
Abstract
Cancer cells have a higher evolutionary potential than normal cells, which commonly leads to medication resistance and a decrease in the efficacy of existing cancer treatments. As a result, discovering new therapeutic drugs is an important priority in the field of oncology. The tumor suppressor protein p53, regulates many cellular activities but is frequently rendered inactive in malignancies due to aberrant overproduction of MDM2 and MDMX. As a result, the method of targeting MDM2 with small-molecule inhibitors to reactivate p53 signalling has gained popularity as a promising approach for anticancer drug development. In this study, we performed a comprehensive structure-based virtual screening of 261,120 compounds from the Asinex database, along with molecular docking, ADMET profiling, and molecular dynamics (MD) simulations using Schrödinger’s Maestro platform, to identify high-affinity MDM2 binders. electronic properties and stability characteristics have been assessed using Density Functional Theory (DFT) computations. The generated lead compounds had favourable pharmacokinetic features and high binding affinities for MDM2, making them suitable scaffolds for further therapeutic study. Overall, our findings lay the groundwork for experimental validation and drive the hunt for next-generation inhibitors of the p53-MDM2 pathway in cancer therapy.
Similar content being viewed by others

Introduction
Cancer, a leading global health concern, is characterized by uncontrolled cell growth and proliferation. It is the world’s second biggest cause of death and the secondary cause of early mortality in 112 nations (before the age of 70)1. By 2050, cancer diagnoses are predicted to exceed 35 million per year, an astounding 77% rise from 2022, highlighting the twin impact of population aging and increased exposure to risk factors associated with socioeconomic changes. Cancer incidence in India is expected to increase from 1.46 million in 2022 to 1.57 million by 20252. Protein-protein interactions (PPIs) regulate a wide range of biological activities, including gene expression, cell development and shape, food intake, signalling cascades, and death. These interactions have a profound impact on tumor genesis, progression, and metastasis, serving as both facilitators and inhibitors of cancer growth3.
The tumor suppressor protein p53, often referred to as “the guardian of the genome,” is critical for maintaining genomic integrity and defending organisms against cancer4,5,6. In breast tissue, p53 maintains metabolic equilibrium throughout pregnancy, protects against latent breast cancer, and performs traditional activities such as genomic protection, DNA repair, and programmed cell death7. TP53, the gene that encodes p53, is the most frequently altered or inactivated gene in human tumors, accounting for nearly half of all malignancies. The mutation rate varies by cancer type, ranging from less than 5% in cervical cancer to over 90% in ovarian and small-cell lung cancers8,9,10. p53 function is controlled by multiple important proteins, including MDM2, MDMX, TSPO (translocator protein), Bcl-2 (B-cell lymphoma-2), and NFAT1 (nuclear factor of activated T-cells 1)8. In 1993, p53 was named the “Molecule of the Year” because of its “exhilarating possibilities for prevention and cure of cancer“11. Thus, Targeting pathways and proteins involved in p53 regulation has substantial implications for cancer research and treatment development. MDM2 is particularly well characterized for its capacity to control p53 expression via a negative feedback mechanism12.
The MDM2 (mouse double minute 2) gene is located on the long arm 13 ~ 14 of chromosome 12 (12q13 ~ 14) and spans 2372 kb. It contains 12 exons and encodes a 498 amino acid protein12. MDM2 and MDMX bind directly to p53, inhibiting its transactivation activity. MDM2 is elevated in a wide range of malignancies and is associated with an adverse prognosis. Gene amplification, single-nucleotide polymorphisms in the promoter region, increased transcription, and enhanced translation of mdm2 may lead to increased degradation and decreased activity of p5313–15. MDM2 also promotes the ubiquitination of p53 and its destruction by proteasomes16. Thus, resolving the interaction between p53 and MDM2 to harness p53 apoptosis-inducing actions has emerged as a promising anticancer method (Fig. 1). Crystallographic studies of MDM2 in complex with p53 have laid the foundation for identifying a wide range of structurally diverse small-molecule inhibitors that disrupt this interaction.
Drug discovery is typically regarded as a time-consuming and expensive endeavor. On average, it can take months or years from the first identification of a possible therapeutic chemical to its commercial approval. To address these issues, pharmacological repurposing or repositioning has emerged as a promising approach to developing novel cancer medicines. This strategy entails repurposing existing, approved drugs—often de-risked compounds—for new therapeutic applications. Drug repurposing has the potential to drastically lower development costs and timeframes for novel cancer medicines17,18,19. Several notable examples demonstrate the success of this approach, including the preclinical use of Raltegravir, an antiviral drug, in cancer therapy20; Astemizole, an antihistamine, in prostate cancer treatment21; Ketorolac, an anti-inflammatory agent, in ovarian cancer therapy22; and Phenothiazines, antipsychotic drugs, in addressing inflammation and cancer23.

Schematic representation of p53 signaling pathway and mechanisms utilized by MDM2 to inhibit p53.
The introduction of computational tools has transformed the drug development landscape, provide significant and insightful resources24 This new technology, known as computer-aided drug design (CADD), has shown to be extremely efficient in terms of lowering drug development time and costs25. CADD refers to a set of computational tools used to find, create, and analyze prospective drug candidates with specific biological features. CADD’s main components include homology modeling, molecular docking, virtual screening (VS) or virtual high-throughput screening (vHTS), quantitative structure-activity relationship (QSAR) analysis, and three-dimensional (3D) pharmacophore mapping. Together, these techniques improve the precision and efficiency of contemporary drug discovery systems25,26,27,28.
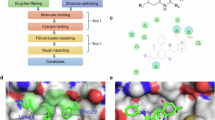
The rapid evolution of lead identification techniques in pharmaceutical research has emphasized the significance of both ligand-based and structure-based computational methods in discovering novel and potent chemical scaffolds designed to disrupt the interaction between the tumor suppressor p53 and its negative regulator MDM2. In this study, To find possible inhibitors, a structure-based drug design technique was used, as illustrated in the lead identification workflow Fig. 2.
