
Abstract
Sindbis virus (SINV) is a zoonotic arbovirus transmitted by mosquitoes and maintained by wild birds with an expanding distribution globally. Despite its importance, surveillance efforts are low or lacking in many areas, especially in Africa. Our study aimed to highlight the epidemiology of SINV in wild birds in a West African country – Nigeria – with implications for human health. Blood samples were collected from wild resident Afrotropical and migrant Palearctic birds over two years. RT-qPCR was used to detect SINV RNA positive samples, followed by confirmatory conventional PCR and Sanger sequencing targeting the non-structural protein gene. Three out of 504 samples (0.6%; 95% CI: 0.12–1.73%) were positive for SINV, all from individuals of a single species, the African Thrush (Turdus pelios). We successfully generated the whole genome sequence of one sample. Phylogenetic analysis revealed it was closely related to strains from Algeria, Spain and Kenya in the SINV-I genotype. The study suggests that SINV is enzootic in the region and that the African Thrush may be a putative reservoir species.
Similar content being viewed by others
Introduction
Sindbis virus (SINV) is an arthropod-borne Alphavirus (family Togaviridae) that causes a febrile illness in humans manifested by rash, arthralgia, fatigue, muscle pain, headache, itching, upper respiratory symptoms and fever1. It was first described from the Sindbis region of Egypt in 1952, when it was isolated from mosquitos in an area where patients presented with febrile illness, rash and arthritis2. A decade later it was detected in humans in South Africa3,4. Since then, the virus has been identified in other parts of the world, including Europe, Asia and Australia1. The disease caused by the virus is known by different names in different geographical areas, such as Pogosta disease in Finland, Karelian fever in Russia, and Ockelbo disease in Sweden5. Although a widespread virus, SINV disease burden has a disjunct distribution, with largest effects in South Africa and Scandinavia5.
SINV is zoonotic and is transmitted through the bite of infected mosquitoes, particularly species of the Culex genus5,6. Wild birds are the main reservoir hosts5, but the virus has been detected through RT-PCR in a range of wildlife (e.g. African Buffalo (Syncerus caffer), Sable antelope (Hippotragus niger), Giraffe (Giraffa camelopardalis), Blesbuck (Damaliscus pygargus phillipsi), and White rhinoceros (Ceratotherium simum)) and domestic vertebrates (sheep, pig, horse) that had neurological infection or unexplained deaths7,8. There is no known human-to-human transmission of the virus9. With wild birds being reservoirs, it is not surprising that they have been implicated in long-distance dispersal, possibly across continents, as the distribution of the virus reflects migratory bird pathways between the Northern and the Southern Hemisphere10,11,12.
SINV is grouped into six genotypes (SINV I-VI) based on the glycoprotein sequence of the E2 gene. The SINV-I genotype strains have been found in Europe, the Middle East and Africa; SINV-II in Australia and Malaysia; SINV-III in India and the Philippines; SINV-IV in Azerbaijan and China; SINV-V in New Zealand; and SINV-VI in south-west Australia5,6. The SINV genotype complex displays significant genetic variability both at the nucleotide level and the amino acid level11,13.
Despite the widespread detection of the virus, the actual disease burden and detailed knowledge of distribution of the virus and susceptible species is still limited. This is especially true in Africa due to under-reporting and limited surveillance efforts in many areas of the continent12,14. Compared to other regions of Africa, there is limited information about SINV from many parts of West Africa. This includes Nigeria, one of the largest and most populous countries in West Africa, where the only mention of SINV comes from a serosurvey of humans in 197815. Furthermore, although many studies in Africa have reported detection and isolation of SINV in mosquitoes1,6,15,16,17, there is lack of information on the status of the virus in wild bird populations and how that influences the epidemiology of the virus in the region.
The ongoing geographic expansion of many arboviruses as a result of climate change is more and more in focus18. SINV has long been known to circulate in Southern and Eastern Africa but recently more infections have been detected outside the previously known distribution range5. However, to the best of our knowledge, there are no circulation records of SINV in either humans, vector mosquitoes, or host bird species in Nigeria, apart from the serology report of Fagbami19 when humans were seropositive to the virus in Southwest Nigeria. Our study therefore, aimed to highlight the epidemiology of SINV in wild birds in Nigeria, setting the stage for further investigation and surveillance. We carried out an intense field sampling project in central Nigeria and report for the first time detection of SINV in African Thrushes (Turdus pelios) and generally in wild birds in Nigeria.
Materials and methods
Ethical considerations
The experimental protocol was approved by the scientific committee of A.P. Leventis Ornithological Research Institute, University of Jos, and the work was carried out in accordance with institutional guidelines and national legislation. The methods are reported according to ARRIVE guidelines. A material transfer agreement on access to genetic resources was approved by the Nigeria Federal Ministry of Environment.
