 5g and 5p complexes remained consistent throughout the simulation, demonstrating a stronger interaction between these molecules and the enzyme, causing a highly stable conformation.

Number of hydrogen bonds made between compound and enzyme during simulation in the presence of 5e (Green), 5g (Brown), apo (Purple), and 5p (Blue).
The most hydrogen bonds in compound 5g were three, and the average was one hydrogen bond. This complex had made hydrogen bonds with co-factor NADPH and Gly225. The complex compound 5e and compound 5p include a maximum hydrogen bond of 3 and keep an average of up to 1.5. The amino acid involved in hydrogen bond formation with compound 5p was co-factor NADPH and Asp181. In the case of compound 5e, the residue involved in hydrogen bond formation was Glu186 and co-factor NADPH87.
Principal component analysis (PCA) study
Principal component analysis (PCA) is often applied to analyze the coordinated motions in complexes, focusing on the C-α atoms94. In this study, PCA was used to assess the conformational sampling of the complexes and apo protein in the simulation. The complexes’ PCA indicated that the first little eigenvectors were important in the total motion. Figure 18 shows the eigenvector plots for the 5e-, 5g-, and 5p-LmPRT1 complexes, as well as the apo protein. The comparison results revealed that the 5e-LmPRT1 complex may have greater conformational diversity, while the 5g- and 5p-LmPRT1 complexes had the fewest motions. Moreover, apo protein showed the greatest conformational variability (Fig. 18).
In addition, the dynamics of complexes were investigated using 2D projection plots of the primary contributing eigenvectors, PC1 and PC2. The superimposed plots revealed that all complexes occupied a region comparable to that of the apo protein (Fig. 19).

Principal component analysis (PCA). The eigenvector index of 5e, 5g, 5p, and apo protein during MD simulations.

Principal component analysis (PCA). PC1 and PC2 of 5e (Blue), 5g (Purple), 5p (Green), and apo protein (Yellow) during MD simulations.
The Ramachandran plot of complexes
The Ramachandran plot shows a graphical illustration of the dihedral angles φ (phi) and ψ (psi) of amino acids in an enzyme structure. These angles illustrate how a polypeptide chain’s amino acids rotate around the bonds that bind them. The figure is applied to determine the permissible and disallowable areas of these dihedral angles according to steric hindrance and other structural restrictions.
The Ramachandran plot is important in determining the stereochemical quality of enzyme structure. Areas with few or no points show energetically unfavorable conformations, whereas areas with many points usually establish and characterize energetically allowed conformations for the enzyme backbone. The plot contains three key areas; the most preferred area: this area corresponds to energetically advantageous mixtures of ψ and ϕ angles, typically related to the permitted conformations of beta sheets and alpha helices. Permitted area: this area contains combinations of torsional angles that are energetically permitted but less public than those in the most preferred area. Outlier area: this area characterizes combinations of ψ and ϕ angles that are energetically rejected. Points in this area indicate potential structural difficulties with the enzyme, such as steric conflicts or unrealistic bond angles and lengths95,96. The Ramachandran plot reported diverse areas of coloring/shading on the ψ-ϕ plot for the complexes (Fig. 20). The most preferred areas (A, B, L) with a percentage of > 90% and permitted areas (a, b,l, p) with a percentage of about 8%.

Ramachandran plot of complexes and apo protein displaying > 90% of amino acids in the core region into active site of LmPRT1 enzyme.
