
Abstract
The examination of the current-voltage properties of protein chains connected to armchair graphene nanoribbon leads is performed using the tight-binding Hamiltonian approach in conjunction with the Landauer–Büttiker formalism. The nonlinear current-voltage behavior for three different conformations of the protein structures is calculated and evaluated in connection with the corresponding transmission probabilities. The characteristics of current-voltage demonstrate fluctuations in relation to variations in temperature or the width of the electrodes, with modifications in the conformations of protein chains also playing a significant role in these phenomena. An increase in coupling is associated with a corresponding rise in electric current, suggesting a strong interaction between the electrodes and the device. This research serves as a benchmark for potential investigations into diseases or for future applications within the realm of nanotechnology.
Similar content being viewed by others


Introduction
A protein is a complex biological macromolecule composed of a sequence of amino acid residues1. Amino acids are linked together in a linear fashion during the process of protein synthesis through the establishment of peptide bonds. This sequence of peptide bonds creates a backbone for the protein, from which the diverse side chains extend. In contrast to the structures of other biological macromolecules, proteins exhibit intricate and irregular configurations. There exists a total of twenty distinct amino acids that serve as the building blocks for all proteins. The linear arrangement of these amino acids is referred to as the primary structure of the protein. Furthermore, under physiological conditions, each protein undergoes a process of folding into a unique three-dimensional (3D) configuration, known as the native tertiary structure. This native conformation is crucial as it dictates the functional capabilities of the protein. The relationship between the amino acid sequence of a protein and its native tertiary structure remains one of the most challenging inquiries in the field of biology. Many computational techniques have been developed to predict protein structure, but few of these methods are rigorous techniques for which mathematical guarantees can be described. Lattice models have demonstrated significant utility in analyzing the complexity of protein structures2. These models can be used to extract essential principles, make predictions, and unify our understanding of many different properties of proteins3.
A fundamental challenge in the development of molecular devices lies in the necessity for a comprehensive understanding of the electron transport characteristics of individual molecules. This knowledge serves as the foundation for the design of nanoelectronic components at the molecular level, enabling the creation of functional elements such as molecular switches, bio-rectifiers, and bio-transistors4,5. The primary emphasis of these studies has been on the electronic technologies required for the production of nanodevices, the development of smaller and faster electro-optical circuits, and the enhancement of genetic management of hereditary diseases. Proteins are frequently regarded as the most promising biomolecules due to their significant role in bioelectronics and their essential function in electron transfer mechanisms within living organisms. Initial investigations into electron transport within proteins commenced with peptides, which are frequently regarded as the portions of proteins characterized by relatively uncomplicated frameworks6,7,8,9,10. Furthermore, quantum transport within proteins plays a critical role in determining the efficiency and velocity of essential biological processes, especially those related to electron transfer11,12. This phenomenon enables particles to circumvent energy barriers via a quantum effect known as tunneling, which is vital for various functions, including photosynthesis, cellular respiration, and enzyme catalysis in living organisms.
The geometric characteristics of proteins pertain to the influence of their 3D conformation and the spatial organization of amino acids on the protein’s architecture, stability, and functionality. These geometric attributes, which encompass hydrogen bonding, van der Waals forces, and the comprehensive folding configuration, determine the manner in which proteins engage with other molecules and execute their biological functions13,14,15. Moreover, analytical approaches such as geometricus and the study of geometric effects in protein interactions employ geometric representations of proteins to examine their structure, forecast the implications of mutations, and elucidate the interactions among proteins. Recent studies emphasize the crucial role of geometric configurations and surface characteristics in determining the behavior of proteins and biomaterials, which in turn affects cellular interactions, tissue regeneration, and the efficacy of drug delivery systems16,17,18,19,20. Investigations focus on the utilization of engineered protein arrangements, altered biomaterial surfaces, and the 3D architecture of scaffolds to influence biological responses. The framework for geometric effects in protein interactions utilizes deep geometric representations of protein complexes to analyze the impact of mutations on binding affinity. It incorporates a graph neural network to derive graphical features and employs a gradient-boosting tree for predicting binding affinity16.
The study of quantum transport in proteins is crucial as it forms the basis of essential biological functions and may influence the advancement of innovative technologies. Quantum phenomena, including electron tunneling, are integral to processes such as energy conversion, electron transfer, and the specific interactions between proteins and their ligands21. These interactions affect a range of functions, from oxygen binding in myoglobin to the operation of light-harvesting complexes22,23,24. It is important to note that the energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) in proteins cannot be directly compared to those of small molecules. In the context of proteins, these levels typically manifest as broad bands instead of distinct energy levels. We note that the protein models utilized in this study encompass thirty-six sub-sites, and we anticipate that the energy levels will exhibit discrete behavior due to the confinement of electrons within limited dimensions. Furthermore, the interaction between proteins and ligands is characterized by the interaction of the protein’s LUMO with the ligand’s HOMO, rather than the reverse, as indicated in21. Numerous vital biological activities, such as energy generation and cellular signaling, are propelled by quantum effects. For instance, electron transfer within protein-based junctions can occur through quantum tunneling rather than merely classical electron movement25. Consequently, proteins are capable of harnessing quantum mechanics to effectively transform energy into usable forms for biochemical reactions, while also functioning as nano-engines that sustain ordered states away from equilibrium. Furthermore, quantum effects can alter the interactions of proteins with other molecules, including small compounds. A notable example is the significance of the quantum mechanical characteristics of the iron atom in myoglobin, which are essential for its ability to bind oxygen26. Thus, a comprehensive understanding of quantum transport in proteins may pave the way for the creation of novel materials and technologies, such as bioelectronic devices. Additionally, quantum mechanical effects can shape the interactions between drugs and proteins, thereby influencing drug design and effectiveness.