Study design
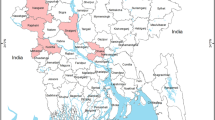
A cross-sectional study was carried out where birds were sampled every month for two years, 2022 and 2023. Fieldwork was conducted in Amurum Forest Reserve (09°53″ N, 08°59″ E) on the Jos Plateau, located 15 km northeast of the city of Jos, in Plateau State Nigeria (Fig. 1). Amurum Forest is about 300-ha large reserve comprising granitic outcrops in scrub Guinea savanna, interspersed with gallery forests and patches of grasslands. It is also the seat of the A.P. Leventis Ornithological Research Institute (APLORI)20. The Amurum Forest Reserve is an Important Bird Area with over 350 bird species recorded20,21, about 36% of the 966 birds occurring in Nigeria22. The reserve serves as wintering and stop-over site for many Palearctic migratory bird species, as well as breeding grounds for resident and intra-African migratory birds23. The diversity of avian assemblages and the close proximity to human settlement that makes it suitable for zoonotic diseases transmission, as well as the presence of potential SINV vectors in the area24,25 informed the choice of the reserve as the study site. Temperature in this area ranges from 8˚C to 38˚C, and mean annual rainfall is 1411mm26. Rainfall varies across the months of the year, with the rainy season lasting for 7 months (April–October) and dry season lasting for 5 months (November–March)27,28. Wild birds were captured weekly using mist nets, and ringed, with appropriate morphometrics recorded29. An average of 10 mist nets of various sizes (9, 12, and 18 m long) were used to capture birds at 12 different locations within and around the reserve (Fig. 1). Nets were opened early in the morning while still dark (between 5:00- 5:30 AM) and bird ringing and blood sampling continued until 11:00 AM29.

Study area map showing sampling points in Amurum Forest Reserve and surrounding areas. The map was created using QGIS (Version 3.28.3, https://qgis.org) and the basemap was sourced from Google Satellite via XYZ Tiles in QGIS and added as a QGIS map layer. Sampling sites were selected based on the observation of abundance of African Thrushes and Palearctic migrant species. The points in Sabon Gari were areas with open lands mixed with farmlands and interspersed with thickets, typical of where to find some Palearctic migrant species, e.g., Common Whitethroat (Curruca communis), Common Nightingale (Luscinia megarhynchos) and Whinchat (Saxicola rubetra). The other points included areas close to gallery forests and pools of water (especially in the dry season), thickets and covers, wooded lands and settlements, typical of where to find African Thrush in abundance. Keys: 1 = Eastern Gully 1, 2 = Eastern gully 2, 3 = Kerker 1, 4 = Kerker 2, 5 = Gidan Babawo 1, 6 = Gidan Babawo 2, 7 = Zarazon, 8 = Pythons’ gully, 9 = Zainab’s gully, 10 = Laminga village, 11 = Sabon Gari village 1, 12 = Sabon Gari village 2.
Blood samples were collected from the brachial veins of birds using 25G needles29,30 and 70µL heparinised micro hematocrit tubes. The target groups were thrushes, robin-chats and chats and Palearctic migratory species, but few samples were collected from other species (Table 1), when they were caught in the nets. The reason for targeting particular species, especially the African Thrush was that thrushes are generally considered as an important amplifying host of SINV, at least in Europe. In Sweden, prevalence of neutralizing antibodies in many thrush species reached 70%31. In other European studies, thrushes recorded high antibody prevalence and many thrush species were found competent for SINV and were potentially able to infect mosquito vectors with the virus, ensuring continuous viral circulation32,33. This role of being an important host for SINV by thrushes is likely the same in Africa as SINV antibody prevalence reached 94% in Olive Thrush in South Africa4. Given these scenarios and the lack of information on SINV occurrence in wild birds in Nigeria, thrushes would be the choice candidate for investigating SINV in wild birds in the area. In addition, the African Thrush is the most common and widespread of the thrushes in this region. Furthermore, its synanthropic nature that enables interaction with humans and animals, its ground feeding behaviour, and preference for thickets and covers that likely predisposes it to mosquito bite all made it our preferred species to start investigating arboviruses, including SINV in birds.
The blood was transferred to Type I Advantec® (Tokyo, Japan) Nobuto blood sampling filter paper and absorbed as dry blood spots34,35 which were placed in labelled Whirl–Pak® Standard Sterilized bags after drying in the field under shade. The bags with the paper strips were then kept in a cooler box until transported to the APLORI molecular ecology lab where they were kept at −20℃ until screening. The samples were shipped on dry ice from Nigeria to the Department of Clinical Microbiology, Umeå University, Sweden for processing. Each dry blood on Nobuto filter paper strip was eluted in 1 mL of phosphate-buffered saline (PBS) containing 0.05% Tween and 0.08% sodium azide in a 2 mL screw-capped tube on a shaking rack at 4℃ for a minimum of 12 h before RNA extraction.