Conclusion
The thiosemicarbazone analogues were prepared through a one-pot, two-step, three-component reaction. The anti-amastigote and anti-promastigote activities of L. tropica and L. major were assessed. Additionally, the cytotoxicity of the analogues was assessed using the murine macrophage cell line J774.A1. All synthesized compounds displayed weak to good anti-leishmanial activity. However, some of the screened analogues displayed low cytotoxicity to macrophage infection, and compounds 5p and 5e showed the highest SI (95.4 and 34.6) against L. major and L. tropica, respectively. Moreover, compounds 5g and 5p were the most potent compounds against amastigote and promastigote forms, respectively, of L. major, with IC50 = 26.7 µM and 12.77 µM, respectively. These two compounds have shown stronger anti-amastigote and anti-promastigote activities compared to other related compounds, and this difference in activity can be attributed to the following structural factors: (I) appropriate electronic substituents, such as methoxy or fluorine groups in the para-phenyl ring, which can enhance π-π interactions and modulate electron density favorably; (II) N-substituents like pyridine-3-yl that can participate in hydrogen bonding or metal coordination, improving specificity and binding affinity to biological targets; (III) structural compatibility that allows efficient cell membrane penetration and interaction with intracellular targets (such as enzymes or nucleic acids); (IV) a well-balanced lipophilicity/hydrophilicity profile, ensuring both good solubility and cellular uptake. In contrast, less active compounds often feature bulky groups, strong electron-withdrawing substituents (e.g., nitro, bromine), or sterically hindered N-substituents, which may reduce their ability to reach or bind effectively to the biological target, leading to diminished anti-amastigote and anti-promastigote activities. Compounds 5e and 5g were the most potent compounds against amastigote and promastigote forms of L. tropica, with IC50 = 92.3 µM and 12.77 µM. Selected compounds to determine induced apoptosis in promastigotes showed that these compounds displayed different early and late apoptosis as well as necrotic effects on promastigotes of L. major and L. tropica. Then, the pharmacokinetic and oral bioavailability features of analogues were considered. The findings exhibited that all compounds had good drug-likeness properties. Furthermore, the most likely sites of the analogues metabolized using the key cytochromes were identified. Next, the in silico cardiotoxicity features of the analogues were analyzed. The pred-hERG results exhibited that analogues do not cause hERG blockage and are not cardiotoxic except for compounds 5m-p. Lastly, MD simulations and docking studies showed that compounds 5e, 5g, and 5p in the LmPTR1 active site are stable.
Data availability
All data generated or analyzed during this study are included in this manuscript and its supplementary information files.
References
Shukla, R., Soni, J., Kumar, A. & Pandey, R. Uncovering the diversity of pathogenic invaders: insights into protozoa, fungi, and worm infections. Front. Microbiol. 15, 1374438 (2024).
Rodrigues, A. C. J. et al. Exploring the leishmanicidal potential of terpenoids: a comprehensive review on mechanisms of cell death. Front. Cell. Infect. Microbiol. 13, 1260448. https://doi.org/10.3389/fcimb.2023.1260448 (2023).
eBioMedicine Leishmania: an urgent need for new treatments. EBioMedicine 87, 104440 https://doi.org/10.1016/j.ebiom.2023.104440 (2023).
Mann, S. et al. A review of leishmaniasis: current knowledge and future directions. Curr. Trop. Med. Rep. 8, 121–132 (2021).
Gupta, O., Pradhan, T., Bhatia, R. & Monga, V. Recent advancements in anti-leishmanial research: synthetic strategies and structural activity relationships. Eur. J. Med. Chem. 223, 113606. https://doi.org/10.1016/j.ejmech.2021.113606 (2021).
Romero, A. H. et al. Design, synthesis, structure-activity relationship and mechanism of action studies of a series of 4-chloro-1-phthalazinyl hydrazones as a potent agent against leishmania Braziliensis. Eur. J. Med. Chem. 127, 606–620. https://doi.org/10.1016/j.ejmech.2017.01.022 (2017).
Zhang, S. X. et al. Global, regional, and National burden of visceral leishmaniasis, 1990–2021: findings from the global burden of disease study 2021. Parasit. Vectors. 18, 157. https://doi.org/10.1186/s13071-025-06796-x (2025).
Herrmann, F. C. et al. In Silico identification and in vitro evaluation of natural inhibitors of leishmania major pteridine reductase I. Molecules 22, 2166 (2017).
