
Abstract
Residual material from fecal immunochemical test (FIT) tubes, commonly used in colorectal cancer screening programs, offers a valuable resource for large-scale gut microbiome studies. With recent advances in sequencing technologies, sequencing the full-length bacterial 16S ribosomal gene is now feasible. In this study, we evaluated the impact of pre-analytical handling conditions on microbiome profiling using FIT samples. Stool samples from eight healthy adults were subjected to various short-term (+ 20 °C) and long-term (–18 °C or –80 °C) storage conditions prior to DNA extraction. We also investigated the effects of sampling variation and the presence of buffer medium. Full-length 16S rRNA gene amplicons were generated and sequenced using Oxford Nanopore Technology to characterize the microbiome composition. Despite variations in sampling and storage conditions, microbiome richness, Shannon diversity, and individual characteristics were preserved, demonstrating the robustness of microbiomes extracted from FIT tubes. However, some variations were noted, such as increased amounts of collagenase-producing bacteria from 0.2 to 0.6% to 1.7–2.6% in samples stored at +20 °C for 4–10 days. Despite unsupervised and varying sampling and storage conditions, the fecal 16S rRNA microbiomes remained representative and robust. These findings support the usability of FIT samples for large-scale population microbiome research.
Similar content being viewed by others
Introduction
Genetic and environmental factors contribute to the significant interindividual variability in human susceptibility to disease. The gut microbiota composition and associated metabolites are influenced by several environmental factors that have garnered increasing attention due to mounting evidence of their substantial impact on host health, disease susceptibility, and recovery1. However, the gut microbiota and host health relationship is complex and bidirectional, as they coevolve and mutually adapt2. The host’s diet, exercise, immune status, chronic illnesses, and other factors (such as age, antibiotics, and environmental conditions) affect gut microbiota composition and metabolite production3.
Analyses of the gut microbiome are becoming “standard tests” used in basic science, population studies, and daily clinical practice4. However, the lack of common standards has made reproducing results from other laboratories challenging for one laboratory5. The preanalytical handling of samples is now a well-recognized source of considerable variation in daily clinical laboratory tests6 and microbiome analysis7. Therefore, guidelines have recently been developed to help standardize preanalytical procedures and workflows, including collection, transport, and storage before analysis, to facilitate the harmonization and commutability of test results through reduced interlaboratory variation8. In clinical practice and research, gut microbiome analyses often require grams of feces to be collected in a transport device and shipped to the laboratory, where the stool sample is preferably homogenized before an aliquot is analyzed9. However, although the collection is noninvasive and practical, the implementation and adherence to stool analyses have been hindered by multiple challenges, including forgetfulness and avoidance10 and difficulties with the stool collection itself11.
Fecal immunochemical test (FIT) based colorectal cancer screening programs also suffer from many of the above challenges, although most programs have a 60–70% uptake rate12. Moreover, several studies have demonstrated the feasibility of extracting sufficient bacterial DNA from residual buffer solutions of 1.5 mL or more for subsequent sequencing of gut microbiomes13,14,15,16,17,18,19. Therefore, FIT sampling tubes could be an inexpensive and easy way to conduct population-based microbiome studies and may also improve the program’s performance and predict, for instance, the risk of complications to surgery. However, unlike “the usual” collection methods for clinical and research microbiome testing, the collection of FIT samples differs in several ways20. First, the samples were not intended for microbiome analysis, as only approximately 10 mg of feces was collected. The participants received only brief written instructions emphasizing the importance of collecting samples from the stool surface and returning them by mail. Neither the time of collection nor the duration of the mail transport is recorded, nor is the temperature at which this is done known. Since these conditions are given when using regular mail services, the focus of the usual verification process had to be turned “upside down.” Instead, the focus had to be on the impact of these factors on sample stability and, consequently, how representative the FIT samples are of the microbiome in the patients.
Several studies have shown that FIT sample DNA seems stable at room temperature in collection buffer13,14,15,16,17,18,19 when 0.5 kilobase (kB) PCRs are performed for microbiome analyses by amplifying the V3–4 regions of the 16S ribosomal RNA (rRNA) gene. The emergence of Oxford Nanopore Technologies (ONT) and other long-read sequencing methods allows routine sequencing of the entire V1–V9 region of the 16S rRNA gene in one read. However, the average size of the DNA fragments isolated from the FIT samples is approximately 0.9 kB17 which is shorter than the 1.6 kB V1–V9 region, resulting in a lower yield of DNA than when the 0.5 kilobase V3–V4 region is sequenced21,22,23. In “routine” clinical and research settings, this is usually compensated for by increasing the amount of DNA extracted.
Therefore, we examined the impact of preanalytical handling of FIT samples (sampling from the surface versus core of the stool, short- and long-term storage at different temperatures, and the impact of the transport medium) on the performance of bacterial 16S rRNA sequencing via ONT. To further investigate the effects of possible preanalytical variation on the biological interpretation of the test results, we, as an example, examined the variation in the detection of bacteria with the potential to produce the enzyme collagenase.
Results
We included eight adult volunteers (five women and three men) in the study, with a median age of 39 years (28–53).
Quality control and sequencing depth
Bacterial DNA was extracted, and microbiomes were sequenced from all samples. One sample was lost during processing (the transport medium sample of Participant #2). The read length of the DNA strands was 1,436 base pairs (1,432–1,440 base pairs), and the quality (Phred) score was 13.3 (13.0–13.7). The DNA concentration in the sampling tubes remained unchanged when stored at −80 °C for up to 400 days (Fig. 1).