RNA extraction and RT-qPCR screening
Viral RNA was extracted using a QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. A 140 μL eluted Dry Blood Spot (DBS) sample volume was used and RNA was eluted in a final volume of 60 μL, collected in a 1.5 mL sterile microcentrifuge tube and stored at − 80 ℃.
SINV RNA was detected using RT-qPCR on a QuantStudio™ 5 System thermocycler (ThermoFisher Scientific, Waltham, USA) using Biosystems qPCRBIO Probe 1-step Go Lo-ROX kit at optimized cycling conditions: 1 cycle at 50 °C for 10 min, 1 cycle at 95 °C for 2 min; 40 cycles at 95 °C for 5 s and 60 °C for 25 s with primers targeting the nonstructural gene one (nsP1); SINV-nsP1F, 5′-GGTTCCTACCACAGCGACGAT-3′, SINV-nsP1R, 5′-TGATACTGGTGCTCGGAAAACA-3′, and SINV-nsP1 probe, 5′-FAM-TTGGACATAGGCAGC GCA-3′36. The final reaction volume was 20 µL, containing 5 µL of template RNA. The SINV Lövånger strain (Accession number: KF737350) was used as a positive control and nuclease free water was used as a negative control in the RT-qPCR.
Hybridization capture and Illumina sequencing
To confirm the RT-qPCR screening, we selected SINV positive samples for whole genome sequencing using the Twist Comprehensive Viral Research Panel (CVRP) (Twist Biosciences, San Francisco, USA). In brief, Illumina TruSeq-compatible libraries were generated using the Twist Library Preparation EF Kit 2.0 with Enzymatic Fragmentation and the Twist Universal Adapter System following the manufacturer’s protocol. Hybridization capture was performed on the generated library using the CVRP and the Twist Target Enrichment Standard Hybridization v1 workflow. The indexed sample library was approximately 8.62 ng/μL and was used in a multiplexed 16-h hybridization capture reaction. Following enrichment, the library was sequenced with 150 bp paired-end reads on the MiSeq platform (Illumina, CA, USA), using the MiSeq Reagent Nano Kit, v2 300-cycle kit. Generated sequenced reads were assembled using Megahit37 and both reads and assemblies were classified using kraken238. The data processing was performed using the workflow manager Snakemake39 together with Bioconda40.
cDNA synthesis, conventional PCR, gel electrophoresis and Sanger sequencing
The enrichment experiments only gave a partial sequence of SINV genome from one of the three RT-qPCR positive samples. We therefore, subjected the three samples to conventional PCR for confirmation of bands on gels and further purification for Sanger sequencing of the non-structural protein (nsp4) gene. The positive RNA samples were converted to cDNA using Thermo Scientific RevertAid RT Kit (Thermo Fisher Scientific, Waltham, USA) using random primers, according to the manufacturer’s instructions. The cDNA synthesis conditions were: first incubation at 25 °C for 5 min, second incubation at 42 °C for 1 h and a termination step at 70 °C for 5 min. The conventional PCR was carried out using forward primer SINV1 (5′-TTTAGCGGATCGGACAATTC-3′) and reverse primer SINV2 (5′-GCGGTGACGAACTCAGTAG-3′) targeting the nsp4 gene of the virus15, and Thermo Fisher Scientific’s Phusion Green Hot Start II High-Fidelity PCR Master Mix. The SINV Lövånger strain (Accession number: KF737350) was used as a positive control and nuclease free water was used as a negative control just like the RT-qPCR. The final reaction volume was 20 µL containing 5 µL of template cDNA. The cycling conditions included a first step of initial denaturation (one cycle) at 98 °C for 30 s, a second step of 35 cycles of Denaturation at 98 °C for 7 s, annealing at 52 °C for 15 s, and extension at 72 °C for 20 s; with a third step of final extension at 72 °C for 7 min for 1 cycle. The PCR products were visualized using gel electrophoresis (1.2% agarose in 1 × TAE with GelRed (Biotium Inc. Hayward, CA, US). Positive samples were purified with ExoSAP-IT kit (Thermo Fisher Scientific) and sequenced using Sanger sequencing by Eurofin Genomics (Germany). Nucleotide sequences were then aligned to available SINV sequences in GenBank using the Basic Local Alignment Search Tool (BLAST) of National Center for Biotechnology Information.