The Glide software was used to do site specific virtual screening (VS) on the Asinex database, which contains 261,120 compounds. The top-ranking compounds with the highest binding affinities were then examined using 100-nanosecond Molecular Dynamics (MD) simulations to determine the protein-ligand complexes’ stability. In silico binding studies demonstrated that the selected hits had higher binding affinities to the active regions of the target protein than the co-crystallized ligand EYH. Furthermore, the majority of the discovered inhibitors are commercially available, allowing the scientific community to easily validate and develop them through experimentation. These findings lay a solid platform for developing these compounds as prospective candidates in the search for effective p53-MDM2 inhibitors.
Materials and methods
Protein preparation
PDB code 6GGN is the X-ray crystal structure of human MDM2 protein acquired from the RCSB PDB database. This structure, with a high resolution of 2.0 Å and an R-value of 0.205, illustrates the crucial role of MDM2, an E3 ubiquitin-protein ligase, in controlling the p53 tumor suppressor pathway. The structure consists of a single macromolecular chain (A) of 491 amino acids that is expressed in Escherichia coli BL21 and has no known mutations. The co-crystallized inhibitor, a pyrazolopyrrolidinone derivative (ID: EYH), binds to p53 in MDM2 and forms critical hydrogen bonds and hydrophobic interactions with residues required for stability. The above configuration effectively inhibits the p53-MDM2 interaction, offering critical insights for developing novel chemotherapy molecules that target the p53-MDM2 pathway29,30.
The downloaded PDB file lacks appropriate bonding and hydrogen atoms, making them inappropriate for further study. Hence, Minimizing the protein in molecular modeling leads to the optimal structural conformation in Maestro’s OPLS4 (Optimized Potentials for Liquid Simulations) force field. Schrödinger’s Protein Preparation Wizard (PPW)31 is used to modify the protein structure by eliminating water molecules beyond 5.0 Å of heteroatom, adjusting bond ordering, inserting missing hydrogen atoms, and creating incomplete loops and side chains. Partial charges are assigned, and disulfide bonds are deliberately created by specifying zero bond orders. The Prime module, which is included in PPW, is used to reconstruct missing loops and side chains for realistic modeling. This complete preparation method improves the protein structure, making it ideal for effective docking simulations32,33.
Receptor grid generation
The minimized protein (6GGN) was subsequently processed for grid generation in the receptor grid generation panel. This stage detects the protein’s active binding pocket and generates a grid that depicts the potential energy landscape, including crucial properties such as electrostatic potential, van der Waals interactions, and hydrogen bonding sites. During grid setup, the ligand from the generated protein structure was removed to make sure that it did not affect the computation or interfere with the ligand-receptor docking process. The receptor grid was created using the default options, with limits on rotatable groups and volume exclusion. The coordinates along the X, Y, and Z axes are − 9.44 Å, −11.29 Å, and − 1.03 Å, respectively. The van der Waals radii were scaled with a factor of 1.0 Å, and partial charge cutoff was used to optimize the binding pocket depiction34,35,36.
Ligand preparation and library design
A total of 261,120 compounds were downloaded from the ASINEX Gold and Platinum Collections using freely available web resources (https://www.asinex.com/screening-libraries-(all-libraries)). These molecules were initially in 2D format37 and unsuitable for docking. To deal with this, all compounds were initially arranged into a new database using Schrödinger’s Phase module. The database was created through ligand preparation using the LigPrep module. filtering ligands based on QikProp characteristics, Lipinski’s Rule of Five, and LigFilter qualities to assure drug similarity. ligand preparation using ligprep to validate the developed hypothesis. LigPrep is crucial in removing structural flaws and producing optimal ligand structures for advanced computational research such as glide docking, phase screening, and molecular dynamics simulations. During this procedure, the ligands were desalted, and tautomers with every possible combination were created38. The Epik module was used to predict the ligands’ ionization states at a pH of 7.0 ± 2.0. The ligand preparation step was carried out utilizing the OPLS4 force field and generated in.phdb format39,40.
Development of e-pharmacophore model
A pharmacophore model depicts the spatial arrangement of chemical characteristics of ligands required for interaction with a target receptor41. In this study, the Phase program was used to construct e-pharmacophores for the protein-ligand complexes42. The Phase module uses six built-in pharmacophore properties for model development: hydrogen bond donor (D), hydrogen bond acceptor (A), hydrophobic (H), positive ionizable (P), negative ionizable (N), and aromatic ring (R). Compounds had to align with at least four sites from the produced e-pharmacophore hypothesis. The Phase fitness score objectively assesses how well ligands align with pharmacophore sites, taking into account vector alignments, volume terms, and RMSD of site matching to ensure an accurate and trustworthy assessment of prospective candidates43. Ligands with fitness ratings above 1.0 were deemed effective inhibitors.

Schematic representation of Lead identification workflow based on the Structure-based drug design.
Structure-based virtual screening by molecular docking
Virtual screening is an effective method for finding dynamic substances with potential therapeutic applications. The basic goal of any virtual screening technique is to reduce the large chemical space of small organic molecules to a manageable number of candidates with a better chance of inhibiting the target protein, hence uncovering prospective therapeutic candidates44. In this investigation, virtual screening was carried out using Glide (Grid-based Ligand Docking with Energetics) within the Schrödinger suite’s Maestro interface. Glide is an effective approach for discovering ligand hits and assisting with lead optimization through structure-based virtual screening using molecular docking45,46. Molecular docking is regarded as a highly reliable computational approach for determining the orientation of ligand molecules within the active site of target proteins and elucidating the specific mechanisms of substrate or inhibitor selection and binding47,48. The technique had three steps of docking: (i) Glide high-throughput virtual screening (HTVS), (ii) Glide SP (standard precision), and (iii) Glide XP (extra precision). Glide SP (Standard Precision) is intended for speedy docking and early virtual screening of large libraries, striking a balance between speed and precision whereas Glide XP (Extra Precision) produces more precise and extensive docking results with better score, but it demands more processing resources49. After screening 59,126 molecules using HTVS the top 20% molecules were chosen for SP docking followed by top 20% molecules were selected for Glide XP docking.