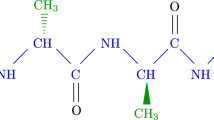
The aim of this study is to investigate electronic quantum transport within a 3D lattice model of protein chains, which are depicted in three distinct conformations (Fig. 1) referred to as \(\mathrm {P_{1}}\), \(\mathrm {P_{2}}\), and \(\mathrm {P_{3}}\)27,28. These models are characterized by a distinct amino acid sequence, exhibiting variations in the distribution of hydrogen bonds; a total of thirteen hydrogen bonds are employed to replicate a mutation associated with a particular disease in a natural protein28. Our analysis concentrates on protein chains comprising thirty-six amino acids28. The \(\mathrm {P_{1}}\) conformation exhibits a straightforward structural arrangement comprised of amino acids linked by covalent (peptide) bonds (Fig. 1a). Conversely, the \(\mathrm {P_{2}}\) and \(\mathrm {P_{3}}\) conformations display a structural arrangement where amino acids are connected by both peptide bonds and non-covalent (hydrogen bond) interactions (Fig. 1b). It is important to highlight that novel strategies addressing the persistent contact challenges in nanodevices frequently utilize carbon allotropes, which enable efficient interaction with macroscopic gold contacts29,30. Moreover, graphene nanoribbons (GNRs) present a simpler alternative for contact materials. The benefits of these contacts include their thermal stability, adaptability in binding various molecules-especially \(\pi -\pi\) stacked aromatic rings-and a decrease in screening effects due to improved gate coupling. Armchair GNR (aGNR) possess an armchair-shaped cross-section at their edges and are commonly referred to as W-aGNR, where W signifies the count of armchair chains along the width. It is expected that aGNRs may demonstrate metallic characteristics based on their width, particularly when \(W=3r+2\), with r being a positive integer31. Consequently, an analysis of electronic transport was performed by placing the protein between two metallic aGNR electrodes. It is essential to recognize that proteins possess electronic characteristics that stem from their amino acid makeup, especially the inclusion of aromatic residues and their capacity to engage in \(\pi\)-interactions. These factors play a significant role in determining the folding and stability of proteins. Conversely, the tight-binding (TB) framework is a theoretical model used to analyze the electronic properties of different materials, employing a Hamiltonian that accounts for several contributing elements. Therefore, this research utilized the TB Hamiltonian model32,33,34,35. Indeed, a TB model of amino acids represents a computational methodology in biophysics that approximates the electronic interactions among amino acids in a protein. This approach utilizes a simplified framework derived from TB theory in solid-state physics, facilitating the assessment of properties such as charge transfer and electronic structure within a substantial biomolecule by concentrating on the most significant interactions between adjacent amino acid residues. Rather than computing the complex specifics of electron distribution, the model emphasizes the principal interaction sites on the protein molecule, conceptualizing them as lattice points with localized orbitals. In order to analyze the quantum transport properties of the protein device utilizing the TB model, we explored the transmission probability (\(\textrm{TP}\)) and current-voltage (\(\mathrm {I-V}\)) characteristics using the Landauer-Büttiker formulation36. This investigation took into account a range of temperatures, varying widths of the aGNR leads (electrodes), and different values of the hopping parameters within the protein chain (device). The electrical current demonstrated a nonlinear behavior, affected by changes in the width and temperature of the leads, which can convert the nonlinear response into a more linear one. Ultimately, the varying hopping parameters modify both the step dimensions in the \(\mathrm {I-V}\) curve and the height and position of the peaks in the \(\textrm{TP}\). An enhancement in coupling correlates with an elevation in electric current, indicating a significant interaction between the leads and the device.
TB Hamiltonian model
The TB Hamiltonian model encompassing the entire system, which includes the protein (\(\hat{{{\mathcal {H}}}}_{\textrm{P}}\)), the aGNR leads denoted by \(\hat{{{\mathcal {H}}}}_{\textrm{E}}\), and their tunneling terms (\(\hat{{{\mathcal {H}}}}_{\textrm{T}}\)), is characterized by the following Hamiltonian:
In the framework of second quantization, the Hamiltonian for the device, \(\hat{{{\mathcal {H}}}}_{\textrm{P}}\), can be expressed in terms of on-site and hopping contributions as follows:
where \(\varepsilon _{\ell }\) displays the on-site energy at grid point (GP) or amino acid \(\ell\) within the protein, while \(c^{\dag }_{\ell }\) and \(c_{\ell }\) represent the creation and annihilation operators for a \(\pi\)-electron at GP \(\ell\), respectively. The term \(t_{\textrm{P}\ell \ell ^{\prime }}\) (\(t^{\prime }_{\textrm{P}\ell \ell ^{\prime }}\)) refers to the nearest-neighbor (nn) hopping amplitude for a \(\pi\)-electron transition from GP \(\ell\) to GP \(\ell ^{\prime }\) of the peptide bond energy (hydrogen bond energy), with \(\mathrm {h.c.}\) indicating the Hermitian conjugate. It would be noteworthy that variations in temperature lead to structural disarray and instabilities within the system27. Therefore, we investigate the effects of temperature alterations on the hopping parameters, particularly concerning fluctuations in twist angles. In this framework, we define \(\theta _{\ell \ell ^{\prime }}\equiv \theta\) as the twist-angle fluctuations occurring between adjacent GP, representing the torsional angle applied along a chemical bond. In the context of a Gaussian distribution for the variable \(\theta\), we get \(\langle \theta \rangle =0\) and \(\langle \theta ^{2}\rangle =k_{\textrm{B}}T/({{\mathcal {I}}}\omega ^{2})\), with \({{\mathcal {I}}}\omega ^{2}\simeq 0.022\;\textrm{eV}\) and \({{\mathcal {I}}}\) shows the reduced moment of inertia associated with the relative rotation of two neighboring amino acids, while \(\omega\) stands for the oscillator frequency corresponding to the vibration mode27. By employing these formulations, one can derive the hopping integrals that are dependent on temperature as \(\cos (\theta )\simeq 1-\theta ^{2}/2\).