Burza, S., Croft, S. L., Boelaert, M. & Leishmaniasis Lancet 392, 951–970 https://doi.org/10.1016/s0140-6736(18)31204-2 (2018).
da Silva, E. A. et al. Potential leishmanicidal of the thiosemicarbazones: A review. Results Chem. 9, 101609. https://doi.org/10.1016/j.rechem.2024.101609 (2024).
Marín, M., López, M., Gallego-Yerga, L., Álvarez, R. & Peláez, R. Experimental structure based drug design (SBDD) applications for anti-leishmanial drugs: A paradigm shift? Med. Res. Rev. 44, 1055–1120. https://doi.org/10.1002/med.22005 (2024).
Mota, W. J. S. et al. Classical and innovative drugs for the treatment of leishmania infections. Discover Public. Health. 21 https://doi.org/10.1186/s12982-024-00247-1 (2024).
Zulfiqar, B., Shelper, T. B. & Avery, V. M. Leishmaniasis drug discovery: recent progress and challenges in assay development. Drug Discov. Today. 22, 1516–1531. https://doi.org/10.1016/j.drudis.2017.06.004 (2017).
Khatoon, S. et al. Novel coumarin-isatin hybrids as potent antileishmanial agents: synthesis, in Silico and in vitro evaluations. Bioorg. Chem. 110, 104816. https://doi.org/10.1016/j.bioorg.2021.104816 (2021).
da Silva, P. R. et al. Novel indol-3-yl-thiosemicarbazone derivatives: obtaining, evaluation of in vitro leishmanicidal activity and ultrastructural studies. Chemico-Biol. Interact. 315, 108899. https://doi.org/10.1016/j.cbi.2019.108899 (2020).
Zahra, S. B. et al. Versatile biological activities of thiosemicarbazones and their metal complexes. J. Mol. Struct. 1322, 140511. https://doi.org/10.1016/j.molstruc.2024.140511 (2025).
Pandeya, S. N. et al. Synthesis and screening for anti-HIV activity of some N-Mannich bases of Isatin derivatives. Chemotherapy 45, 192–196. https://doi.org/10.1159/000007182 (1999).
Tsimberidou, A. M., Alvarado, Y. & Giles, F. J. Evolving role of ribonucleoside reductase inhibitors in hematologic malignancies. Expert Rev. Anticancer Ther. 2, 437–448. https://doi.org/10.1586/14737140.2.4.437 (2002).
Li, W. et al. Micelles loaded with puerarin and modified with triphenylphosphonium cation possess mitochondrial targeting and demonstrate enhanced protective effect against isoprenaline-induced H9c2 cells apoptosis. Int. J. Nanomed., 8345–8360 (2019).
Linciano, P. et al. Aryl thiosemicarbazones for the treatment of trypanosomatidic infections. Eur. J. Med. Chem. 146, 423–434. https://doi.org/10.1016/j.ejmech.2018.01.043 (2018).
Ma, X. & Zhang, W. Recent developments in one-pot Stepwise synthesis (OPSS) of small molecules. Iscience 25 (2022).
Dömling, A., Wang, W. & Wang, K. Chemistry and biology of multicomponent reactions. Chem. Rev. 112, 3083–3135. https://doi.org/10.1021/cr100233r (2012).
Graziano, G. et al. Multicomponent Reaction-Assisted drug discovery: A Time- and Cost-Effective green approach speeding up identification and optimization of anticancer drugs. Int. J. Mol. Sci. 24, 6581 (2023).
Kang, L. et al. Structure–activity relationship investigation of coumarin–chalcone hybrids with diverse side-chains as acetylcholinesterase and butyrylcholinesterase inhibitors. Mol. Divers. 22, 893–906 (2018).
Takeuchi, H. et al. CCL21 chemokine regulates chemokine receptor CCR7 bearing malignant melanoma cells. Clin. Cancer Res. 10, 2351–2358. https://doi.org/10.1158/1078-0432.Ccr-03-0195 (2004).