Drug-likeness and ADMET predictions
Risk assessment is important in the early phases of drug discovery because it evaluates the safety profile and biological behavior of small-molecule ligands. Drug-likeness screening is an important phase in this procedure, which seeks to find compounds with desirable pharmacokinetic and pharmacodynamic properties50. A large proportion of drug candidates fail clinical trials due to poor pharmacokinetics and poor ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties. To address these issues, the ADMET characteristics of possible drug candidates were evaluated using Maestro’s QikProp module51. QikProp helps to evaluate drug-likeness by predicting several physicochemical and pharmacokinetic properties. These include molecular weight, hydrogen bond donor and acceptor counts, the anticipated octanol/water partition coefficient (QlogPo/w), polar surface area (PSA), human oral absorption percentage, and apparent Caco-2 cell permeability (QPPCaco2)52,53. Following these predictions, additional validation in vitro investigations are required to fully assess the compounds’ therapeutic potential and safety profile54.
Calculation of binding free energy by using MM-GBSA
Binding affinities reported by docking calculations may be unreliable as independent criteria for ranking molecules55. The Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) method is a more precise way to estimate ligands’ relative binding affinities to a target. This technique not only predicts binding free energy with greater precision, but it also provides useful insights by breaking it down into interaction and desolvation components56. In this study, the MM-GBSA approach, which is implemented in Prime, was used to rescore docked ligand poses. These docked postures served as inputs for the energy minimization of protein-ligand complexes (Ecomplex), unbound proteins (Eprotein), and free ligands (Eligand)57. The binding free energy (ΔGbind) was determined with the following equation:
Molecular dynamics (MD) simulation
The function and dynamics of protein-ligand complexes have historically been investigated using molecular dynamics (MD) simulations. Unlike biological processes that entail the dissolution of proteins and ligands in water, molecular docking cannot fully reproduce these phenomena. To acquire a better understanding of the protein-ligand complex’s stability, a 100 ns MD simulation was run with Desmond58. The complexes were prepared for MD simulation by solvating them within Simple Point Charge (SPC) water model. The simulation grid box was neutralized by adding cations (Na⁺) and anions (Cl⁻). Throughout the simulation, the Martyna-Tobias-Klein barostat and Nose-Hoover thermostat methods kept the pressure (1 atm) and temperature (300 K) constant. The simulation was run for about 100 ns using the NPT ensemble. To analyze the dynamic behavior of the protein-ligand complex, RMSD was calculated for overall stability and RMSF for residue-specific variations59,60.
DFT studies
The Density Functional Theory (DFT) technique has been widely used to study the electronic structural features of molecules61. In this study, the DFT method was used to examine the electronic properties of compounds found using the E-pharmacophore model and conformations created using virtual screening, while also exploring receptor-ligand interactions using quantum chemistry techniques. The bound ligand conformations were subjected to DFT computations using the Schrödinger software suite’s Jaguar module. Geometric optimization was performed using a hybrid DFT approach that included Becke’s three-variable exchange potential and the Lee-Yang-Parr functional (B3LYP) at the 6-31G** basis set level62,63. Vibrational frequency calculations verified that the optimized geometries are actual minima, free of imaginary vibrational frequencies. The following electronic properties were computed and analyzed: frontier molecular orbital energies (EHOMO and ELUMO), energy gap (ΔE), absolute hardness (η), global softness (σ), electron affinity (EA), ionization potential (χ), chemical potential (µ), and electrophilicity index (ω)64.
Result & discussion
Pharmacophore identification
A three-dimensional pharmacophore model was created using the Phase module of Schrödinger program to represent a wide range of structural variations and biological activity. The approach began with the creation of a hypothesis based on the protein-ligand combination of 6GGN and it’s co-crystal ligand EYH. The final pharmacophore model included two Hydrophobic (H9 & H10), one Acceptor (A6), and three aromatic ring (R16, R17 & R18), making a total 6 characteristics that were strategically placed to meet the binding site’s structural and functional criteria. (Fig. 3)

Using PHASE Module (A) e-Pharmacophore Hypothesis (B) e-Pharmacophore Hypothesis with co-crystaline Ligand. The red ball symbolizes a hydrogen bond acceptor, the green ball denotes a hydrophobic group, and the brown rings reflect aromatic properties.
Ligand database screening
The derived e-pharmacophore hypothesis was used to filter the Phase database for promising compounds with structural compatibility and positive interaction characteristics. During the ligand database screening, 59,126 compounds were found to match at least four of the six characteristics specified by the hypothesis.
ADMET studies
Predicting the pharmacokinetic and toxicological characteristics of possible lead compounds in the early stages of drug discovery is an important method for reducing future problems. A drug candidate’s potential can be examined in silico by evaluating its physicochemical features, drug-likeness, and ADMET (absorption, distribution, metabolism, excretion, and toxicity) characteristics. In this study, the ADMET profiles of the top ten lead compounds which 2D structure are given in Fig. 4 were examined using Maestro’s QikProp module. Initially, drug-likeness qualities and conformity with Lipinski’s Rule of Five were evaluated and described in Table 1, along with comparisons to a standard drug. A molecule is druggable if it meets the following criteria: logP ≤ 5, molecular weight (MW) ≤ 500 Da, hydrogen bond acceptors (HBA) ≤ 10, hydrogen bond donors (HBD) ≤ 5, and number of rotatable bonds (nRB) ≤ 10. All ten lead compounds followed Lipinski’s Rule of Five, indicating their appropriateness as potential drug candidates. Effective oral bioavailability is essential for any drug candidate and all ten lead compounds demonstrate excellent oral absorption characteristics. The high absorption percentage indicates that these substances will be efficiently absorbed and then eliminated from the human body after exerting their therapeutic effects. These findings emphasize the selected leads’ potential for further development as orally administrable medicines.