The second quantization representation of the leads’ Hamiltonian (\(\hat{{{\mathcal {H}}}}_{\textrm{E}}\)) can be formulated as:
where \(\lambda\) corresponds to the left (\(\textrm{L}\)) and right (\(\textrm{R}\)) leads, \(\epsilon ^{A}_{\lambda i}\) (\(\epsilon ^{B}_{\lambda i}\)) serves as the on-site energy of sub-site A (B) inside the Bravais lattice unit cell (BLUC) i of either the lead \(\textrm{L}\) or lead \(\textrm{R}\), \(a^{\dag }_{\lambda i}\) (\(b_{\lambda j}\)) points to the corresponding \(\pi\)-electron creation (annihilation) operator of sub-site A (B) in the BLUC i (j), and \(t_{\lambda ij}\) plays the role of the hopping term from a sub-site inside the BLUC i to its nn sub-sites inside the BLUC j of lead \(\lambda\). The tunneling component of the Hamiltonian (\(\hat{{{\mathcal {H}}}}_{\textrm{T}}\)) is farther decomposed as tunneling term from left electrode to the protein chain (\(\hat{{{\mathcal {H}}}}_{\textrm{LP}}\)) and tunneling from the device to the right lead (\(\hat{{{\mathcal {H}}}}_{\textrm{PR}}\)), both terms can be shown as the following form:
the term \(t_{\textrm{LP}i\ell }\) (\(t_{\textrm{PR}\ell i}\)) implies the hopping contribution from the BLUC i of lead \(\textrm{L}\) to the \(\ell\)-th GP of protein (the \(\ell\)-th GP of protein to the BLUC i of lead \(\textrm{R}\)). In addition, the switching between devices and leads may occur from either sub-site a or b. However, in every term of Eq. (4), it is depicted by just one of these sub-sites. It would be noted that prior to the application of the bias voltage, V, we assume that all on-site energies throughout the system are set to zero, effectively establishing a reference point for energy levels. Subsequently, the on-site energies in lead \(\textrm{L}\) are assigned a value of \(\mu _{\textrm{L}}\), representing the relevant chemical potential, while those in lead \(\textrm{R}\) are designated as \(\mu _{\textrm{R}}\). This configuration results in the relationship \(\mu _{\textrm{L}} - \mu _{\textrm{R}}=V\). Furthermore, the on-site energies of the GPs within the protein exhibit a linear variation from \(\mu _{\textrm{L}}\) to \(\mu _{\textrm{R}}\) across the points. We operate within a framework in which the physical constants \(\{e,\hbar ,k_{\textrm{B}}\}\) are standardized to unity.
Quantum transport formulation
Green’s functions play a crucial role in numerous applications within quantum mechanics, particularly in the analysis of transport phenomena, where they facilitate the examination of particle behavior in intricate systems. The definition of the Green’s function is intrinsically linked to the Hamiltonian operator of the system, and its characteristics are directly associated with the energy spectrum of the system. The Green’s function associated with the system, encompassing both the leads and the device within the device’s subspace, can be articulated as follows36:
in this context, \(E={{\mathcal {E}}}+\textrm{i}0^{+}\), where \({{\mathcal {E}}}\) performs as the energy levels, \({\varvec{I}}\) remarks the identity matrix. Also, the expression \({\varvec{H}}_{\textrm{P}}-{\varvec{\varSigma }}_{\textrm{L}}(E)-{\varvec{\varSigma }}_{\textrm{R}}(E)\) is referred to as the effective Hamiltonian for the device, with the self-energies of the left (\(\textrm{L}\)) and right (\(\textrm{R}\)) aGNR electrodes being characterized by this formulation37:
in which \({\varvec{g}}_{\lambda }(E)=(E{\varvec{I}}-{\varvec{H}}_{\lambda })^{-1}\) stands for the surface Green’s function36 and \({\varvec{H}}_{\lambda }\) displays the Hamiltonian of the electrode \(\lambda\). The Green’s function \({\varvec{G}}\) and \({\varvec{g}}\) are derived through a recursive method36. Typically, the self-energies of molecules connected to the electrodes are computed through the non-equilibrium Green’s function formalism, a robust approach for analyzing electronic transport in nanoscale systems. This approach proves to be particularly advantageous in the simulation of device, rendering direct computation of the complete Green’s function computationally prohibitive. In non-equilibrium Green’s function calculations pertaining to electron transport, surface Green’s functions are conventionally employed to represent the electrodes. These functions encapsulate the electronic structure of the semi-infinite electrodes, thereby effectively linking the device to the leads. Consequently, this approach necessitates the treatment of the leads as a semi-infinite solid with a surface region that is coupled to the electrode. The calculation generally entails determining the Green’s function for the electrode followed by that for the surface region, utilizing a TB Hamiltonian and matrix manipulations that streamline computations by concentrating on the interactions among neighboring atomic orbitals. The Hamiltonian is expressed in matrix form, and the surface Green’s function is computed through matrix algebra. Subsequently, the surface Green’s function is obtained by taking into account the coupling between the device and the electrode regions. Specifically, the surface Green’s function at the device is computed using Eq. (5). In this equation, the self-energies are derived from the Green’s functions of the electrodes, denoted as \({\varvec{g}}_{\lambda }\), along with the coupling terms between the device and the electrodes, represented as \({\varvec{H}}_{\lambda }\). To derive the surface Green’s function from a contact Hamiltonian matrix, a recursive method can be employed37. The transmission probability, referred to as \(\textrm{TP}\), is expressed via the Fisher-Lee relation, a fundamental equation in the field of quantum transport physics that establishes a direct relationship between the electrical conductance of a system and its transmission characteristics36:
The Landauer-Büttiker formula serves as a pivotal equation within the realm of mesoscopic physics, fundamentally determining the electric current traversing a system by utilizing the transmission probabilities of electrons. This formulation facilitates the examination of quantum transport phenomena in various devices. The electrical current traversing the protein can be described by this formula36:
wherein
hints the Fermi-Dirac distribution function for lead \(\lambda\), and T marks the temperature of both electrodes.