Jaafar, A. et al. Synthesis, characterization, antifungal and antibacterial activities evaluation of copper (II), zinc (II) and cadmium (II) chloride and bromide complexes with new (E)-1-(3,4-dimethoxybenzylidene)-4-methylthiosemicarbazone ligand. Appl. Organomet. Chem. 34, e5988. https://doi.org/10.1002/aoc.5988 (2020).
Rashdan, H. R. M., Abdelrahman, M. T., Shehadi, I. A., El-Tanany, S. S. & Hemdan, B. A. Novel Thiadiazole-Based molecules as promising inhibitors of black Fungi and pathogenic bacteria: in vitro antimicrobial evaluation and molecular Docking studies. Molecules 27, 3613 (2022).
Coxon, G. D. et al. Synthesis, antitubercular activity and mechanism of resistance of highly effective thiacetazone analogues. PLOS ONE. 8, e53162. https://doi.org/10.1371/journal.pone.0053162 (2013).
Lata, D. et al. Thiosemicarbazone platinacycles with tertiary phosphines. Preparation of novel heterodinuclear platinum–tungsten complexes. Polyhedron 41, 30–39. https://doi.org/10.1016/j.poly.2012.04.015 (2012).
Serda, M. et al. Investigation of the biological properties of (hetero)aromatic thiosemicarbazones. Molecules 17, 13483–13502. https://doi.org/10.3390/molecules171113483 (2012).
Rodrigues, C. et al. Coordination of nitro-thiosemicarbazones to ruthenium(II) as a strategy for anti-trypanosomal activity improvement. Eur. J. Med. Chem. 45, 2847–2853. https://doi.org/10.1016/j.ejmech.2010.03.005 (2010).
Batista, D. G. J. et al. Manganese(II) complexes with N4-methyl-4-nitrobenzaldehyde, N4-methyl-4-nitroacetofenone, and N4-methyl-4-nitrobenzophenone thiosemicarbazone: investigation of in vitro activity against trypanosoma Cruzi. Polyhedron 29, 2232–2238. https://doi.org/10.1016/j.poly.2010.04.023 (2010).
Olanrewaju, A. A. et al. Synthesis, in-vitro and in-silico antibacterial and computational studies of selected thiosemicarbazone-benzaldehyde derivatives as potential antibiotics. SN Appl. Sci. 5, 213. https://doi.org/10.1007/s42452-023-05429-1 (2023).
Santos, R. F. et al. Therapeutic effect of oral Quercetin in hamsters infected with leishmania Viannia Braziliensis. Front. Cell. Infect. Microbiol. 12, 1059168 (2023).
Batista, S. A. A. et al. Synthesis and comparison of antileishmanial and cytotoxic activities of S-(–)-limonene benzaldehyde thiosemicarbazones with their R-(+)-analogues. J. Mol. Struct. 1179, 252–262 (2019).
Saraei, S. et al. In vitro evaluation of the potent antileishmanial activity of Ferula tabasensis alone or in combination with shark cartilage extract against the standard Iranian strain of leishmania major (MRHO/IR/75/ER). Iranian J. Pharm. Res. IJPR 22 (2023).
Molaei, S. et al. One-pot synthesis of polyhydroquinoline-1, 2, 3-triazole hybrids in deep eutectic solvent as anti-leishmanial agents and molecular modeling studies. J. Biomol. Struct. Dyn. 42, 4834–4850 (2024).
Fehling, H. et al. High content analysis of Macrophage-Targeting Eh PIb-Compounds against cutaneous and visceral leishmania species. Microorganisms 9, 422 (2021).
Sifontes-Rodríguez, S., Escalona-Montaño, A. R., Sánchez-Almaraz, D. A., Pérez-Olvera, O. & Aguirre-García, M. M. Detergent-free parasite transformation and replication assay for drug screening against intracellular leishmania amastigotes. J. Microbiol. Methods. 215, 106847 (2023).