Tiling PCR and nanopore sequencing
After confirming the presence of SINV through Sanger sequencing, we designed a tiled sequencing primer panel based on publicly available SINV genomes to increase the likelihood of generating full genome sequences from the three RT-qPCR positive samples. Complete SINV genomes were downloaded from the BV-BRC public repository on November 12, 202441. We excluded duplicate genome sequences and experimentally evolved SINV lineages. Subsequently, we aligned SINV genome sequences belonging to genotype I (n = 87, excluding MF409177) using MAFFT (v7.508)42 with the auto setting (see supplementary table S1 for full list of included accession numbers). The resulting multiple sequence alignment was then used as input for varVAMP (v1.2.1)43 to generate tiled sequencing primers, with the following parameters: consensus nucleotide threshold of 1, maximum ambiguous nucleotides per primer set to 2, optimal amplicon length of 1200 bp, maximum amplicon length of 1500 bp, and an overlap of 200 bp between amplicons.
We further evaluated primer specificity by mapping the primers and the Illumina reads generated during enrichment to the closest matching SINV reference strain available at the time (OK644705), identified using Mash Screen (v2.3)44. Mapping was performed with Minimap2 (v2.24-r1122)45,46 and SAMtools (v1.16.1)47, and visualized in IGV48. We manually corrected potential mismatches between the mapped reads and primer sequences by introducing additional ambiguous nucleotides into the primers (See supplementary table S2 for primer sequences). Additionally, we designed primer SINV_10_RIGHT_EXT to improve genome coverage. We ordered all primers from Eurofins Genomics (Ebersberg, Germany) and combined them into two separate equimolar 10 µM non-overlapping pools (Pool 1 and Pool 2, Supplementary table S2).
The tiled amplicon sequencing protocol was adapted from Quick et al.49, with a few differences. We used RNA from the SINV positive samples, SINV strain Lövånger (KF737350, positive control), and nuclease free water (negative control) and reverse-transcribed them into cDNA using ProtoScript II first strand synthesis kit (New England Biolabs, Ipswich, MA, USA), following the manufacturer’s protocol. A volume of 2.5 µl cDNA was used as template for the tiling PCR, which was conducted in two separate reactions with 600 nM of primer pool 1 and 500 nM of primer pool 2, respectively, in a total volume of 25 µl 1X Q5 Hot Start High-Fidelity Master Mix (New England Biolabs). The thermal cycling conditions were as follows: 98 °C for 30 s, followed by 40 cycles at 98 °C for 15 s, 63 °C for 30 s, 72 °C for 90 s, with a final extension at 72 °C for 2 min. Samples were held at 4 °C until purified using 1X Ampure XP beads (Beckman-Coulter, Brea, CA, USA) according to manufacturer’s protocol. Only the sample that had previously generated sequence data during the enrichment experiment produced amplicons and was subsequently advanced, along with the controls, for sequencing.
We combined and prepared equal amounts of amplicons from pool 1 and pool 2 for sequencing using Native Barcoding Kit 24 V14 (SQK-NBD114.24, Oxford Nanopore Technologies, Oxford, UK), as per the protocol. The libraries were sequenced on a MinION flow cell (FLO-MIN114). Primer pair SINV_10_LEFT and SINV_10_RIGHT_EXT, along with primer pairs that showed poor tiling coverage when pooled, were used in singleplex PCRs and resequenced alongside the PCR amplicons generated by the two primer pools.
We performed basecalling and demultiplexing using Dorado (v0.9.0 + 9dc15a8; Oxford Nanopore Technologies) with super accuracy (sup). We filtered demultiplexed reads misplaced due to barcode bleeding by mapping the reads to OK644705 and KF737350 using minimap2. We then generated consensus sequences from the remaining reads using Artic (v1.5.7; https://github.com/artic-network/fieldbioinformatics) with reference genome KF737350 and basecalling model r1041_e82_400bp_sup_v500.
Phylogenetic analysis
We aligned the consensus sequences of the whole genome generated from tiled amplicon sequencing with publicly available complete SINV genomes (BV-BRC, January 23, 2025) and the Whataroa virus genome (NC_016961.1) using MAFFT (n = 114, Supplementary table S1). A maximum likelihood tree was constructed using IQ-TREE (v2.3.6)50 with ModelFinder51, 1000 ultrafast bootstrap replicates52, and NC_016961.1 as the outgroup. We visualized the phylogenetic tree using iTol (v7)53.
Results
We screened dry blood spots from 504 birds of 29 species (20 Afrotropical species and 9 Palearctic migratory species; Table 1 and 2). A total of 3 (0.6%; 95% CI: 0.12–1.73%) were positive for SINV from RT-qPCR screening (Table 1 and 2). The cycle threshold (Ct) values for the three positive samples were 28.47, 37.35 and 38.55, respectively. All three positive samples were collected from adult African Thrushes (Turdus pelios).
