
Abstract
Tomato brown rugose fruit virus (ToBRFV) is a single-stranded positive-sense RNA virus that targets tomato and pepper plants and is causing significant damage to crops in some regions of the world. ToBRFV is a highly contagious virus that is stable and rapidly spreads by mechanical methods and seeds. As a result, it may spread both locally and over long distances, and it is now recognized as a pandemic in plants. This study investigates the effectiveness of the systems CRISPR-Cas12a and CRISPR-Cas9, in conjugation with recombinase polymerase amplification (RPA), to detect ToBRFV in tomato plant samples collected from the field. The trans-cleavage activity of both nucleases, Cas12a and Cas9, was exploited to process a probe labelled with fluorescein and biotin to be resolved on a lateral flow device, thereby enabling a visual readout. We were able to detect the RNA genome of the virus in about 1 h at a low constant temperature. These results could pave way to offer a rapid, sensitive, and specific method for on-site detection of ToBRFV.
Similar content being viewed by others

Introduction
Plant viruses pose a significant challenge to agriculture, which are responsible for almost half of all new plant diseases, thereby leading to an estimated annual economic loss exceeding $30 billion1. The tomato brown rugose fruit virus (ToBRFV), a new member species of the genus Tobamovirus, causes significant damage to tomato crops in some regions of the world2. ToBRFV is related to several other viruses, including tobacco mosaic virus (TMV), tomato mosaic virus (ToMV), tomato mottle mosaic virus (ToMMV), cucumber green mottle mosaic virus (CGMMV), and odontoglossum ringspot virus (ORSV)3,4,5,6. ToBRFV is a single-stranded positive-sense RNA virus that targets tomato and pepper plants initially identified in 2014 in greenhouses in Israel and Jordan7. Despite the long-standing resistance of the Tm-22 gene against Tobamoviruses, ToBRFV has proven to overcome all known resistance genes in tomatoes8,9. This virus has been found to naturally infect tomato (Solanum lycopersicum) and pepper (Capsicum annuum)7,8,10. Tomato cultivars carrying the Tm-1, Tm-2, or Tm-22 Tobamovirus resistance (R) gene are susceptible to infection by ToBRFV8,11,12. Additionally, it can infect pepper cultivars that have the L alleles (L1 or L2); and in pepper plants that encode for the L3 and L4 alleles, it can cause a hypersensitivity reaction (HR)13. Tomatoes and peppers, similar to other crops, are consistently targeted by pests and pathogens. Primary pathogens are Cucumoviruses, Begomoviruses, Potyviruses, Tospoviruses, and Tobamoviruses14,15. Infection with these viruses diminishes crop yield, degrades the quality and marketability of the fruits, and results in significant economic losses14,15,16,17. The ToBRFV appears to be spreading rapidly, as the list of countries affected by ToBRFV has grown swiftly. To date, it has reached more than 45 countries worldwide18,19,20,21,22,23,24.
The virus genome consists of a single-stranded positive-sense RNA molecule with specific open reading frames (ORFs) encoding essential proteins for viral replication. Notably, the production of a helicase and methyltransferase protein (p126) and an RNA-dependent RNA polymerase protein (p183) play crucial roles in the viral life cycle (Fig. 1)7,25.
Traditional diagnostic methods for viral infections involve enzyme-linked immunosorbent assay (ELISA) tests for viral protein detection or polymerase chain reaction (PCR) techniques for genome amplification26. To detect ToBRFV, various serological and molecular techniques have been employed. Serological approaches, such as ELISA, are popular due to their ease of use and affordability19,27though they offer limited sensitivity and specificity. Molecular techniques such as PCR28,29 have been developed to address these limitations, and even isothermal methods such as loop-mediated isothermal amplification (LAMP)29 have been put forward. These molecular methods provide high specificity and sensitivity for detecting ToBRFV and are helpful for field diagnostics, vital for early virus detection and management. However, reverse transcription PCR (RT-PCR) involves a lengthy assay time (approximately 2 h), requires skilled personnel, and relies on costly laboratory equipment. While LAMP-based techniques have shortened amplification times and have reduced instrumental dependence, they tend to be prone to false positives, thereby compromising their use for effective field applications. Notwithstanding, Cao et al. used reverse transcription recombinase-aided amplification (RT-RAA) to identify ToBRFV in a lateral flow assay (LFA)30.