2D Structure of hit molecules identified from ASINEX dataset.
Receptor-based virtual screening
Following the formulation of an E-pharmacophore scheme, Glide was used to conduct structure-based virtual screening (VS) on the 59,126 hits. At each stage of the screening procedure, from HTVS to SP to XP, a smaller subset of molecules proceeded, resulting in better binding accuracy. Finally, the top 10 hits with XP Glide values less than − 8.600 kcal/mol were chosen for further analysis. The fitness scores for the top 10 hits ranged between 1.2 and 1.8.
The study used Serdemetan and MI-773 as reference drugs because of their well-established efficacy and clinical importance. However, our hit compounds had higher binding affinities, with XP G-scores of −8.606 kcal/mol or lower, surpassing those of MI-773 (−6.54 kcal/mol) and Serdemetan (−5.71 kcal/mol). Additionally, the re-docked co-crystal ligand EYH also has a higher binding affinity (−6.552 kcal/mol) than the detected hits.
Insights into the ligand interactions
Figures 5 and 6 shows the 2D and 3D interaction patterns of the top three MDM2 inhibitors, as well as the standard MI-773, Serdemetan, and the co-crystal ligand EYH. Unique colors are used to represent various types of interactions which are also given in Figs. 5 and 6. Table 2 summarizes each compound’s docking scores, MM-GBSA, and ineraction parameters such as H-bond interactions, hydrophobic interactions, pi-pi stacking interactions, No of positive charge interaction, and No of polar interaction. The examined ligands had good to exceptional binding affinities to the MDM2 target, with binding energies ranging from − 9.086 to −8.606 kcal/mol. Compounds L9, L3, and L4 showed the lowest binding energies against MDM2 active site residues (− 9.086, − 8.853, and − 8.820 kcal/mol, respectively). Interestingly, compound L9 has the best docking score and a large MM-GBSA energy of −61.76 kcal/mol, showing potent binding. The MM-GBSA values range from − 73.68 kcal/mol (compound L4) to −39.12 kcal/mol (compound L7), indicating variances in binding energy contributions.The interaction measurements include the number of hydrophobic amino acid contacts (11–16), hydrogen bonds (0–3), π-π stacking interactions (0–2), charged interactions (0–1), and polar interactions (1–4). Compound L4 has the maximum number of hydrophobic contacts (16) and MM-GBSA energy (−73.68 kcal/mol), indicating significant hydrophobic stabilization. Compound L6 has the most H-bond interaction (one with LYS94 and two with HIE96). The majority of compounds showed hydrogen bonding and π-π stacking interactions, emphasizing their importance in binding. Polar and charged interactions contribute to the binding profile, which varies amongst compounds. Serdemetan and MI773, established MDM2 inhibitors, serve as reference compounds and co-crystal ligand EYH, with docking scores of −5.713, −6.545, and − 6.552 kcal/mol, respectively, and lower binding energy values than the Hit molecuels.

2D and 3D Docking representation of the top 3 Leading p53-MDM2 Inhibitors (L3, L4 & L5).

2D and 3D Docking representation of reference compound (EYH: Co-crystal ligand, MI773 & Serdemetan).
Binding free energy calculation using MM-GBSA method
The top hit compounds with high Glide scores were further investigated with MM-GBSA to determine their binding energies and find the most promising inhibitors. The MM-GBSA binding affinity values for these hits ranged from − 73.68 to −49.12 kcal/mol, much greater than reference drug’s binding affinity of −51.14 kcal/mol for MI-773, −38.68 kcal/mol for serdemetan and comparative with co-crystal ligand EYH’s binding affinity of −60.70 kcal/mol.
Molecular dynamics simulation analysis
Molecular dynamics (MD) simulation is a computational technique that uses complex algorithms to predict and assess the stability of molecular systems. In this study, MD simulations were used to test the stability of receptor-ligand complexes, confirm anticipated binding modes, and analyze putative interactions discovered earlier using Glide XP docking. MD simulations have the advantage of accurately replicating biological settings, allowing for a dynamic picture of molecular interactions. The top ten hits from structure-based virtual screening were subjected to 100 ns MD simulations. Important MD parameters, such as root mean square deviation (RMSD) and root mean square fluctuation (RMSF), as well as 2D ligand-protein interaction diagrams and the fraction of interactions with active site residues, were obtained from the simulated trajectories. These findings gave important insights into the complexes’ stability and binding processes at near-physiological circumstances.
Protein-ligand root mean square deviation (RMSD)
Root Mean Square Deviation (RMSD) quantifies the changes in conformation of a protein and ligand throughout MD simulations, offering insights into structural stability and variability. The RMSD of the protein backbone across a 100 ns simulation timeframe is valuable for assessing the dynamic characteristics of apo-proteins in comparison to protein-ligand complexes. The analysis (Fig. 7) of ligand RMSD reveals that compounds L3, L4 and L5 exhibit minimal deviation from their original positions, suggesting stable binding within the active site.

Protein-ligand RMSD of L3, L4, L5 and MI773 (S2).