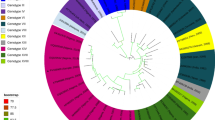
Conventional PCR confirmed two out of the three RT-qPCR positive samples but only one sample gave clear band to use for sequencing. Sanger sequence of SINV nsp4 gene from the sample that produced a clear and sharp band on gel (this had the lowest cycle threshold value [Ct = 28.47] from RT-qPCR) has been deposited in GenBank (Accession number: PQ666553). Result of BLAST revealed its close identity with SINV strains of genotype I from Kenya, Algeria and Spain with 98.69%, 98.60% and 98.60% identities respectively. Tiled amplicon sequencing yielded the whole genome sequence for one of the three RT-qPCR positive samples (the one that showed sharp band on gel and had the lowest Ct value with RT-qPCR). The length of the genome was 11,532 bp. The result of phylogenetic analysis of this whole genome sequence revealed that this isolate was in clade D of SINV genotype 1 group and clustered with Kenyan, Algerian and Spanish sequences of mosquito isolates (Fig. 2), just like the nsp4 gene sequence.

A maximum likelihood tree constructed from complete SINV genomes. Genome AF429428 is shown as the root, with major clades collapsed (except Clade D). The Whataroa virus genome (NC_016961.1) was used as the outgroup but is not shown.
Discussion
Knowledge about SINV in West Africa is limited compared to other regions in Africa. Our study provides the first evidence of SINV occurrence in wild birds in Nigeria and the broader region of West Africa. This finding highlights the need for improved arbovirus surveillance targeting a range of potential hosts in the region, as well as investigations into the epidemiology of SINV and other arboviruses and their ecological drivers. Although the prevalence of SINV was low in this study, detecting viral RNA indicates active circulation of the virus. However, it is worthy of note that the period of viremia of SINV is short, typically between 2–5 days31. As such it may be difficult to track the true extent of its occurrence in the bird population. A broadened approach would be to combine virus detection from reservoir hosts with vector screening, as well as the use of serology to explore the history of infection and get an idea of how widespread the virus is in bird and human populations. Unfortunately, we did not sample mosquitoes in this study, but the detection of SINV in avian blood samples implies the likely presence of the vectors in the area. Moreover, other studies that involved mosquito sampling have reported potential SINV vectors such as Culex quinquefasciatus to be abundant in the nearby city of Jos24,25. Therefore, sampling and characterizing mosquito populations in this area should be a priority in future studies.
Given the under-reporting of SINV in Africa in general and West Africa in particular, we cannot apriori know what is expected regarding the epidemiological status of the virus. An evident first question to ask is whether the detection of SINV here is the result of spillover infections introduced by wintering Palearctic birds that may be coming from SINV endemic regions in Europe, or that SINV is endemic, but undetected in the study area. To start with, it is interesting to note that the only species positive for the virus was the African Thrush, a common resident and intra-African migratory species that occurs in sub-Saharan Africa. This species is from the same genus of thrushes (Turdus) that in previous studies have been identified as important amplifying hosts of SINV in Europe and Africa4,31,54. Hence, biologically the African Thrush could potentially serve a similar role in Africa. At Amurum, African Thrushes are present year-round, but seeing an influx of individuals during the months of February-April is likely a result of intra-African migration of birds heading for breeding areas in the Sudan and Sahel savanna. We did not detect SINV RNA in any of the 28 individuals of 9 Palearctic migratory bird species that were sampled. The sample of each species (average of 3 samples per species) is too small to draw firm conclusion on their role as source of SINV in the area. One can note, however, that SINV-positive samples were all from the dry season, with detections in March which coincides with the period of influx of the African Thrush. Palearctic migrants arrive in Amurum in the autumn, usually in late August/early September, and consists of a pulse of species aiming for wintering areas further south, and another set of species, such as Common Whitethroat (Curruca communis), Garden Warbler (Sylvia borin), Tree Pipit (Anthus trivialis) and Eurasian Wryneck (Jynx torquilla) that winter in the Amurum area. Given that SINV is an acute viral infection, requiring an arthropod vector for transmission, detections in March means that if Palearctic birds are responsible for a recent introduction, it would still have required multiple rounds of bird-vector infections to be maintained until our detections in African Thrushes. Moreover, passing the Mediterranean Sea and the Saharan desert is physiologically taxing, and if infection lowers the bird’s condition even slightly the risk of succumbing during migration is high55. Another factor to consider is the speed of migration. Long-distance Palaearctic passerine migrants generally adopt a migration strategy with several legs of migration, interspersed with stays in stopover sites where fat stores are replenished. This means that it can take more than a month and several stopovers for a passerine bird to reach the wintering ground56, and given the short viremic period (2–5 days) of SINV31, viremia may wane significantly before the bird reaches the African wintering ground. Considering these facts, we hypothesize that SINV is more likely an undetected enzootic infection in the studied avifauna. Given the zoonotic potential of the virus, its presence in African Thrushes represents a public health concern as the bird is associated with human settlement. Moreover, previous studies have linked SINV outbreaks and increased seroprevalence in human to spatial and temporal fluctuation in the population of wild birds32,54. Particularly, Lundström et al.54 reported that areas with higher densities of birds corresponded with increased human cases of SINV infection in Sweden. Experimental infection of birds with SINV revealed that thrushes were highly competent, developing viremia sufficient to infect mosquitoes31,33. Their African counterpart, the African Thrush as well as other species may have similar status, thereby maintaining the virus in the local area. Therefore, there is need for a more widespread sampling effort not only in birds, but also in mosquitoes, as well as an awareness among public health officials that SINV is a putative infection in the human population.