Some bacteria and archaea contain an innate immune system called clustered regularly interspaced short palindromic repeats (CRISPR)31which has resulted very versatile to be exploited in biotechnology. The use of CRISPR systems can add a higher level of specificity in the detection32. This kind of approaches have shown potential for identifying viral pathogens by targeting specific sequences within their genomes. The CRISPR-Cas12a and CRISPR-Cas13a/d systems have been exploited for the detection of RNA and DNA viral pathogens in plants33,34,35,36. The main difference between CRISPR type V (Cas12) and type VI (Cas13) systems is that DNA is targeted in former while RNA is targeted in the latter. Both nucleases have non-specific collateral cleavage upon target recognition. This collateral activity involves cleaving non-targeted single-stranded DNA (ssDNA) for Cas12 or single-stranded RNA (ssRNA) for Cas13. In particular, these systems have been already exploited to detect ToBRFV27,37,38. They have relied on producing a fluorescent signal upon specific recognition of the virus. For in situ applications, it would be convenient nonetheless to exploit LFA devices to monitor the output of the CRISPR reactions.
The trans-cleavage activity of the CRISPR-Cas9 system has been recently disclosed39adding this system to the toolbox of CRISPR diagnostics. Our work aims to investigate the use of the CRISPR-Cas12a and CRISPR-Cas9 systems to detect ToBRFV on an LFA device. By leveraging these innovative techniques, we present a rapid and accurate method for on-site identification of ToBRFV infections in tomato plants, offering a practical solution for monitoring and managing viral diseases in agricultural settings.
Results
Detection of ToBRFV with CRISPR-Cas12a
Custom RPA primers were designed to target a conserved sequence of ToBRFV. We considered the gene coding for the coat protein of the virus (Fig. 1). This region is convenient because it is included in all possible sub-genomic RNAs. Using samples collected from tomato (S. lycopersicum L.) plants infected with ToBRFV, we applied PCR with those RPA primers and subsequent gel electrophoresis to confirm the presence of the virus.

Phenotypic effects of ToBRFV-infected plants and classical detection. (A) Genome architecture of ToBRFV. Each number indicates where an open reading frame (ORF) begins and ends in a representative Iranian ToBRFV isolate (OP557566, 6393 bases). (B) Images of S. lycopersicum L. plants infected by ToBRFV. (i) Mock-treated plants; and (ii) infected plants showing necrosis. C) Gel electrophoresis image showing the expected viral product upon RT-PCR (total RNA was used as starting material).
Then, a Cas12a-dependent CRISPR RNA (crRNA) was designed to target the resulting DNA amplicon. We ensured the presence of a suitable protospacer adjacent motif (PAM) for Cas12a recognition (TTTV). Cas12a was a commercial preparation from Lachnospiraceae bacterium and the crRNA was produced by in vitro transcription. Activation of Cas12a triggered collateral trans-cleavage of an ssDNA reporter molecule labelled with fluorescein and biotin, whose sequence reads TTATT. The cleavage of this molecule set apart fluorescein and biotin so that it could be measured in an LFA device. A reaction buffer previously used to detect plant viruses with this nuclease was considered33. Using the RPA product generated from the tomato plant samples, we demonstrated that the CRISPR-Cas12a assay coupled to LFA is suitable to detect ToBRFV with sufficient sensitivity and specificity with a visual readout (Fig. 2).
Detection reaction signals were robust to variations in Cas12a concentration, from 50 to 200 nM, and increased over time, from 30 to 120 min (Fig. S1). Signals at 50 nM and 2 h showed the highest visual signal-to-noise ratio, but with 1 h it was sufficient to obtain a significant signal. Moreover, we assessed the specificity of the test by using TMV-infected plant samples (Fig. S1).

Detection of ToBRFV with CRISPR-Cas12a. (A) Scheme of the detection in which the RPA product (dsDNA) generated from the viral genome is recognized by a programmed ribonucleoprotein (formed by L. bacterium Cas12a and ToBRFV-targeting crRNA) that in turn cleaves an ssDNA probe [labelled with fluorescein (F) and biotin (B)]. A commercial LFA strip is used to reveal the detection. (B) Image of the LFA strips when detecting ToBRFV in tomato plant samples (performed in triplicate). ToBRFV-derived dsDNA generated by RPA. (C) Quantification of test line intensity (bars represent means and standard deviations, n = 3). *Statistically significant change by a t-test, P < 0.05.