The leads are frequently characterized by k-modes, which denote the permissible electronic states or wave functions within the leads. These leads are effectively modeled as reservoirs containing distinct electron states or k-modes, which correspond to the propagating electron waves. In essence, the k-modes represent the various pathways through which electrons can traverse the leads. The recursive technique subsequently assesses how these diverse pathways (k-modes) from the leads affect the device, thereby embedding the leads’ impact into the device’s operational characteristics. This impact is encapsulated in the self-energy, which is crucial for comprehending the transport phenomena within the system. To compute the self-energies presented in Eq. (7), it is necessary to identify the wave numbers that correspond to the energy levels of the electrons38. These quantum numbers can be derived from the energy spectrum of aGNR electrodes, which is obtained from the solution of the Schrödinger equation31 utilizing the Hamiltonian outlined in Eq. (3):
the notation \(k_{\lambda m}\) provides the wave numbers that correspond to the \(\pi\)-electron energy levels in each lead, where m takes values from 1 to W, with W indicating the width of the leads. Moreover, the on-site energies for the sub-sites A and B within lead \(\lambda\) are denoted by \(\epsilon _{\lambda }\). The parameter \(t_{0}\) is equivalent to \(t_{\lambda ij}\), and the function \(h(k_{\lambda m})\) is defined accordingly:
the variable \(l=3a_{0}\) reads the length of the BLUC for each lead, while \(a_{0}=0.142\; \textrm{nm}\) indicates the interatomic distance. Additionally, \(\alpha _{m}=m\pi /(W+1)\), and \(k_{\lambda m}\) can be obtained from Eqs. (11) and (12) as follows:
in which \(\xi _{\lambda }=-\zeta _{\lambda }/4\) and \(\zeta _{\lambda }\) is defined by:
the variable \(\epsilon _{\lambda }\) has been substituted with \(\mu _{\lambda }\), as detailed in ending paragraph of “TB Hamiltonian model” section. As we conclude this section, we highlight that the the density of states (DOS) of the device can be written as: \(D({{\mathcal {E}}})=-\textrm{Tr Im}{{\varvec{G}_{\textrm{P}}}(E)}/\pi\).
Results and discussion
The formalism established in the preceding section is employed to examine the influence of various protein conformations, specifically designated as \(\mathrm {P_{1}}\), \(\mathrm {P_{2}}\), and \(\mathrm {P_{3}}\). This investigation also considers the effects of temperature variations and electrode’s width on the electron quantum transport properties of the protein chains. Assuming that the aGNR leads are of a uniform type, the parameter \(t_{0}\) is assigned a value of \(-2.8\;\textrm{eV}\)31, while the hopping terms between leads and the device are defined as \(t_{\textrm{LP} i\ell }=t_{\textrm{PR}\ell i}\equiv t^{\prime \prime }=-0.3\;\textrm{eV}\). Additionally, the hopping term within the device is set according to the values reported in Ref.27, specifically \(t_{\textrm{P}\ell \ell ^{\prime }}\equiv t_{p}=-0.3\;\textrm{eV}\). Furthermore, the energy associated with hydrogen bonds represents relatively weak intermolecular interactions when contrasted with peptide bonds, as illustrated in Fig. 1b by dashed lines, so we set \(t^{\prime }_{\textrm{P}\ell \ell ^{\prime }}=-0.03\;\textrm{eV}\). Although hydrogen bonds are relatively weak, their abundance and the specific arrangement of these bonds among amino acids in the chain can effectively stabilize and influence the spatial structure of proteins. In this context, we examine thirteen connections that are instrumental in stabilizing these conformational configurations27,28. For the analysis, three configurations of \(t_{p}\) are established across thirty-six GP, with \(t_{p}\) values varying within the range of \(-0.3\le t_{p}\le -0.26\). Furthermore, the lead widths’ are defined as \(W=26,32,41\), which exhibit metallic characteristics. It is imperative to observe that proteins are frequently examined and preserved at reduced temperatures, as this practice diminishes their molecular activity and minimizes the likelihood of alterations or degradation in their structure and function39. Specifically, lowering the temperature decreases molecular motion, thereby decelerating the rate of destabilizing interactions. This preservation method enables proteins to sustain their native conformation and functionality for extended durations. Conversely, elevated temperatures result in increased kinetic energy, causing proteins to vibrate more intensely, which can disrupt stabilizing interactions such as hydrogen bonds, ionic bonds, and hydrophobic interactions, ultimately leading to unfolding or denaturation; thus, the chosen temperatures are typically kept at minimal levels. When converting the dimensionless the temperatures of the leads, i.e. \(T=0.001\), \(T=0.005\), and \(T=0.0075\) into Kelvin, the corresponding values are approximately 3.48 K, 17.4 K, and 26.1 K, respectively.