Tavakoli, P., Ghaffarifar, F., Delavari, H. & Shahpari, N. Efficacy of manganese oxide (Mn2O3) nanoparticles against leishmania major in vitro and in vivo. J. Trace Elem. Med. Biol. 56, 162–168 (2019).
Razzaghi-Asl, N., Sepehri, S., Ebadi, A., Miri, R. & Shahabipour, S. Molecular Docking and quantum mechanical studies on biflavonoid structures as BACE-1 inhibitors. Struct. Chem. 26, 607–621 (2015).
Sepehri, S. et al. Biological evaluation, and molecular Docking studies of novel 4-[4-Arylpyridin-1(4H)-yl]benzoic acid derivatives as Anti-HIV-1 agents. Chem. Biodivers. 14, e1700295. https://doi.org/10.1002/cbdv.201700295 (2017). Synthesis.
Keivanloo, A., Sepehri, S., Bakherad, M. & Eskandari, M. Click synthesis of 1, 2, 3-Triazoles‐Linked 1, 2, 4‐Triazino [5, 6‐b] indole, antibacterial activities and molecular Docking studies. ChemistrySelect 5, 4091–4098 (2020).
Sepehri, S. et al. Synthesis, evaluation of the cytotoxicity, apoptosis induction in AGS cell line and gene expression and molecular modeling studies of novel tetrahydropyrimidine derivatives. Arab. J. Chem. 17, 105448. https://doi.org/10.1016/j.arabjc.2023.105448 (2024).
Zelenin, K. N. et al. Ring-Chain tautomerism of N-Substituted thiosemicarbazones. Tetrahedron 49, 1257–1270. https://doi.org/10.1016/S0040-4020(01)85816-6 (1993).
Uda, M. & Kubota, S. Ring-chain tautomerism of acetone N-methylated thiosemicarbazones. J. Heterocycl. Chem. 16, 1273–1275. https://doi.org/10.1002/jhet.5570160635 (1979).
Siles, R., Chen, S. E., Zhou, M., Pinney, K. G. & Trawick, M. L. Design, synthesis, and biochemical evaluation of novel Cruzain inhibitors with potential application in the treatment of chagas’ disease. Bioorg. Med. Chem. Lett. 16, 4405–4409. https://doi.org/10.1016/j.bmcl.2006.05.041 (2006).
Linciano, P. et al. Aryl thiosemicarbazones for the treatment of trypanosomatidic infections. Eur. J. Med. Chem. 146, 423–434. https://doi.org/10.1016/j.ejmech.2018.01.043 (2018).
Eldehna, W. M. et al. Synthesis, in vitro biological evaluation and in Silico studies of certain Arylnicotinic acids conjugated with Aryl (thio)semicarbazides as a novel class of anti-leishmanial agents. Eur. J. Med. Chem. 179, 335–346. https://doi.org/10.1016/j.ejmech.2019.06.051 (2019).
Fonseca Nayara, C. et al. Synthesis of a Sugar-Based thiosemicarbazone series and Structure-Activity relationship versus the parasite cysteine proteases rhodesain, cruzain, and Schistosoma mansoni cathepsin B1. Antimicrob. Agents Chemother. 59, 2666–2677. https://doi.org/10.1128/aac.04601-14 (2015).
Magalhaes Moreira, D. R. et al. Conformational restriction of Aryl thiosemicarbazones produces potent and selective anti-Trypanosoma Cruzi compounds which induce apoptotic parasite death. Eur. J. Med. Chem. 75, 467–478. https://doi.org/10.1016/j.ejmech.2014.02.001 (2014).
Manzano, J. I. et al. Arylthiosemicarbazones as antileishmanial agents. Eur. J. Med. Chem. 123, 161–170. https://doi.org/10.1016/j.ejmech.2016.07.014 (2016).
Tenório, R. P. et al. Synthesis of thiosemicarbazone and 4-thiazolidinone derivatives and their in vitro anti-Toxoplasma gondii activity. Bioorg. Med. Chem. Lett. 15, 2575–2578. https://doi.org/10.1016/j.bmcl.2005.03.048 (2005).