Phylogenetic analysis of the nsP4 gene, and the whole genome showed a close relationship between the African Thrush SINV sequence and mosquito strains from Algeria, Kenya and Spain1,57,58 all of which are members of the genotype 1 (SINV-I) known to circulate in Europe and Africa causing serious disease burden in humans5. The similarity of our strain to strains from other African locales supports the presence of more widely distributed African SINV strains rather than introduced European lineages. Furthermore, the closely related Spanish strain has been suggested to originate in Africa in relatively recent time58. Therefore, one can hypothesize that SINV distribution could be driven by seasonal intra-African bird migration, facilitating its movement across subregions. Migratory birds have been implicated in SINV spread within and across continents, including the supposed first transmission from Africa to Europe1,6,11. However, like other bird-borne arboviruses, the ecological dynamics of SINV in endemic settings are influenced by the assemblage of bird species, vector populations, and seasonal variations in both host and vector abundance5,54, factors that as of yet are poorly studied in our study area.
Regarding vectors and seasonality of the virus detection in this study, it is interesting to note the time of virus detection was in the dry season, when water is scarce and streams are reduced to pools. Such water-scarce condition can foster concentration of birds and mosquitoes around limited water bodies, thereby potentially increasing virus transmission in local patches. Evidently, more research is needed to characterize ecology and epidemiology in this system.
Conclusion
Here for the first time, we confirm the presence of the SINV in African Thrush in Nigeria and the broader West Africa, highlighting the potential role of wild birds in its local transmission. Because mosquitoes are the principal vectors of SINV, detecting the virus in birds indicates an active vector-borne cycle. We recommend adopting a One Health surveillance framework that integrates wildlife (avian), vector (mosquito), and human sampling. This coordinated, multi-sectorial approach will help clarify the SINV ecology, support early detection of human infection, and guide effective prevention and control strategies for both wildlife and public health.
Data availability
Sequences of the nsp4 gene as well as the whole genome are deposited in Genbank with accession numbers PQ666553 and PV075212, respectively. All other data is found in the article or in the electronic supplementary files.
References
Azar, S. R., Campos, R. K., Bergren, N. A., Camargos, V. N. & Rossi, S. L. Epidemic alphaviruses: Ecology, emergence, and outbreaks. Microorganisms 8, 1167 (2020).
Taylor, R. M., Hurlbut, H. S., Work, T. H., Kingston, J. R. & Frothingham, T. E. Sindbis virus: A newly recognized arthropod-transmitted virus. Am. J. Trop. Med. Hyg. 4, 844–862 (1955).
Malherbe, H., Strickland-Cholmley, M. & Jackson, A. L. Sindbis virus infection in man: Report of a case with recovery of virus from skin lesions. S. Afr. Med. J. 37, 547–552 (1963).
McIntosh, B. M., McGillivray, G. M., Dickinson, D. B. & Malherbe, H. Illness caused by Sindbis and West Nile viruses in South Africa. S. Afr. Med. J. 38, 291–294 (1964).
Adouchief, S., Smura, T., Sane, J., Vapalahti, O. & Kurkela, S. Sindbis virus as a human pathogen: Epidemiology, clinical picture, and pathogenesis. Rev. Med. Virol. 26, 221–241 (2016).
Sigei, F. et al. Evolutionary analyses of Sindbis virus strains isolated from mosquitoes in Kenya. Arch. Virol. 163, 2465–2469 (2018).
Steyn, J. et al. Zoonotic alphaviruses in fatal and neurologic infections in wildlife and nonequine domestic animals, South Africa. Emerg. Infect. Dis. 26, 1182–1191. https://doi.org/10.3201/eid2606.191179 (2020).
van Niekerk, S. et al. Sindbis and Middelburg old world alphaviruses associated with neurologic disease in horses, South Africa. Emerg. Infect. Dis. 21, 2225–2229. https://doi.org/10.3201/eid2112.150132 (2015).