In Fig. 2, the band structure (BS) and the corresponding density of states (DOS) at a constant temperature of \(T=0.001\) are illustrated for a specific conformation (\(\mathrm {P_{1}}\)). It is crucial to acknowledge that while a traditional BS plot, which relies on the premise of an infinite periodic lattice, cannot be directly utilized for finite systems, we can still explore the energy levels of such finite protein structures in a way that emulates the BS. This exploration can be conducted by plotting the energy levels as a function of a relevant parameter, such as the site index, thus providing a representation of the band-like features present within this finite system. The confinement of electrons to limited dimensions leads to the quantization of energy levels. As a result, instead of forming a continuous band, the allowed energy states are converted into discrete values, resembling distinct steps or individual levels. The figure demonstrates that the curves display symmetry around the zero-energy point (the Fermi level), which is a result of the particle-hole symmetry inherent in the Hamiltonian model. In a system exhibiting particle-hole symmetry, the energy levels of electrons and holes are symmetrically arranged, with the HOMO denoting the highest energy level occupied by electrons and the LUMO indicating the lowest energy level that is unoccupied by electrons. Additionally, for a molecule characterized by particle-hole symmetry, the HOMO may correspond to a bonding orbital, while the LUMO would represent its associated antibonding orbital. Consequently, in systems with particle-hole symmetry, the energy levels of electrons and holes are interconnected in a particular manner. When an electron is excited from the HOMO to the LUMO, it leaves a hole in the HOMO, which acts as a positively charged particle. The energy necessary to generate this electron-hole pair is equivalent to the energy difference between the HOMO and LUMO. Moreover, this symmetry suggests that the HOMO and LUMO can be viewed as analogous to the valence and conduction band edges in a solid-state system, respectively. The existence of particle-hole symmetry streamlines this process by ensuring that the energy levels are reflected around a central point, thereby facilitating the identification of the HOMO and LUMO. According to the BS or DOS, the HOMO is positioned at \(-0.022\;\textrm{eV}\) (relative to the Fermi level), while the LUMO is likely at \(0.022\;\textrm{eV}\), resulting in a HOMO-LUMO gap of \(0.044\;\textrm{eV}\). The corresponding HOMO and LUMO levels for \(\mathrm {P_{2}}\) and \(\mathrm {P_{3}}\) are \(0.02\;\textrm{eV}\), which are lower than the values for the \(\mathrm {P_{1}}\) conformation. A reduced HOMO-LUMO gap indicates a smaller energy difference, facilitating electron movement between the orbitals. This can enhance chemical reactivity and the potential for binding interactions. Conversely, a larger HOMO-LUMO gap implies increased stability, as it necessitates more energy for electrons to transition and engage in reactions.
The influence of electrode’s width is illustrated in Fig. 3a–c under a constant lead temperature of \(T=0.001\), considering three device conformations, \(\{\mathrm {P_{1}},\mathrm {P_{2}},\mathrm {P_{3}}\}\), and three distinct lead widths’, \(W=26,32,41\). The \(\textrm{TP}\) curves are presented in the insets of the panels (a), (b), and (c) for configurations \(\mathrm {P_{1}}\), \(\mathrm {P_{2}}\), and \(\mathrm {P_{3}}\), respectively, while the corresponding \(\mathrm {I-V}\) characteristics are depicted in the main parts of panels (a), (b), and (c). It is observed that an increase in lead’s width results in a decrease in the peak heights’ of the \(\textrm{TP}\) curves across the three device configurations. The observed phenomenon can arise from the creation of additional pathways for electron movement in the leads, in contrast to the narrower configurations. This phenomenon can be attributed to the enhanced availability of vertical pathways along the ribbon, stemming from the overlap of the normally nonhybridized \(p_{z}\) orbitals. Such overlap increases the likelihood of deviations in the orientation of a portion of the electrons from horizontal trajectories aligned with the x-axis to newly accessible pathways in the y-direction (vertical direction). In essence, the overlapping nonhybridized \(p_{z}\) orbitals divert some of the electrons’ mobility away from the axial direction along the ribbon, favoring perpendicular movement instead. Consequently, the increased potential for these transverse movements logically leads to a reduction in longitudinal displacements, thereby resulting in diminished peak heights in the \(\textrm{TP}\) curves. As illustrated in the insets of Fig. 3a–c, this effect becomes more pronounced with increasing ribbon width, aligning with the previous reasoning, as the addition of lateral bonds provides greater opportunities for electron traversal, consequently reducing the axial component of motion. Furthermore, as depicted in the Fig. 3a–c, the \(\mathrm {I-V}\) curves exhibit significant non-linearity, characterized by sharp increases in conductance followed by nearly flat plateaus, which diminish in height as the electrode width expands. The observed phenomenon can be attributed to the proportional relationship between the electrical current and \({{\mathcal {T}}}({{\mathcal {E}}})\) as described by the Fisher-Lee formula in Eq. (7), which results in a reduction in the magnitudes of the corresponding \(\mathrm {I-V}\) curves. In other words, the likelihood of transmission is intrinsically linked to the \(\mathrm {I-V}\) characteristics, as an increased \(\textrm{TP}\) results in a greater current flow within the device. Additionally, it is typically observed that an increase in V leads to a corresponding rise in the electrical current within the device, which remains relatively low near the zero bias. Also, the scarcity of available states at the zero point contributes to the low electrical current observed in this region. Moreover, the \(\mathrm {I-V}\) curve exhibits a relatively stable behavior between the two peaks of \(\textrm{TP}\), whereas it demonstrates a distinct transition from one step to another in the presence of a peak within \(\textrm{TP}\). In summary, expanding the electrode’s width typically diminishes the influence of voltage on the \(\mathrm {I-V}\) curve, particularly when the effective surface area of the electrode is enhanced. This phenomenon occurs because larger electrodes exhibit reduced impedance, thereby necessitating less voltage to achieve a specific current. Furthermore, enlarging the surface area of an electrode can facilitate a greater flow of current for a specified voltage; however, this may also distribute the current across a wider area, which could lead to a decrease in current density at particular points.