Detection of ToBRFV with CRISPR-Cas9
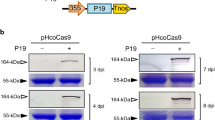
Parallel experiments were conducted using the CRISPR-Cas9 system, knowing the ability of this other nuclease to process poly(T) strands. Within the previously used DNA amplicon, the PAM recognized by Cas9 (NGG) was also found. Thus, we designed a crRNA to target it. The main difference with respect to the Cas12a-dependent crRNA is that, in this case, the spacer is located in the 5’ end. Moreover, Cas9 works with a bimolecule RNA, including a trans-activating crRNA (tracrRNA). Cas9 was a commercial preparation from Streptococcus pyogenes and, as before, the crRNA and tracrRNA were in vitro transcribed. In this case, we used a poly(T) probe of 18 nt (T18) labelled with fluorescein and biotin to be processed upon activation of Cas9. Following the RPA step, we confirmed that the CRISPR-Cas9 assay coupled to LFA is also suitable to detect ToBRFV in tomato plant samples with a visual readout (Fig. 3). While the assay displayed sufficient sensitivity and specificity, in comparison to the CRISPR-Cas12a assay, the intensity of the test line here showed lower dynamic range.
Detection reaction signals with Cas9 could be increased by reducing the amount of probe in the reaction (from 100 to 50 nM; Fig. S2). Moreover, we analyzed the performance of the test upon variations in Cas9 concentration, from 50 to 200 nM, and incubation times, from 30 to 120 min. As before, signals at 50 nM and 2 h showed the highest visual signal-to-noise ratio, but this time with 30 min it was sufficient to obtain a significant signal. These results suggest roughly comparable, yet condition-dependent trans-cleavage activity of both Cas9 and Cas12a. Besides, we assessed a specific detection by challenging the system with TMV-infected plant samples (Fig. S2). Nonetheless, a faint band was noticed in the test line in the negative case, so further screening of primers and crRNAs might be performed to enhance the specific detection ability with Cas9.

Detection of ToBRFV with CRISPR-Cas9. (A) Scheme of the detection in which the RPA product (dsDNA) generated from the viral genome is recognized by a programmed ribonucleoprotein (formed by S. pyogenes Cas9 and ToBRFV-targeting crRNA) that in turn cleaves an ssDNA probe [labelled with fluorescein (F) and biotin (B)]. A commercial LFA strip is used to reveal the detection. (B) Image of the LFA strips when detecting ToBRFV in tomato plant samples (performed in triplicate). ToBRFV-derived dsDNA generated by RPA. (C) Quantification of test line intensity (bars represent means and standard deviations, n = 3). *Statistically significant change by a t-test, P < 0.05.
Discussion
The findings of this study suggest that both the CRISPR-Cas12a and CRISPR-Cas9 systems are instrumental to have a practical and rapid approach to detect ToBRFV in infected plant tissues using LFA devices. The trans-cleavage activity of both Cas12a and Cas9 was exploited to process appropriate ssDNA probes. The current study demonstrates that accurate detection can be achieved swiftly, within a 1.5-h timeframe, starting directly from harvested leaves and maintaining a constant low temperature of 37–39 °C. These methods are applicable to a diverse range of plant species and plant pathogens, thereby representing a promising and expedient approach for next-generation agricultural practices.
In the current study, the ToBRFV genome amplification was conducted using the RPA method, but nothing prevents using other isothermal approaches, such as LAMP. Indeed, Bernabé-Orts et al. utilized LAMP coupled to CRISPR-Cas12a for the detection of ToBRFV27. While RPA requires only two oligonucleotides and operates at 37–42 °C, LAMP uses six oligonucleotides and requires higher incubation temperatures of 60–65 °C, so its usage is more demanding in the field. The CRISPR step is important to add specificity to the detection. On the other hand, Nanopore sequencing represents an alternative approach40as the devices have increased portability. However, they still require a laptop and more sophisticated sample handling, so CRISPR-Cas detection systems are more suitable for rugged environments.