The RMSD analysis of compound L3 shows that it starts off with a stable conformation, marked by a low RMSD of approximately 0.75 Å at t = 0 ns. Throughout the simulation, the RMSD gradually rises to around 1.25 Å by 10 ns, suggesting minor structural changes have occurred. From 10 to 60 ns, the RMSD remains consistently around 1.25 Å, with slight variations, indicating that the protein has reached a state of equilibrium. Nevertheless, between 60 and 70 ns, there is a temporary increase in RMSD (~ 2.00 Å), signifying conformational changes. Following 70 ns, the RMSD levels off with slight fluctuations, reflecting a structurally stable system with occasional variations. The ligand RMSD for compound L3 varies slightly across the simulation, ranging from 1.5 Å to 5.6 Å. The ligand initially binds fairly stable, but after 60 ns, the RMSD temporarily increases to around 5.6 Å may indicating conformational adjustments event. Nevertheless, after 70 ns, the ligand achieves stability with modest variations, showing that it is still attached while making small positional changes inside the binding pocket.
For the standard L4 the protein RMSD reveals that an early phase of stabilization during the first 20 ns, with an average variation of roughly 1.0 to 1.3 Å, showing a relatively stable protein structure. During 20–70 ns, the RMSD stabilise with slightly change between 0.6 to1.25 Å. However, after 70 ns its decrese to 0.4 Å suggest minor struture changes may occurs.
The protein RMSD analysis for compound L5 suggest that the structure is initially stable, displaying an RMSD of approximately 0.95 Å at t = 0 ns. During the first 10–20 ns, the RMSD gradually increases and settles between 1.25 and 1.50 Å, suggesting minor structural changes. In the range from 60 to 80 ns, there is a moderate degree of variability, with the RMSD reaching around 2.10 Å, which indicates temporary conformational shifts. Even so, after 80 ns, the RMSD levels off with slight fluctuations, signifying that the system has achieved a relatively stable state. Similarly, the ligand’s RMSD remains within a reasonable range 2.5–3.5 Å during the simulation, indicating that the ligand maintains binding stability at the active site despite minor variations. During the first 60–70 ns, the RMSD gradually increases between 3.00 and 4.00 Å, suggesting minor structural changes and after 70 ns struture achieved a stable state. Perhaps, our data show that L5 maintains a stable contact with the protein, with minor conformational changes happening throughout simulation.
The protein RMSD analysis for Standard S2 show that the structure is initially stable, marked by a low RMSD of approximately 0.90 Å at t = 0 ns. Throughout the simulation, the RMSD gradually rises to around 1.8 Å by 15 ns, suggesting minor structural changes have occurred. From 10 to 60 ns, the RMSD remains consistently between 1.5 Å to 1.8 Å, with slight variations, indicating that the protein has reached a state of equilibrium. In the range from 60 to 80 ns, there is a moderate degree of fluctuations, with the RMSD gradually increae and reaching around 2.5 Å, which indicates temporary conformational shifts. After 80 ns, the RMSD stabilizes with slight changes, indicating that the system is in a relatively stable state. whereas the ligand exhibited significant dynamic behavior, with RMSD fluctuations ranging from 1.5 Å to 4.8 Å. This substantial movement shows that the ligand is highly flexible and that conformational change may occur within the binding site. The marked differences in ligand RMSD could imply that the ligand analyzes a broader range of binding configurations or moves more freely.
Root means square fluctuation (RMSF)
The root mean square fluctuation (RMSF) analysis provides insights into the structural flexibility of the protein-ligand complex by measuring atomic fluctuations throughout time. The peak in the RMSF figure represented the protein’s variations during the simulation procedure, with lower RMSF values indicating fewer conformational changes65. Figure 8 represent RMSF graph for L3, L4, L5 & standard compound MI773 (S2).

RMSF of L3, L4, L5 & MI773 (S2).
Lower RMSF values were primarily observed in central residues (residue indices ~ 35–85) throughout all four complexes, indicating stable interactions within the protein’s core. All compounds showed intermediate fluctuation patterns, with slightly higher mobility around residue indices 20–30 and 50–60. L3 and L4 had reduced fluctuations, with RMSF values typically under 1.5 Å throughout the structure, indicating a more rigid and stable binding conformation. S2 had stronger RMSF peaks (up to ~ 3.9 Å) at the ends, indicating more local flexibility or weaker binding stability. Overall, these findings suggest that compound L4 promotes the most stable protein conformation, which may indicate a more beneficial binding relationship than L3, L5, and S2.
Protein ligand contacts
Figure 9 shows the statistical analysis of key protein-ligand interactions found during the molecular dynamics (MD) simulation, including hydrogen bonds, hydrophobic interactions, ionic interactions, and water bridges.

protein-ligand interactions of L3, L4, L5 & MI773 (S2).
Interactions between L3 and protein are made up of hydrogen bonds, water bridges and hydrophobic interactions. The residues TYR 67, GLN 72, and LYS 94 form H-bonds with L3, indicating polar interactions between the ligand’s functional groups and the side chains of these amino acids. GLN 72 forms hydrogen bonds and water bridges of up to 0.8 Å. In addition to hydrogen bonds, TYR 67 and LYS 94 form hydrophobic interactions and water bridges with the ligand.
Compound L5’s protein-ligand interactions were predominantly affected by three key residues GLN18, ILE19, and GLN24 that played an important role in the complex’s stability via hydrogen bonding. Hydrogen bonds at ILE19 were discovered to be as near as 0.44 Å, with further reinforcement via hydrophobic interactions and water bridges reaching up to 0.5 Å. Hydrogen bonding interactions at GLN18 and GLN24 extended up to 0.20 Å, whereas additional water bridges raised interactions to a high value of 0.3 Å. These molecular interactions helped to stabilize the protein-ligand complex.
As observed in the interaction fraction plot for L4, GLN24 exhibited both hydrogen bonding and water bridge interactions, contributing to a total interaction fraction of approximately 0.20. Other residues did not exhibit significant hydrogen bonding, indicating that classical hydrogen bonds have a modest role in the stability of L4 binding. Among all the residues implicated in interactions, HIS96 had the highest interaction fraction, primarily through hydrophobic contacts, with additional stabilizing from water bridges. Ten residues, on the other hand, showed minimal to no interactions throughout the simulation, implying that they do not contribute to ligand stability.