The impact of three distinct conformations of the device is illustrated in Fig. 4, with fixed parameters of \(W=26\) and \(T=0.001\). As depicted in Fig. 4, variations in the conformations lead to alterations in both the height and position of the peaks within the \(\textrm{TP}\) curves, which in turn affect the dimensions and heights of the steps observed in the \(\mathrm {I-V}\) curves. This phenomenon can be explained by the varying values of the hopping terms within the system, which result in an increased number of charge transport pathways for proteins \(\mathrm {P_{2}}\) and \(\mathrm {P_{3}}\). As a result, the step-like features of the \(\mathrm {I-V}\) curve display altered heights and lengths, accompanied by a heightened steepness in each step. Despite both proteins \(\mathrm {P_{2}}\) and \(\mathrm {P_{3}}\) having equivalent quantities of hydrogen bonds, protein \(\mathrm {P_{3}}\) exhibits a higher saturation current compared to protein \(\mathrm {P_{2}}\). This difference can be attributed to the strategic distribution of hydrogen bonds throughout the structure of protein \(\mathrm {P_{3}}\)27. The current-voltage characteristics presented in this discussion demonstrate considerable potential for distinguishing between different protein profiles, including both normal and mutant variants.
The influence of finite temperature on the \(\mathrm {I-V}\) curves is illustrated in Fig. 5a–c for a fixed value of \(W=26\) and three distinct conformations of protein chains, \(\{\mathrm {P_{1}},\mathrm {P_{2}},\mathrm {P_{3}}\}\), in panels (a), (b), and (c), respectively. It is evident that as the temperature of the leads rises, the step-like characteristics of the \(\mathrm {I-V}\) relationship evolve40, tending towards a linear configuration with increasing temperature. This phenomenon can be understood as a typical broadening of the fermionic distribution at elevated temperatures; the energy of electrons migrating from the lead to the device also increases, facilitating their movement across the gaps between the peaks of the \(\textrm{TP}\) (the insets of the Figs. 3 and 6). Consequently, the \(\mathrm {I-V}\) curve transitions from a step-like form to one that exhibits linear behavior. Furthermore, the observed linearity in the \(\mathrm {I-V}\) curve can be attributed to temperature-related alterations in carrier concentration and energy distribution, which simplify the \(\mathrm {I-V}\) relationship. This phenomenon is mainly a result of enhanced thermalization of carriers and the expansion of energy levels, contributing to a more linear device behavior. At elevated temperatures, the energy states within the device, exhibit increased broadening. This broadening results in less distinct energy levels, facilitating easier movement of carriers between them. Consequently, this phenomenon often leads to a more linear \(\mathrm {I-V}\) characteristic, as the transitions between energy levels become less pronounced. Conversely, at reduced temperatures, quantum effects become increasingly significant, resulting in more intricate and nonlinear I-V characteristics. As temperature rises, electrons and other carriers are more frequently thermalized, acquiring sufficient energy to move more freely, thus leading to a more classical system behavior. This thermalization process can diminish some of the quantum effects responsible for the observed nonlinearities.
In the realm of electronic transport, the degree of coupling between the leads and the device plays a crucial role in facilitating the flow of electrons. Figure 6 illustrates the \(\mathrm {I-V}\) curve for a constant temperature of \(T=0.001\) and an electrode width of \(W=26\), showcasing three distinct coupling strengths (hopping terms between the leads and the device) at \(t^{\prime \prime }=0.25,0.30,0.35\;\textrm{eV}\) for the device configuration \(\mathrm {P_{1}}\). Figure 6 illustrates that an increase in coupling correlates with a rise in electric current, indicating a significant interaction between the leads and the device. This phenomenon occurs because the energy levels of the device become more closely aligned with those of the leads, facilitating a more efficient electron transfer process. Typically, the disparity in energy levels between the leads and the device presents a barrier that electrons must surmount for transfer to occur. Specifically, for effective electron transfer, the energy levels of the protein molecule must align with those of the leads, a condition that is significantly affected by the strength of the coupling. A higher hopping energy signifies a stronger coupling between the leads and the device, which enhances electron transfer efficiency, whereas weak coupling results in an increased barrier for electron transfer. Consequently, a lower hopping value, indicative of reduced coupling, complicates the movement of electrons between the electrode and the protein chain.