Yet, the use of CRISPR systems still faces some limitations for field deployment, such as the requirement for specialized reagents, including purified nucleases and RNAs. There is in addition the need for cold chain storage, which hinders their practical use in remote or resource-limited settings, unless working with samples that can be rehydrated41. Moreover, many CRISPR diagnostics rely on nucleic acid amplification steps, which complicate portability and increase the risk of contamination. Further work should focus on developing amplification-free strategies for plant virus detection42.
In sum, we here successfully detected ToBRFV infections in tomato plants. As this study serves as a proof of concept, it is anticipated that these methods will be extended to other crops such as peppers, corn, and potatoes soon. Crucially, CRISPR-Cas systems are also being used as RNA interference tools to fight diseases and to develop more resilient and productive plant kinds43,44. In this sense, it is thought that integrated breeding, engineering, and diagnostic programs based on CRISPR-Cas would transform agriculture and contribute to addressing the issue of supplying enough food.
Materials and methods
Plant inoculation
The Iranian isolate of ToBRFV, Accession No. OP557566.1 (kindly provided by Dr. K. Bananej, IRIPP, Tehran), was used for mechanical inoculating four-week-old S. lycopersicum L. plants. An aliquot of 100 mg of frozen infected tissues was ground in a ball mill and homogenized in 20 volumes of ice-cold 50 mM phosphate buffer, pH 7.4 [3.1 g NaH2PO4.H2O and 10.9 g Na2HPO4 (anhydrous) in ultra-pure water] using an iced mortar and pestle. The homogenate was then centrifuged at 4 ºC, and crude extract was obtained from the supernatant. Typically, up to 1 µg RNA per mg of tissue was recovered. A cotton swab soaked in the extract was used to spread the virus using carborundum onto the adaxial side of a plant leaf. A photoperiod of 12 h/day and 12 h/night was maintained in a growth chamber at 25 °C45.
Preparation of tomato leaf samples
Systemic tissues (approximately 100 mg) were collected from non-inoculated upper leaves at 10 days post-inoculation (dpi). These aliquots were purified using 1 mL of ice-cold RNX-Plus (SinaClon, Karaj, Iran) according to the manufacturer’s protocol. Also, different dilutions [by a factor of 1010 with ribonuclease-free (DEPC) water] of these original samples were prepared. Following the manufacturer’s instructions, complementary DNA (cDNA) was synthesized from extracted RNA using the SinaClon cDNA Synthesis Kit. Furthermore, cDNAs were stored in a refrigerator at -80 °C until use.
ToBRFV detection by PCR
The presence of the virus within the inoculated plants was confirmed by RT-PCR (Eppendorf Mastercycler) followed by gel electrophoresis. 1 µL cDNA was mixed with 200 µM dNTPs (Thermo), 500 nM of forward and reverse primers, and 0.2 U/µL Phusion polymerase (Thermo) in a volume of 20 µL. Reactions were incubated at 98 °C for 30 s for denaturation, followed by 35 cycles of amplification at 98 °C for 10 s and 72 °C for 30 s (manufacturer’s 2-step protocol). The forward primer reads AGTAGATGACGCAACGGTGGCTATAAGGAG and the reverse primer TACGTGCCTACGGATGTGTATGAACCATAC. These primers were either used for PCR or RPA.
Guide RNAs
The spacer of the Cas12a-dependent crRNA used in this work reads CAAUGGUCCUCUGCACCUGC, and the spacer of the Cas9-dependent crRNA AUCUCAAGAUGCAGGUGCAG. The crRNAs were synthesized through in vitro transcription using the TranscriptAid T7 High Yield Transcription kit (Thermo) with DNA templates obtained from IDT. Subsequently, the crRNAs were purified using RNA Clean and Concentrator spin columns (Zymo) and quantified using a NanoDrop spectrophotometer.
ToBRFV amplification using RPA
The TwistAmp Basic kit (TwistDX) was used for isothermal amplification of the ToBRFV genome. Forward and reverse primers (480 nM) were combined with 29.5 µL rehydration buffer to reach a total volume of 45.5 µL (adjusted with RNase-free water). The TwistAmp Basic reaction pellet was resuspended with this volume, and 22.8 µL was used for each reaction with 1 µL cDNA. The reaction was initiated by adding 14 mM magnesium acetate. Reactions were incubated at 39 ºC for 30 min (Eppendorf Thermomixer) following manufacturer’s instructions.