Dodd, R. H., Ouannes, C., Robert-Gero, M. & Potier, P. Hybrid molecules: growth Inhibition of leishmania donovani promastigotes by thiosemicarbazones of 3-carboxy-.beta.-carbolines. J. Med. Chem. 32, 1272–1276. https://doi.org/10.1021/jm00126a021 (1989).
Pervez, H., Manzoor, N., Yaqub, M. & Khan, K. M. 5-Nitroisatin-derived thiosemicarbazones: potential antileishmanial agents. J. Enzyme Inhib. Med. Chem. 29, 628–632. https://doi.org/10.3109/14756366.2013.836641 (2014).
Temraz, M. G. et al. Anti-leishmanial click modifiable thiosemicarbazones: design, synthesis, biological evaluation and in Silico studies. Eur. J. Med. Chem. 151, 585–600. https://doi.org/10.1016/j.ejmech.2018.04.003 (2018).
Feitosa, L. M. et al. New pyrazolopyrimidine derivatives as leishmania amazonensis arginase inhibitors. Bioorg. Med. Chem. 27, 3061–3069. https://doi.org/10.1016/j.bmc.2019.05.026 (2019).
Mendes, E. P. et al. Evaluation of novel chalcone-thiosemicarbazones derivatives as potential anti-Leishmania amazonensis agents and its HSA binding studies. Biomolecules 9, 643 (2019).
Taha, M. et al. Molecular hybridization conceded exceptionally potent quinolinyl-oxadiazole hybrids through phenyl linked Thiosemicarbazide antileishmanial scaffolds: in Silico validation and SAR studies. Bioorg. Chem. 71, 192–200 (2017).
Naseer, M. M. et al. Novel Thiosemicarbazones of Coumarin Incorporated Isatins: Synthesis, Structural Characterization and Antileishmanial Activity.
Blau, L. et al. Design, synthesis and biological evaluation of new Aryl thiosemicarbazone as antichagasic candidates. Eur. J. Med. Chem. 67, 142–151. https://doi.org/10.1016/j.ejmech.2013.04.022 (2013).
Khoshbakht, A., Shiran, J. A., Miran, M. & Sepehri, S. Synthesis and evaluation of in vitro antioxidant, anticancer, and antibacterial properties of new benzylideneiminophenylthiazole analogues. BMC Chem. 18, 173 (2024).
Schmitt, M. et al. Synthesis, Antileishmanial activity and in Silico studies of aminoguanidine hydrazones (AGH) and thiosemicarbazones (TSC) against leishmania Chagasi amastigotes. Med. Chem. (2021).
Ansari, M., Montazeri, M., Daryani, A., Farshadfar, K. & Emami, S. Synthesis and in vitro anti-Toxoplasma gondii activity of a new series of Aryloxyacetophenone thiosemicarbazones. Mol. Divers. 24, 1223–1234 (2020).
Olsen, L., Montefiori, M., Tran, K. P. & Jørgensen, F. S. SMARTCyp 3.0: enhanced cytochrome P450 site-of-metabolism prediction server. Bioinformatics 35, 3174–3175. https://doi.org/10.1093/bioinformatics/btz037 (2019).
Saaby, L. & Brodin, B. A. Critical view on in vitro analysis of P-glycoprotein (P-gp) transport kinetics. J. Pharm. Sci. 106, 2257–2264. https://doi.org/10.1016/j.xphs.2017.04.022 (2017).
Bagchi, S. et al. In-vitro blood-brain barrier models for drug screening and permeation studies: an overview. Drug. Des. Devel. Ther. 13, 3591–3605. https://doi.org/10.2147/dddt.s218708 (2019).
Asgari, M. S. et al. Magnetic silica nanoparticle-supported copper complex as an efficient catalyst for the synthesis of novel Triazolopyrazinylacetamides with improved antibacterial activity. Chem. Heterocycl. Compd. 56, 488–494. https://doi.org/10.1007/s10593-020-02685-6 (2020).
de Viana, O. Silico structural insights and potential inhibitor identification based on the benzothiazole core for targeting leishmania major pteridine reductase 1. ACS Omega. 10, 306–317 (2024).