Conclusion
In conclusion, the TB Hamiltonian model was utilized in conjunction with the Landauer-Büttiker framework to investigate the quantum transport characteristics of protein chains connected to aGNR leads, focusing on the \(\mathrm {I-V}\) characteristics and \(\textrm{TP}\). Under constant temperature conditions, the electrical current displayed a nonlinear response, marked by distinct steps in the \(\mathrm {I-V}\) curve. The dimensions of these steps can be associated with the amplitude and spacing of the peaks identified in the \(\textrm{TP}\). This behavior is modulated by changes in electrode temperatures, which can shift the nonlinear response towards a more linear profile. This effect can be interpreted as a typical broadening of the fermionic distribution at higher temperatures; the energy of electrons transitioning from the lead to the device also increases, thereby enhancing their movement across the gaps between the peaks of the \(\textrm{TP}\). Furthermore, the \(\mathrm {I-V}\) characteristics are influenced by the lead widths. The observed effects may result from the establishment of additional pathways for electron transport in the leads, as opposed to narrower configurations. Ultimately, variations in the hopping parameters modify both the dimensions of the steps in the \(\mathrm {I-V}\) curve and the height and position of the peaks in the \(\textrm{TP}\). An increase in coupling is associated with a rise in electric current, signifying a substantial interaction between the leads and the device. This occurs because the energy levels of the device become more closely aligned with those of the leads, promoting a more efficient electron transfer process. If the electronic transport properties are anticipated to directly influence protein structure, our findings pave the way for novel research pathways regarding biological implications.

(a) A diagrammatic representation is provided, illustrating the left (\(\textrm{L}\)) and right (\(\textrm{R}\)) armchair graphene nanoribbon (aGNR) leads, alongside the protein chain (\(\mathrm {P_{1}}\)) composed of thirty-six bases. The dashed lines signify the Bravais lattice unit cell (BLUC) of the leads, with A and B indicating the carbon sub-sites within the electrodes, while V denotes the applied voltage bias. (b) A schematic depiction of the conformations of \(\mathrm {P_{2}}\) (on the left) and \(\mathrm {P_{3}}\) (on the right) is included, with hydrogen bonds represented by dashed lines.

The DOS for protein \(\mathrm {P_{1}}\) along with the corresponding BS is presented.

The \(I-V\) curves are presented for a constant temperature, \(T=0.001\), examining three varying widths of \(W=26, 32, 41\), alongside three unique conformations of the protein chain, \(\{\mathrm {P_{1}},\mathrm {P_{2}},\mathrm {P_{3}}\}\). The insets are shown the associated \(\textrm{TP}\) diagrams.

The \(I-V\) curves are shown for a constant value of lead width, \(W=26\), with a specified temperature of \(T=0.001\), and for three distinct conformations of the protein chain, \(\{\mathrm {P_{1}},\mathrm {P_{2}},\mathrm {P_{3}}\}\). The inset illustrates the corresponding \(\textrm{TP}\) diagrams.

The \(I-V\) curves are illustrated for a fixed value of electrode width, \(W=26\), encompassing three different conformations of the protein chain, \(\{\mathrm {P_{1}},\mathrm {P_{2}},\mathrm {P_{3}}\}\), and three amounts of temperature, \(T=0.001, 0.005, 0.0075\).

The \(I-V\) curves are presented for a fixed lead width of \(W=26\), at a designated temperature of \(T=0.001\), under three different levels of coupling strength, and for the \(\mathrm {P_{1}}\) conformation of the protein chain.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Bhopatkar, A. A., Uversky, V. N. & Rangachari, V. Disorder and cysteines in proteins: A design for orchestration of conformational see-saw and modulatory functions. Prog. Mol. Biol. Transl. Sci. 174, 331 (2020).
Hart, W. E., & Newman, A. Handbook of Computational Molecular Biology (ed. Aluru, S.) 1st Ed. (Chapman and Hall/CRC, 2005).
Dill, K. A. et al. Principles of protein folding: A perspective from simple exact models. Protein Sci. 4, 561 (1995).
Elbing, M. et al. Single-molecule diode. Proc. Natl. Acad. Sci. 102, 8815 (2005).
Del Valle, M., Gutiérrez, R., Tejedor, C. & Cuniberti, G. Tuning the conductance of a molecular switch. Nat. Nanotechnol. 2, 176 (2007).
Zhang, X. Y., Shao, J., Jiang, S. X., Wang, B. & Zheng, Y. Structure-dependent electrical conductivity of protein: Its differences between alphadomain and beta-domain structures. Nanotechnology 26, 125702 (2015).
Prytkova, T. R., Kurnikov, I. V. & Beratan, D. N. Coupling coherence distinguishes structure sensitivity in protein electron transfer. Science 315, 622 (2007).
Giese, B., Graber, M. & Cordes, M. Electron transfer in peptides and proteins. Curr. Opin. Chem. Biol. 12, 755 (2008).
Skourtis, S. S. & Beratan, D. N. Theories of structure function relationships for bridge-mediated electron transfer reactions. Adv. Chem. Phys. 106, 377 (1999).
Regan, J. J. & Onuchic, J. N. Electron-transfer tubes. Adv. Chem. Phys. 107, 497 (1999).
Panitchayangkoon, G., Voronine, D. V., Abramavicius, D. & Engel, G. S. Direct evidence of quantum transport in photosynthetic light-harvesting complexes. Proc. Natl. Acad. Sci. 108, 20908 (2011).
Fereiro, J. A., Yu, X., Pecht, I. & Cahen, D. Tunneling explains efficient electron transport via protein junctions. Proc. Natl. Acad. Sci. 115, E4577 (2018).