CRISPR-Cas12a reaction
A mix of Cas12a from L. bacterium (NEB) and crRNA was incubated for 30 min for ribonucleoprotein formation. 1 µL RPA product was mixed with the CRISPR-Cas12a ribonucleoprotein preparation (50 nM), ssDNA probe (200 nM) in NEBuffer 2.1 [10 mM tris-HCl, 50 mM NaCl, 10 mM MgCl2, and 100 µg/mL BSA (pH 7.9); NEB] to make a total volume of 20 µL. Reactions were incubated for 1 h at 37 ºC (Eppendorf Thermomixer). The ssDNA probe was FAM-TTATT-B7, where FAM is fluorescein and B7 biotin. To further assess the detection ability of the method, different nuclease concentrations (50, 100, and 200 nM) and reaction times (30, 60, and 120 min) were assayed with 1 µL RPA product. Besides, TMV-infected Nicotiana tabacum plant samples45 were collected to perform a specificity assay. In particular, 1 µL RPA product from TMV-derived cDNA with the ToBRFV-targeting primers was used in a CRISPR-Cas12a reaction with the ToBRFV-targeting crRNA.
CRISPR-Cas9 reaction
The bimolecule crRNA-tracrRNA was constituted by hybridizing an equimolar amount of both RNAs, incubating them 3 min at 95 ºC and then slowly reducing the temperature to 10 ºC during 30 min (Eppendorf Mastercycler). A mix of Cas9 from S. pyogenes (IDT) and crRNA-tracrRNA was incubated for 30 min for ribonucleoprotein formation. 1 µL RPA product was mixed with the CRISPR-Cas9 ribonucleoprotein preparation (200 nM), ssDNA probe (50 or 100 nM) and COLUMBO46 reaction buffer [1x Tris/Acetate/EDTA (TAE) buffer pH 8.5 (Invitrogen), 0.05% Tween 20, and 12.5 mM MgCl2] to make a total volume of 20 µL. Reactions were incubated for 1 h at 37 °C (Eppendorf Thermomixer). The ssDNA probe was FAM-TTTTTTTTTTTTTTTTTT-B7. To further assess the detection ability of the method, different nuclease concentrations (50, 100, and 200 nM) and reaction times (30, 60, and 120 min) were assayed with 1 µL RPA product. Besides, TMV-infected Nicotiana tabacum plant samples45 were collected to perform a specificity assay. In particular, 1 µL RPA product from TMV-derived cDNA with the ToBRFV-targeting primers was used in a CRISPR-Cas9 reaction with the ToBRFV-targeting crRNA.
Lateral flow assay
CRISPR-Cas reaction volumes were mixed with 80 µL GenLine Dipstick buffer (Milenia) for 1:5 dilution and the lateral flow strip (HybriDetect, Milenia) was dipped into the reaction tube. Images were captured with a smartphone after 2 min. Image quantification of the band intensity in the test line was done with Fiji software47.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Yang, X., Li, Y. & Wang, A. Research advances in potyviruses: from the laboratory bench to the field. Annu. Rev. Phytopathol. 59, 1–29 (2021).
Oladokun, J. O., Halabi, M. H., Barua, P. & Nath, P. D. Tomato brown rugose fruit disease: current distribution, knowledge and future prospects. Plant. Pathol. 68, 1579–1586 (2019).
Dombrovsky, A., Tran-Nguyen, L. T. T. & Jones, R. A. C. Cucumber green mottle mosaic virus: rapidly increasing global distribution, etiology, epidemiology, and management. Annu. Rev. Phytopathol. 55, 231–256 (2017).
Broadbent, L. Epidemiology and control of tomato mosaic virus. Annu. Rev. Phytopathol. 14, 75–96 (1976).
Scholthof, K. B. G. Tobacco mosaic virus: a model system for plant biology. Annu. Rev. Phytopathol. 42, 13–34 (2004).
Zheng, Y. X., Shen, B. N., Chen, C. C. & Jan, F. J. Odontoglossum ringspot virus causing flower crinkle in Phalaenopsis hybrids. Eur. J. Plant. Pathol. 128, 1–5 (2010).
Salem, N. et al. A new tobamovirus infecting tomato crops in Jordan. Arch. Virol. 161, 503–506 (2016).