Possart, K., Herrmann, F. C., Jose, J. & Schmidt, T. J. In Silico and in vitro search for dual inhibitors of the trypanosoma brucei and leishmania major pteridine reductase 1 and dihydrofolate reductase. Molecules 28, 7526 (2023).
Dello Iacono, L., Di Pisa, F. & Mangani, S. Crystal structure of the ternary complex of leishmania major pteridine reductase 1 with the cofactor NADP+/NADPH and the substrate folic acid. Struct. Biol. Crystall. Commun. 78, 170–176 (2022).
Ullah, W. et al. Computational investigation of turmeric phytochemicals targeting PTR1 enzyme of leishmania species. Heliyon 10 (2024).
das Neves, G. M., Kagami, L. P., Gonçalves, I. L. & Eifler-Lima, V. L. Targeting pteridine reductase 1 and dihydrofolate reductase: the old is a new trend for leishmaniasis drug discovery. Future Med. Chem. 11, 2107–2130 (2019).
Panecka-Hofman, J. & Poehner, I. Structure and dynamics of pteridine reductase 1: the key phenomena relevant to enzyme function and drug design. Eur. Biophys. J. 52, 521–532 (2023).
Kaur, J., Dube, D., Ramachandran, R., Singh, P. & Singh, N. Thianthrene is a novel inhibitor of leishmania donovani pteridine reductase 1 (PTR1). J. Mol. Biochem. 1 (2012).
Sabt, A., Eldehna, W. M., Ibrahim, T. M., Bekhit, A. A. & Batran, R. Z. New antileishmanial Quinoline linked Isatin derivatives targeting DHFR-TS and PTR1: design, synthesis, and molecular modeling studies. Eur. J. Med. Chem. 246, 114959 (2023).
Shtaiwi, A. Thiadiazine-thiones as inhibitors of leishmania pteridine reductase (PTR1) target: investigations and in Silico approach. J. Biomol. Struct. Dyn. 42, 8588–8597 (2024).
Khalilzadeh, M. et al. Synthesis, biological assessment, and computational investigations of Nifedipine and monastrol analogues as anti-leishmanial major and anti-microbial agents. Mol. Divers. 27, 2555–2575 (2023).
Rezaei, A. R., Saberi, S. & Sepehri, S. Synthesis, antileishmanial activity and molecular Docking study of a series of dihydropyridine derivatives. Polycycl. Aromat. Compd. 43, 4640–4653 (2023).
Bora, K., Sarma, M., Kanaujia, S. P. & Dubey, V. K. Dual-target drugs against leishmania donovani for potential novel therapeutics. Sci. Rep. 13, 18363 (2023).
Nath, M., Bhowmik, D., Saha, S., Nandi, R. & Kumar, D. Identification of potential inhibitor against leishmania donovani mitochondrial DNA primase through in-silico and in vitro drug repurposing approaches. Sci. Rep. 14, 3246 (2024).
Prasanna, P., Joshi, T., Pant, M., Pundir, H. & Chandra, S. Evaluation of the inhibitory potential of valproic acid against histone deacetylase of leishmania donovani and computational studies of valproic acid derivatives. J. Biomol. Struct. Dyn. 41, 5447–5464 (2023).
Gariganti, N. et al. Design, synthesis, anticancer activity of new amide derivatives derived from 1, 2, 3-triazole-benzofuran hybrids: an insights from molecular docking, molecular dynamics simulation and DFT studies. J. Mol. Struct. 1273, 134250 (2023).
Hassan, A. H. et al. Bestatin analogs-4-quinolinone hybrids as antileishmanial hits: design, repurposing rational, synthesis, in vitro and in Silico studies. Eur. J. Med. Chem. 250, 115211 (2023).