Sanvictores, T. & Farci, F. Biochemistry, Primary Protein Structure (StatPearls Publishing, 2025).
Schweizer, J., Loose, M., Bonny, M. & Schwille, P. Geometry sensing by self-organized protein patterns. Proc. Natl. Acad. Sci. 109, 15283 (2012).
Durairaj, J., Akdel, M., de Ridder, D. & van Dijk, A. D. J. Geometricus represents protein structures as shapemers derived from moment invariants. Bioinformatics 36, i718 (2020).
Liu, X., Luo, Y., Li, P., Song, S. & Peng, J. Deep geometric representations for modeling effects of mutations on protein-protein binding affinity. PLoS Comput. Biol. 17, e1009284 (2021).
Mendes, G. G. et al. Genetic functionalization of protein-based biomaterials via protein fusions. Biomacromolecules 25, 4639 (2024).
Tunuhe, A. et al. Protein-based materials: Applications, modification and molecular design. BioDesign Res. 7, 100004 (2025).
Pan, X. & Kortemme, T. Recent advances in de novo protein design: Principles, methods, and applications. J. Biol. Chem. 296, 100558 (2021).
Rumpler, M., Woesz, A., Dunlop, J. W. C., van Dongen, J. T. & Fratzl, P. The effect of geometry on three-dimensional tissue growth. J. R. Soc. Interface 5, 1173 (2008).
Pang, X., Zhou, L., Zhang, L. et al. Two rules on the Protein-Ligand interaction. Nat. Prec. (2008).
Usselman, R. J. et al. The quantum biology of reactive oxygen species partitioning impacts cellular bioenergetics. Sci. Rep. 6, 38543 (2016).
Fereiro, J. A., Yu, X., Pecht, I. & Cahen, D. Tunneling explains efficient electron transport via protein junctions. Proc. Natl. Acad. Sci. 115, E4577 (2018).
Melkikh, A. V. Why does a cell function? New arguments in favor of quantum effects. Biosystems 245, 105311 (2024).
Georgiev, D. D. & Glazebrook, J. F. Quantum transport and utilization of free energy in protein \(\alpha\)-helices. Adv. Quantum Chem. 82, 253 (2020).
Kiss, R. et al. Structural basis of small molecule targetability of monomeric tau protein. ACS Chem. Neurosci. 9, 2997 (2018).
Sarmentoa, R. G., Frazaob, N. F. & Macedo-Filhoc, A. Electronic transport on the spatial structure of the protein: Three-dimensional lattice model. Phys. Lett. A 381, 276 (2017).
Gutin, A. M., Abkevich, V. & Shakhnovich, E. A protein engineering analysis of the transition state for protein folding: Simulation in the lattice model. Fold Des. 3, 183 (1998).
Paez, C. J., Schulz, P. A., Wilson, N. R. & Römer, R. A. Robust signatures in the current-voltage characteristics of DNA molecules oriented between two graphene nanoribbon electrodes. New J. Phys. 14, 093049 (2012).
Leonard, F. & Talin, A. A. Electrical contacts to one- and two-dimensional nanomaterials. Nat. Nanotechnol. 6, 773 (2011).
Wakabayashi, K., Sasaki, K., Nakanishi, T. & Enoki, T. Electronic states of graphene nanoribbons and analytical solutions. Sci. Technol. Adv. Mater. 11, 054504 (2010).
Mousavi, H. & Grabowski, M. Nonlinear electron transport across short DNA segment between graphene leads. Solid State Commun. 279, 30 (2018).
Mousavi, H., Khodadadi, J. & Grabowski, M. Electronic properties of long DNA nanowires in dry and wet conditions. Solid State Commun. 222, 42 (2015).
Mousavi, H., Jalilvand, S., Sani, S. S., Hartman, J. A. L. & Grabowski, M. Electronic properties of different configurations of double-strand DNA-like nanowires. Solid State Commun. 319, 113974 (2020).
Jalilvand, S., Sepahvand, R. & Mousavi, H. Electronic behavior of randomly dislocated RNA and DNA nanowires: A multi-model approach. Eur. Phys. J. 137, 928 (2022).
Datta, S. Quantum Transport: Atom to Transistor (Cambridge University Press, 2005).
Lewenkopf, C. H. & Mucciolo, E. R. The recursive Green’s function method for graphene. J. Comput. Electron. 12, 203 (2013).
Schomerus, H. Effective contact model for transport through weakly-doped graphene. Phys. Rev. B 76, 045433 (2007).
Zheng, J., Guo, N., Huang, Y., Guo, X. & Wagner, A. High temperature delays and low temperature accelerates evolution of a new protein phenotype. Nat. Commun. 15, 2495 (2024).
Mukhopadhyay, S. et al. Solid-state protein junctions: Cross-laboratory study shows preservation of mechanism at varying electronic coupling. iScience 23, 101099 (2020).
Funding
This study is in no way funded by any organization and is an independent investigation carried out exclusively by the author of the study.
Author information
Authors and Affiliations
Contributions
The study has been exclusively conducted by the author, Hamze Mousavi, who has been responsible for all aspects including the conceptualization, findings, preparation of materials, data collection and analysis, coding, interpretation of results, and the drafting of the original manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mousavi, H. Quantum transport in protein chains. Sci Rep 15, 26703 (2025). https://doi.org/10.1038/s41598-025-11140-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-11140-5
Keywords
This article is cited by
-
Quantum charge transport in DNA and RNA systems coupled to nanoribbon electrodes
Discover Nano (2026)
-
Protein-DNA interaction in tight-binding paradigm
Scientific Reports (2025)