The Standard S2 exhibits protein-ligand interactions primarily via three H-bonds with residues GLN 18, LEU 54, and GLN 72. GLN 18 has a low interaction fraction, while LEU 54 and GLN 72 play a greater part, forming hydrogen bonds up to 0.9 Å and 0.6 Å, respectively. Water bridges stabilize the complex by extending ligand-residue interactions at GLN 72 to 0.8 Å.
Density function theory analysis
DFT calculations were used to determine the electronic properties of 10 lead compounds. Table 3 provides the statistical information for these molecular characteristics while Fig. 10 shows contour maps of the HOMO and LUMO orbitals for the top three candidates.

Contour maps of the frontier molecular orbitals (HOMO and LUMO) of the top three p53-MDM2 inhibitors L3, L4 & L5 with standard compound (EYH: Co-crystal ligand, MI773 & Serdemetan).
The energy gap (ΔE) determines the stability and reactivity of compounds, with higher gaps indicating greater stability. All 10 lead compounds had relatively significant energy gaps, indicating their great stability. Compound L9 has the largest energy gap at 5.4079 eV, with EHOMO and ELUMO values of −6.1287 eV and − 0.7208 eV, respectively. Compound L1 had the narrow energy gap of 4.1114 eV, with EHOMO and ELUMO values of −5.6354 eV and − 1.5239 eV, respectively, making it slightly least stable and high reactive of the ten lead molecules.
The electrophilicity index (ω) helps determine a molecule’s overall electrophilic nature. A greater electrophilicity index indicates a stronger ability to receive electrons, making the molecule more reactive to nucleophiles. The ionization potential (IP) is the energy needed to remove an electron from a molecule. A higher IP indicates increased stability and a lesser tendency to lose lectrons. In contrast, a smaller IP suggests a greater potential to lose electrons. Electron affinity (EA) and electronegativity (χ) values measure a compound’s capacity to attract electrons. Compound L1 had the highest electron affinity (EA = 1.5239 eV), indicating a robust electron-accepting capability. This suggests that compound L1 could behave as a strong electron acceptor in chemical processes. Global hardness (η) and softness (σ) indicate molecule polarizability and reactivity. A higher global hardness indicates better stability and less reactivity, whereas a lower global hardness (higher global softness) indicates more reactivity. A narrower energy gap between HOMO and LUMO is frequently associated with increased global softness and responsiveness.
Conclusion and future prospects
Many human malignancies have aberrant MDM2 overexpression, which allows cancer cells to bypass cell cycle control and promote unregulated growth and spread. As a result, altering the p53-MDM2 binding relationship is a promising strategy for creating anticancer agents. To discover new MDM2 inhibitors, we used a comprehensive computational strategy that included structure-based virtual screening, molecular docking, ADMET profiling, molecular dynamics (MD) simulations, Density Functional Theory (DFT) calculations. The candidates that were examined had higher binding affinities (−9.086 to −8.606 kcal/mol) than the reference inhibitor. They exhibited acceptable pharmacokinetic features and met druggability standards. Additionally, molecular dynamic simulations demonstrated that all of the selected hits L3, L4 & L5 formed stable protein-ligand complexes throughout the simulation time. All ten molecules showed HOMO–LUMO gap values that are similar to reference, MI-773 and the co-crystallized ligand EHY, indicating comparable electronic characteristics and the possibility of similar reactivity and stability. To enhance this promising strategy, more in-vitro and in-vivo research are needed to ensure rigorous validation. This study not only highlights the possibility of repurposing existing medications as p53-MDM2 inhibitors, but it also paves the way for the creation and development of novel analogues based on these scaffolds, providing new paths for battling this fatal disease.
Data availability
The ASINEX Gold and Platinum Collections were obtain from using freely available web resources (https://www.asinex.com/screening-libraries-(all-libraries)).
References
Society American Cancer. Global cancer facts and figures. American Cancer Society 1–48 at. (2024).
Sathishkumar, K., Chaturvedi, M., Das, P., Stephen, S. & Mathur, P. Cancer incidence estimates for 2022 & projection for 2025: result from National Cancer registry programme, India. Indian J. Med. Res. 156, 598 (2022).
Sowmya, G. & Ranganathan, S. Protein-Protein interactions and prediction: A comprehensive overview. Protein Pept. Lett. 21, 779–789 (2013).
Vogelstein, B., Lane, D. & Levine, A. J. Surfing the p53 network. Nature 408, 307–310 (2000).
Toledo, F. & Wahl, G. M. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat. Rev. Cancer. 6, 909–923 (2006).
Vousden, K. H. & Lane, D. P. p53 in health and disease. Nat. Rev. Mol. Cell. Biol. 8, 275–283 (2007).
Moulder, D. E., Hatoum, D., Tay, E., Lin, Y. & McGowan, E. M. The roles of p53 in mitochondrial dynamics and cancer? metabolism: the pendulum between survival and death in breast cancer?? Cancers (Basel). 10, 189 (2018).
Joerger, A. C. & Fersht, A. R. The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annu. Rev. Biochem. 85, 375–404 (2016).
Sabapathy, K. & Lane, D. P. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 15, 13–30 (2018).
Comprehensive genomic characterization. Defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008).
Koshland, D. E. Molecule of the year. Sci. (80-). 262, 1953–1953 (1993).
Zhu, H. et al. Targeting p53–MDM2 interaction by small-molecule inhibitors: learning from MDM2 inhibitors in clinical trials. J. Hematol. Oncol. 15, 91 (2022).
Quintás-Cardama, A. et al. p53 pathway dysfunction is highly prevalent in acute myeloid leukemia independent of TP53 mutational status. Leukemia 31, 1296–1305 (2017).