Gylfe, Å. et al. Mosquito borne Sindbis virus infection and long-term illness. Emerg. Infect. Dis. 24, 1141–1142 (2018).
Hubálek, Z. An annotated checklist of pathogenic microorganisms associated with migratory birds. J. Wildl. Dis. 40, 639–659 (2004).
Lundström, J. O. & Pfeffer, M. Phylogeographic structure and evolutionary history of Sindbis virus. Vector Borne Zoonotic Dis. 10, 889–907 (2010).
Ling, J. et al. Introduction and dispersal of Sindbis virus from Central Africa to Europe. J. Virol. 93, e00134-e219 (2019).
Michie, A. et al. Genomic analysis of Sindbis virus reveals uncharacterized diversity within the Australasian region, and support for revised SINV taxonomy. Viruses 16, 7 (2024).
Storm, N. et al. Human cases of Sindbis fever in South Africa, 2006–2010. Epidemiol. Infect. 142, 234–238 (2014).
Ochieng, C. et al. Mosquito-borne arbovirus surveillance at selected sites in diverse ecological zones of Kenya; 2007–2012. Virol. J. 10, 140 (2013).
Crabtree, M., Sang, R., Lutomiah, J., Richardson, J. & Miller, B. Arbovirus surveillance of mosquitoes collected at sites of active Rift Valley fever virus transmission: Kenya, 2006–2007. J. Med. Entomol. 46, 961–964 (2009).
Ayhan, N. et al. Detection and isolation of Sindbis virus from field-collected mosquitoes in TimimounAlgeria. Viruses 14, 894. https://doi.org/10.3390/v14050894 (2022).
Whitehorn, J. & Yacoub, S. Global warming and arboviral infections. Clin. Med. (Lond.) 19, 149–152 (2019).
Fagbami, A. Human arthropod-borne virus infections in Nigeria: Serological and virological investigations in Shaki, Oyo State. J. Hyg. Epidemiol. Microbiol. Immunol. 22, 184–189 (1978).
Vickery, J. & Jones, P. J. A new ornithological institute in Nigeria. Bull. Afr. Bird Club 9, 61–62 (2002).
Ezealor, E. A. Nigeria. In Fishpool, L. D. C. & Evans, M. I. (eds) Important Bird Areas in Africa and Associated Islands—Nigeria: Priority Sites for Conservation 673–692 (Pisces Publications, 2001). https://datazone.birdlife.org/userfiles/file/IBAs/AfricaCntryPDFs/Nigeria.pdf
Lepage, D. Checklist of the birds of Nigeria. Avibase – Bird Checklists of the World. https://avibase.bsc-eoc.org/checklist.jsp?list=howardmoore®ion=NG (2025).
Ibrahim, M. I. Renewable natural resource (RNR) draft action plans for Laminga Community Forest (Amurum). Nigerian Conserv. Found. 2, 2–5 (2002).
Anyanwu, G.I. et al. Mosquito breeding sites : distribution and relative abundance of species in the Jos Plateau, Nigeria, Med. Ento. Zool., 1999, Volume 50, Issue 3, Pages 243–249, Released on J-STAGE August 09, 2016, Online ISSN 2185–5609, Print ISSN 0424–7086, https://doi.org/10.7601/mez.50.243
Njunwa, K. J. & Irving-Bell, R. J. Evaluation of resting sites of Culex quinquefasciatus and Anopheles gambiae s.l. in an urban-rural transect in Jos, Nigeria. East Afr. Med. J. 93, 394–401 (2016).
Payne, R. B. A new species of firefinch Lagonosticta from northern Nigeria and its association with the Jos Plateau indigobird Vidua maryae. Ibis 140, 368–381 (1998).
Guntul, T. K., Oche, C. Y. & Madaki, M. Climate records of Jos Plateau (Univ, 2007).
Wuyep, S. Z. & Madaki, D. H. Climate change, rainfall trends, and variability in Jos Plateau. J. Appl. Sci. 20, 76–82 (2020).
Nwaogu, C. J., Cresswell, W. & Tieleman, B. I. Geographic variation in baseline innate immune function does not follow variation in aridity along a tropical environmental gradient. Sci. Rep. 10, 5909 (2020).
Wilhelmsson, P., Jaenson, T. G. T., Olsen, B., Waldenström, J. & Lindgren, P.-E. Migratory birds as disseminators of ticks and the tick-borne pathogens Borrelia bacteria and tick-borne encephalitis (TBE) virus: a seasonal study at Ottenby Bird Observatory in South-eastern Sweden. Parasites Vectors 13, 607 (2020).
Hesson, J. C. et al. Temporal variation in Sindbis virus antibody prevalence in bird hosts in an endemic area in Sweden. PLoS ONE 11, e0162005. https://doi.org/10.1371/journal.pone.0162005 (2016).