Tewari, D. et al. Screening of potential inhibitors of leishmania major N-myristoyltransferase from Azadirachta indica phytochemicals for leishmaniasis drug discovery by molecular docking, molecular dynamics simulation and density functional theory methods. Journal Biomol. Struct. Dyn, 1–18 (2023).
Bhowmik, D. et al. Evaluation of potential drugs against leishmaniasis targeting catalytic subunit of leishmania donovani nuclear DNA primase using ligand based virtual screening, Docking and molecular dynamics approaches. J. Biomol. Struct. Dyn. 39, 1838–1852 (2021).
Ranjan, P. & Dubey, V. K. Identification of potential anti-leishmanial compounds from natural sources against citrate synthase enzyme using structure-based drug designing. J. Mol. Struct. 1295, 136556 (2024).
Ibrahim, T. M. et al. Tetrahydrobenzo [h] Quinoline derivatives as a novel chemotype for dual antileishmanial-antimalarial activity graced with antitubercular activity: design, synthesis and biological evaluation. Eur. J. Med. Chem. 257, 115534 (2023).
Hardy, L., Matthews, W., Nare, B. & Beverley, S. Biochemical and genetic tests for inhibitors ofleishmaniapteridine pathways. Exp. Parasitol. 87, 158–170 (1997).
Boakye, A., Gasu, E. N., Mensah, J. O. & Borquaye, L. Computational studies on potential small molecule inhibitors of leishmania pteridine reductase 1. J. Biomol. Struct. Dyn.. 41, 12128–12141 (2023).
Goyzueta-Mamani, L. D. et al. Targeting leishmania infantum Mannosyl-oligosaccharide glucosidase with natural products: potential pH-dependent Inhibition explored through computer-aided drug design. Front. Pharmacol. 15, 1403203 (2024).
Vemula, D., Mohanty, S. & Bhandari, V. Repurposing of food and drug admnistration (FDA) approved library to identify a potential inhibitor of trypanothione synthetase for developing an antileishmanial agent. Heliyon 10 (2024).
Sakyi, P. O. et al. Targeting leishmania donovani sterol methyltransferase for leads using pharmacophore modeling and computational molecular mechanics studies. Inf. Med. Unlocked. 37, 101162 (2023).
Singh, P., Kumar, V., Lee, K. W. & Hong, J. C. Discovery of novel allosteric SHP2 inhibitor using Pharmacophore-Based virtual screening, molecular docking, molecular dynamics simulation, and principal component analysis. Pharmaceuticals 17, 935 (2024).
Jayavel, P., Ramasamy, V., Amaladoss, N., Renganathan, V. & Shupeniuk, V. I. A facile synthesis, characterization, DFT, ADMET and in-silico molecular Docking analysis of novel 4-ethyl acridine-1, 3, 9 (2, 4, 10H)-trione. Chem. Phys. Impact. 8, 100476 (2024).
Solis, A. D. Deriving high-resolution protein backbone structure propensities from all crystal data using the information maximization device. PLoS One. 9, e94334 (2014).
Acknowledgements
Supports of this project by Ardabil University of Medical Sciences are welcomed. The funding Number is IR.ARUMS.REC.1401.180.
Author information
Authors and Affiliations
Contributions
J.A.S. M.B. and Z.M. conducted the literature review, synthesized the compounds, and executed the experimental procedures and analyzed the spectra. M.R.R. and N.S. performed molecular docking. S.M. conducted the literature review, biological activity of the compounds, and executed the experimental procedures. Y.A.Z. performed figures and graphical abstract. S.S. designed and ran the characterization experiments, analyzed the data, and wrote the original draft as well as supervised the work, and all authors reviewed and edited the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Molaei, S., Shiran, J.A., Shakour, N. et al. Synthesis of thiosemicarbazone Schiff base derivatives as anti-leishmanial agents and molecular dynamics simulations insights. Sci Rep 15, 24867 (2025). https://doi.org/10.1038/s41598-025-10545-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-10545-6