Momand, J. The MDM2 gene amplification database. Nucleic Acids Res. 26, 3453–3459 (1998).
Shangary, S. & Wang, S. Small-Molecule inhibitors of the MDM2-p53 Protein-Protein interaction to reactivate p53 function: A novel approach for Cancer therapy. Annu. Rev. Pharmacol. Toxicol. 49, 223–241 (2009).
DeVine, T. & Dai, M. S. Targeting the Ubiquitin-Mediated proteasome degradation of p53 for Cancer therapy. Curr. Pharm. Des. 19, 3248–3262 (2013).
Oprea, T. I. et al. Drug repurposing from an academic perspective. Drug Discov Today Ther. Strateg. 8, 61–69 (2011).
Park, K. A review of computational drug repurposing. Transl Clin. Pharmacol. 27, 59 (2019).
Pushpakom, S. et al. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 18, 41–58 (2019).
Alburquerque-González, B. et al. The FDA-Approved antiviral raltegravir inhibits Fascin1-Dependent invasion of colorectal tumor cells in vitro and in vivo. Cancers (Basel). 13, 861 (2021).
Garcia-Quiroz, J. & Camacho, J. Astemizole: an old Anti-histamine as a new promising Anti-cancer drug. Anticancer Agents Med. Chem. 11, 307–314 (2011).
Guo, Y. et al. A novel Pharmacologic activity of ketorolac for therapeutic benefit in ovarian Cancer patients. Clin. Cancer Res. 21, 5064–5072 (2015).
Rácz, B. & Spengler, G. Repurposing antidepressants and phenothiazine antipsychotics as efflux pump inhibitors in Cancer and infectious diseases. Antibiotics 12, 137 (2023).
Tiwari, A. & Singh, S. Computational approaches in drug designing. in Bioinformatics 207–217Elsevier, (2022). https://doi.org/10.1016/B978-0-323-89775-4.00010-9
Yu, W. & MacKerell, A. D. Computer-Aided drug design methods. in 85–106 (2017). https://doi.org/10.1007/978-1-4939-6634-9_5
Surabhi, S., Singh, B. & COMPUTER AIDED DRUG DESIGN: AN OVERVIEW. J. Drug Deliv Ther. 8, 504–509 (2018).
Veselovsky, A. & Ivanov, A. Strategy of Computer-Aided drug design. Curr. Drug Target. -Infectious Disord. 3, 33–40 (2003).
Hassan Baig, M. et al. Computer aided drug design: success and limitations. Curr. Pharm. Des. 22, 572–581 (2016).
Vaupel, A. et al. In vitro and in vivo characterization of a novel, highly potent p53-MDM2 inhibitor. Bioorg. Med. Chem. Lett. 28, 3404–3408 (2018).
In vitro and in vivo characterization of a novel, highly potent p53-MDM2 inhibitor. at (2018). https://doi.org/10.2210/pdb6ggn/pdb
Yadav, S., Ahamad, S., Gupta, D. & Mathur, P. Lead optimization, pharmacophore development and scaffold design of protein kinase CK2 inhibitors as potential COVID-19 therapeutics. J. Biomol. Struct. Dyn. 41, 1811–1827 (2023).
Madhavi Sastry, G., Adzhigirey, M., Day, T., Annabhimoju, R. & Sherman, W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 27, 221–234 (2013).
Shelley, J. C. et al. Epik: a software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 21, 681–691 (2007).
Hasan, Y. & Al-hamashi, A. Identification of Selisistat derivatives as SIRT1-3 inhibitors by in Silico virtual screening. Turkish Comput. Theor. Chem. 8, 1–11 (2024).
Halgren, T. A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 49, 377–389 (2009).
Sahayarayan, J. J. et al. In-silico protein-ligand Docking studies against the Estrogen protein of breast cancer using pharmacophore based virtual screening approaches. Saudi J. Biol. Sci. 28, 400–407 (2021).
Perez, J. J. Managing molecular diversity. Chem. Soc. Rev. 34, 143 (2005).
Chen, I. J. & Foloppe, N. Drug-like bioactive structures and conformational coverage with the ligprep/confgen suite: comparison to programs MOE and catalyst. J. Chem. Inf. Model. 50, 822–839 (2010).
Roos, K. et al. OPLS3e: extending force field coverage for Drug-Like small molecules. J. Chem. Theory Comput. 15, 1863–1874 (2019).
Lu, C. et al. OPLS4: improving force field accuracy on challenging regimes of chemical space. J. Chem. Theory Comput. 17, 4291–4300 (2021).
Schaller, D. et al. Next generation 3D pharmacophore modeling. WIREs Comput. Mol. Sci 10, e1468. https://doi.org/10.1002/wcms.1468 (2020).
Salam, N. K., Nuti, R. & Sherman, W. Novel method for generating Structure-Based pharmacophores using energetic analysis. J. Chem. Inf. Model. 49, 2356–2368 (2009).
Dixon, S. L. et al. PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 20, 647–671 (2006).
Vyas, V. & Virtual Screening A fast tool for drug design. Sci. Pharm. 76, 333–360 (2008).
Chen, Z. et al. Pharmacophore-based virtual screening versus docking-based virtual screening: a benchmark comparison against eight targets. Acta Pharmacol. Sin. 30, 1694–1708 (2009).
Halgren, T. A. et al. Glide: A new approach for rapid, accurate Docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 47, 1750–1759 (2004).
Sharma, B., Bhattacherjee, D., Zyryanov, G. V. & Purohit, R. An insight from computational approach to explore novel, high-affinity phosphodiesterase 10A inhibitors for neurological disorders. J. Biomol. Struct. Dyn. 41, 9424–9436 (2023).