Kurkela, S. et al. Sindbis virus infection in resident birds, migratory birds, and humans, Finland. Emerg. Infect. Dis. 14, 41–47. https://doi.org/10.3201/eid1401.070510 (2008).
Lundström, J. O. et al. Viremia in three orders of birds Anseriformes, Galliformes and Passeriformes) inoculated with Ockelbo virus. J. Wildl. Dis. 29, 189–195 (1993) (PMID: 8387608).
Nobuto, K. Toxoplasmosis in animal and laboratory diagnosis. Jpn. Agric. Res. Q. 1, 11–18 (1966).
Dusek, R. J., Hall, J. S., Nashold, S. W., TeSlaa, J. L. & Ip, H. S. Evaluation of Nobuto filter paper strips for the detection of avian influenza virus antibody in waterfowl. Avian Dis. 55, 674–676 (2011).
Sane, J. et al. Development and evaluation of a real-time RT-PCR assay for Sindbis virus detection. J. Virol. Method. 179, 185–188 (2012).
Li, D., Liu, C. M., Luo, R., Sadakane, K. & Lam, T. W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Köster, J. & Rahmann, S. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics 28, 2520–2522 (2012).
Grüning, B. et al. Bioconda: Sustainable and comprehensive software distribution for the life sciences. Nat. Methods 15, 475–476 (2018).
Olson, R. D. et al. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): A resource combining PATRIC, IRD, and ViPR. Nucleic Acids Res. 51, D678–D689 (2022).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Fuchs, J. et al. varVAMP: Automated pan-specific primer design for tiled full genome sequencing and qPCR of highly diverse viral pathogens. bioRxiv https://doi.org/10.1101/2024.05.08.593102 (2024).
Ondov, B. D. et al. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 17, 132 (2016).
Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Li, H. New strategies to improve Minimap2 alignment accuracy. Bioinformatics 37, 4572–4574 (2021).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Robinson, J. T. et al. Integrative Genomics Viewer. Nat. Biotechnol. 29, 24–26 (2011).
Quick, J. et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 12, 1261–1276 (2017).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Hoang, D. T. et al. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522 (2018).
Letunic, I. & Bork, P. Interactive Tree of Life (iTOL) v6: Recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 52, W78–W82. https://doi.org/10.1093/nar/gkae268 (2024).
Lundström, J. O. et al. Prevalence of Sindbis virus-neutralizing antibodies among Swedish passerines indicates that thrushes are the main amplifying hosts. J. Med. Entomol. 38, 289–297 (2001).
Newton, I. Migration mortality in birds. Ibis https://doi.org/10.1111/ibi.13316 (2024).
Tapia-Harris, C., Izang, A. & Cresswell, W. Migratory routes, breeding locations and multiple non-breeding sites of Common Whitethroats Curruca communis revealed by geolocators. PLoS ONE 17(9), e0274017. https://doi.org/10.1371/journal.pone.0274017) (2022).
Koskei, E. et al. Isolation and phylogenetic characterization of arboviruses circulating among phlebotomine sandflies in parts of North Rift, Kenya. Front. Virol. 4, 1289258. https://doi.org/10.3389/fviro.2024.1289258 (2024).
Gutiérrez-López, R. et al. First isolation of the Sindbis virus in mosquitoes from southwestern Spain reveals a new recent introduction from Africa. One Health 20, 100947. https://doi.org/10.1016/j.onehlt.2024.100947 (2025).
Acknowledgements
We thank Yakhat Barshep for facilitating the material transfer agreement on access to genetic resources. We also extend our gratitude to Joy Ishong, Stephen Ezekiel and Monday Okpanachi for assisting during the sample collection and storage. The APLORI management offered great support to this project. We received field sampling materials and sample storage equipment from APLORI. This is contribution no 232 from A.P. Leventis Ornithological Research Institute. We acknowledge the kind reception given by the Head of Department and staff of Clinical Microbiology, Umeå University, during the lab work.
Funding
Open access funding provided by Linnaeus University. Open access funding provided by Linnaeus University. Formas Early Career Research Grant (2020–01056) and VR– The Swedish Research Council – Viruses and Pandemics (2023–02568).
Author information
Authors and Affiliations
Contributions
Conceptualization: JW, OWL, UO, DAM; methodology and software: DAM, JAI, OWL; formal analysis: EK, DAM; writing—original draft preparation: DAM; review and editing: OWL, JW, JN, AS, UO ED, JAI, LI; supervision and project administration: JW, OWL; funding acquisition and resources: JW and OWL. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Matthew, D.A., Karlsson, E., Izang, J.A. et al. First detection of Sindbis virus in wild birds in Nigeria. Sci Rep 15, 24621 (2025). https://doi.org/10.1038/s41598-025-10556-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-10556-3