Luria, N. et al. A new Israeli tobamovirus isolate infects tomato plants harboring Tm-22 resistance genes. PLoS One. 12, e0170429 (2017).
Hak, H. & Spiegelman, Z. The tomato brown rugose fruit virus movement protein overcomes Tm-22 resistance in tomato while attenuating viral transport. Mol. Plant-Microbe Interact. 34, 1024–1032 (2021).
Salem, N. M., Cao, M. J., Odeh, S., Turina, M. & Tahzima, R. First report of tobacco mild green mosaic virus and tomato brown rugose fruit virus infecting Capsicum annuum in Jordan. Plant. Dis. 104, 601 (2020).
Chanda, B., Gilliard, A., Jaiswal, N. & Ling, K. S. Comparative analysis of host range, ability to infect tomato cultivars with Tm-22 gene, and real-time reverse transcription PCR detection of tomato brown rugose fruit virus. Plant. Dis. 105, 3643–3652 (2021).
Salem, N., Jewehan, A., Aranda, M. A. & Fox, A. Tomato brown rugose fruit virus pandemic. Annu. Rev. Phytopathol. 61, 137–164 (2023).
Fidan, H. et al. Robust molecular detection of the new tomato brown rugose fruit virus in infected tomato and pepper plants from Turkey. J. Integr. Agric. 20, 2170–2179 (2021).
Moury, B. & Verdin, E. Viruses of pepper crops in the Mediterranean basin: a remarkable stasis. In Advances in Virus Research, vol. 84 127–162 (Elsevier, 2012).
Hanssen, I. M. & Lapidot, M. Major tomato viruses in the Mediterranean basin. In Advances in Virus Research, vol. 84 31–66 (Elsevier, 2012).
Zhang, S., Griffiths, J. S., Marchand, G., Bernards, M. A. & Wang, A. Tomato brown rugose fruit virus: an emerging and rapidly spreading plant RNA virus that threatens tomato production worldwide. Mol. Plant. Pathol. 23, 1262–1277 (2022).
Jones, R. A. C. & Naidu, R. A. Global dimensions of plant virus diseases: current status and future perspectives. Annu. Rev. Virol. 6, 387–409 (2019).
Beris, D. et al. First report of tomato brown rugose fruit virus infecting tomato in Greece. Plant. Dis. 104, 2035 (2020).
Alfaro-Fernández, A., Castillo, P., Sanahuja, E., Rodríguez-Salido, M. C. & Font, M. I. First report of tomato brown rugose fruit virus in tomato in Spain. Plant. Dis. 105, 515 (2021).
Camacho-Beltrán, E. et al. Occurrence of tomato brown rugose fruit virus infecting tomato crops in Mexico. Plant. Dis. 103, 1440 (2019).
Ling, K. S., Tian, T., Gurung, S., Salati, R. & Gilliard, A. First report of tomato brown rugose fruit virus infecting greenhouse tomato in the united States. Plant. Dis. 103, 1439 (2019).
Yan, Z. Y. et al. First report of tomato brown rugose fruit virus infecting tomato in China. Plant. Dis. 103, 2973 (2019).
Fidan, H., Sarikaya, P. & Calis, O. First report of tomato brown rugose fruit virus on tomato in Turkey. New. Dis. Rep. 39, 588–2044 (2019).
Hamborg, Z. & -R. Blystad, D. First report of tomato brown rugose fruit virus in tomato in Norway. Plant. Dis. 106, 2004 (2022).
Ishibashi, K. & Ishikawa, M. Replication of tobamovirus RNA. Annu. Rev. Phytopathol. 54, 55–78 (2016).
Rubio, L., Galipienso, L. & Ferriol, I. Detection of plant viruses and disease management: relevance of genetic diversity and evolution. Front. Plant. Sci. 11, 1092 (2020).
Bernabé-Orts, J. M., Hernando, Y. & Aranda, M. A. Toward a CRISPR-based point-of-care test for tomato brown rugose fruit virus detection. PhytoFrontiers™ 2, 92–100 (2022).
Nourinejhad Zarghani, S. et al. Applicability of different methods for quantifying virucidal efficacy using MENNO florades and tomato brown rugose fruit virus as an example. Plants 12, 894 (2023).