Sharma, B. & Purohit, R. Enhanced sampling simulations to explore Himalayan phytochemicals as potential phosphodiesterase-1 inhibitor for neurological disorders. Biochem. Biophys. Res. Commun. 758, 151614 (2025).
Enyedy, I. J. & Egan, W. J. Can we use Docking and scoring for hit-to-lead optimization? J. Comput. Aided Mol. Des. 22, 161–168 (2008).
Chukwuemeka, P. O. et al. Targeting dysregulation signatures of p53-MDM2 interactions to identify newer small molecule inhibitors for cancer therapy: Insight from computational direction. at (2021). https://doi.org/10.21203/rs.3.rs-217761/v1
Ahamad, S., Kanipakam, H., Birla, S., Ali, M. S. & Gupta, D. Screening Malaria-box compounds to identify potential inhibitors against SARS-CoV-2 mpro, using molecular Docking and dynamics simulation studies. Eur. J. Pharmacol. 890, 173664 (2021).
Haider, K. et al. Design, synthesis, biological evaluation, and in Silico studies of 2-aminobenzothiazole derivatives as potent PI3Kα inhibitors. Arch Pharm. (Weinheim) 355 (10), 2200146. https://doi.org/10.1002/ardp.202200146 (2022).
Ioakimidis, L., Thoukydidis, L., Mirza, A., Naeem, S. & Reynisson, J. Benchmarking the reliability of qikprop. Correlation between experimental and predicted values. QSAR Comb. Sci. 27, 445–456 (2008).
Perkin, V. O., Antonyan, G. V., Radchenko, E. V. & Palyulin, V. A. Web services for the prediction of ADMET parameters relevant to the design of neuroprotective drugs. in 465–485 (2023). https://doi.org/10.1007/978-1-0716-3311-3_16
Sirous, H., Chemi, G., Campiani, G. & Brogi, S. An integrated in Silico screening strategy for identifying promising disruptors of p53-MDM2 interaction. Comput. Biol. Chem. 83, 107105 (2019).
Hayes, J. M. et al. Kinetics, in Silico docking, molecular dynamics, and MM-GBSA binding studies on prototype indirubins, KT5720, and staurosporine as phosphorylase kinase ATP‐binding site inhibitors: the role of water molecules examined. Proteins Struct. Funct. Bioinforma. 79, 703–719 (2011).
Abdullah, J. A. et al. Synthesis, characterization and in-silico assessment of novel Thiazolidinone derivatives for cyclin-dependent kinases-2 inhibitors. J. Mol. Struct. 1223, 129311 (2021).
Bowers, K. J. et al. Molecular dynamics—Scalable algorithms for molecular dynamics simulations on commodity clusters. in Proceedings of the ACM/IEEE conference on Supercomputing - SC ’06 84 (ACM Press, New York, New York, USA, 2006). 84 (ACM Press, New York, New York, USA, 2006). (2006). https://doi.org/10.1145/1188455.1188544
P., G. & M. K., K. Docking studies and molecular dynamics simulation of Triazole benzene sulfonamide derivatives with human carbonic anhydrase IX Inhibition activity. RSC Adv. 11, 38079–38093 (2021).
Huang, C., Li, C., Choi, P. Y. K., Nandakumar, K. & Kostiuk, L. W. A novel method for molecular dynamics simulation in the isothermal–isobaric ensemble. Mol. Phys. 109, 191–202 (2011).
Ong, C. B., Ng, L. Y. & Mohammad, A. W. A review of ZnO nanoparticles as solar photocatalysts: synthesis, mechanisms and applications. Renew. Sustain. Energy Rev. 81, 536–551 (2018).
Jana, S. & Singh, S. K. Identification of selective MMP-9 inhibitors through multiple e-pharmacophore, ligand-based pharmacophore, molecular docking, and density functional theory approaches. J. Biomol. Struct. Dyn. 37, 944–965 (2019).
Ahamed, J. I. et al. A combined experimental and DFT computations study of novel (E)-3-(benzofuran-2-yl)-2-(thiophen-2-yl)acrylonitrile(TACNBNF): insight into the synthesis, single crystal XRD, NMR, vibrational spectral analysis, in vitro antioxidant and in Silico molecular d. J. Mol. Struct. 1202, 127241 (2020).
Ismael, M., Abdel-Mawgoud, A. M. M., Rabia, M. K. & Abdou, A. Design and synthesis of three Fe(III) mixed-ligand complexes: exploration of their biological and phenoxazinone synthase-like activities. Inorganica Chim. Acta. 505, 119443 (2020).
Ahmad, S. et al. Molecular dynamics simulation and docking studies reveal NF-κB as a promising therapeutic drug target for COVID-19. at (2021). https://doi.org/10.21203/rs.3.rs-469785/v2
Acknowledgements
The authors are thankful to the Department of Chemistry, Gujarat University, Ahmedabad, Gujarat, India, for providing the necessary Schrödinger software facilities. UGC-Info net and INFLIBNET Gujarat University are acknowledged for providing other e-sources.
Funding
One of the authors (Dushyant D. Kotadiya) is thankful to CSIR NET Senior Research Fellowship (Grant No: 09/0070(13660)/2022-EMR-I) for financial support.
Author information
Authors and Affiliations
Contributions
Dushyant D Kotadiya : Performed Computational experiments, Writing original draft preparation including figures and Conceptualization. Apurva Prajapati: Computational study. Dharmesh Patel, Pooja Thakur, Ruchi Nair: Created Figures, Reviewing, Editing. Hitesh D Patel: Supervision, Writing- Reviewing and Editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kotadiya, D.D., Prajapati, A., Patel, D.A. et al. In silico identification of prospective p53-MDM2 inhibitors from ASINEX database using a comprehensive molecular modelling approach. Sci Rep 15, 34687 (2025). https://doi.org/10.1038/s41598-025-10589-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-10589-8