Rizzo, D. et al. Rapid and sensitive detection of tomato brown rugose fruit virus in tomato and pepper seeds by reverse transcription loop-mediated isothermal amplification assays (real time and visual) and comparison with RT-PCR end-point and RT-qPCR methods. Front. Microbiol. 12, 640932 (2021).
Cao, Y. et al. Rapid and visual field diagnosis of tomato brown rugose fruit virus using reverse transcription recombinase aided amplification (RT RAA) combined with lateral flow strips (LFS). Crop Prot. 173, 106355 (2023).
Jiang, F. & Doudna, J. A. CRISPR–Cas9 structures and mechanisms. Annu. Rev. Biophys. 46, 505–529 (2017).
Kaminski, M. M., Abudayyeh, O. O., Gootenberg, J. S., Zhang, F. & Collins, J. J. CRISPR-based diagnostics. Nat. Biomed. Eng. 5, 643–656 (2021).
Marqués, M. C. et al. Diagnostics of infections produced by the plant viruses TMV, TEV, and PVX with CRISPR-Cas12 and CRISPR-Cas13. ACS Synth. Biol. 11, 2384–2393 (2022).
Aman, R., Mahas, A., Marsic, T., Hassan, N. & Mahfouz, M. M. Efficient, rapid, and sensitive detection of plant RNA viruses with one-pot RT-RPA–CRISPR/Cas12a assay. Front. Microbiol. 11, 610872 (2020).
Mahas, A. et al. LAMP-coupled CRISPR–Cas12a module for rapid and sensitive detection of plant DNA viruses. Viruses 13, 466 (2021).
Jiao, J. et al. Field detection of multiple RNA viruses/viroids in Apple using a CRISPR/Cas12a-based visual assay. Plant. Biotechnol. J. 19, 394–405 (2021).
Hak, H. et al. Rapid on-site detection of crop RNA viruses using CRISPR/Cas13a. J. Exp. Bot. 2024, erae495 (2024).
Alon, D. M., Hak, H., Bornstein, M., Pines, G. & Spiegelman, Z. Differential detection of the tobamoviruses tomato mosaic virus (ToMV) and tomato brown rugose fruit virus (ToBRFV) using CRISPR-Cas12a. Plants 10, 1256 (2021).
Chen, J. et al. Trans-nuclease activity of Cas9 activated by DNA or RNA target binding. Nat. Biotechnol. https://doi.org/10.1038/s41587-024-02255-7 (2024).
Sun, K. et al. Nanopore sequencing technology and its application in plant virus diagnostics. Front. Microbiol. 13, 939666 (2022).
Pardee, K. et al. Rapid, low-cost detection of Zika virus using programmable biomolecular components. Cell 165, 1255–1266 (2016).
Li, H. et al. Amplification-free crispr/cas detection technology: challenges, strategies, and perspectives. Chem. Soc. Rev. 52, 361–382 (2023).
Zhu, H., Li, C. & Gao, C. Applications of CRISPR–Cas in agriculture and plant biotechnology. Nat. Rev. Mol. Cell. Biol. 21, 661–677 (2020).
Schindele, P., Wolter, F. & Puchta, H. Transforming plant biology and breeding with CRISPR/Cas9, Cas12 and Cas13. FEBS Lett. 592, 1954–1967 (2018).
Besati, M. et al. Optimization and characterization of essential oil-loaded nanoemulsions for enhanced antiviral activity against tobacco mosaic virus (TMV) in tobacco plants. Ind. Crops Prod. 220, 119253 (2024).
Márquez-Costa, R. et al. Multiplexable and biocomputational virus detection by CRISPR-cas9-mediated strand displacement. Anal. Chem. 95, 9564–9574 (2023).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 9, 676–682 (2012).
Acknowledgements
A portion of this research was supported by the Spanish Ministry of Science, Innovation and Universities and AEI (PDC2022-133941-I00, co-financed by NextGenerationEU/PRTR).
Author information
Authors and Affiliations
Contributions
M.R. S. and H.R. designed research, M.B. and M.R.S. performed research, A.A. and M.F. helped to analyze data, and M.B. and M.R.S. wrote the paper. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Besati, M., Safarnejad, M.R., Aliahmadi, A. et al. Detection of tomato brown rugose fruit virus through CRISPR-Cas12a and CRISPR-Cas9 systems. Sci Rep 15, 25638 (2025). https://doi.org/10.1038/s41598-025-11825-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-11825-x