A scatter plot of the DNA concentration following extraction as a function of storage duration at −80 °C before the bacterial DNA was extracted. The straight line is a Deming regression line.
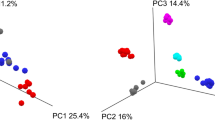
The median number of reads for all the samples was 116,691 (range 1,956–602,613). Participant #3 had the lowest number of reads (43,644; range 4,943–107,291), and Participant #5 had the highest (254,414; range 30,062–578,435). Most samples (N = 143) had fewer than 300,000 reads. The accuracy of the Basic Local Alignment Search Tool (BLAST) alignment was 94.6% (91.1–96.4%). The samples with the most reads appeared to be randomly distributed across different sequencing runs (Fig. 2). The rarefaction curves illustrate the sequencing depth of each sample (Fig. 3). The PCoA plot revealed that all participants clustered differently (Fig. 4).

Bar plot of the number of reads in all samples, shown for each participant and divided into the five subgroups (surface vs. core, short-term storage (+ 20 °C), long-term storage (–18 °C or –80 °C), or buffer vs. water). One sample from Participant #2 was lost, hence the zero read count. The scale on the y-axis is fixed in all plots to make comparing the participants easier.

Rarefaction curves of all participants. The five subgroups (surface vs. core, short-term storage (+20 °C), long-term storage (–18 °C or –80 °C), or buffer vs. water) are colored separately (colors similar to the corresponding bars in Fig. 2). The scale on the y-axis is fixed in all plots to make comparing the participants easier.

Principal Coordinates Analysis plot (Canberra distance) of all samples. The Axis.1 depicts the dimension with the most significant variation (in percent) of the samples and the Axis.2 depicts the dimension of the second-most variation (in percent). Differences in beta diversity between the participants were calculated using the permutational analysis of variance (PERMANOVA) test.
Preanalytical variation
Sampling site
No significant variation in species richness, alpha diversity, or relative abundance was detected between surface and core fecal samples (Supplementary Table S1 and Supplementary Fig. S1a-b).
Short-term storage (+20 °C)
We found no differences in richness or alpha diversity (Supplementary Table S1). The relative abundance visualization revealed minor changes at the phylum and genus levels during the ten days of storage at room temperature, although none were significant (Supplementary Fig. S2a-b).
Long-term storage (–18 °C or –80 °C)
There were no significant differences in richness or alpha diversity when the samples were stored at −18 °C or −80 °C (Supplementary Table S1). The relative abundance visualizations revealed that several phyla and genera presented minor variations among the samples stored for 1, 3, and 6 months, but none of the changes were significant (Supplementary Fig. S3a-b).
Impact of the transport medium
We found no difference in richness or alpha diversity between the buffer and water samples (Supplementary Table S1). The visualization of the relative abundance could indicate slight changes in the Bacillota/Bacteroidota ratio and the amounts of Blautia and a few other genera (Supplementary Fig. S4a-b). However, the relative abundance calculations revealed no differences between the groups.
In summary, the PCoA plot revealed that although there were minor differences within each participant’s samples, the most significant differences were observed between different individuals. Moreover, the PCoA confirmed that the intraindividual variation in the fecal microbiome between different sampling and storage methods was insignificant; thus, interindividual variations were preserved in the analyses.
Bacteria with the potential to produce collagenases
The subgroup of bacteria with the potential to produce collagenase (according to a recent study24) constituted 0.2–0.6% of most samples containing the FIT buffer medium (Fig. 5). However, their abundance increased to 1.7–2.6% after the samples were stored for four days or more at +20 °C. In contrast, Bacteroides thetaiotaomicron and Clostridium perfringens constituted approximately the same proportion in all the short-term storage samples. The fraction made up of Enterococcus faecalis increased over time (Fig. 6a). Enterococcus faecalis was present in samples where the buffer medium had been replaced with sterile water, but was absent in the FIT buffer medium samples (Fig. 6b). In the surface vs. core samples or long-term storage samples, the amount of Enterococcus faecalis was minor and did not seem to vary (Supplementary Fig. S5–S6).

Bar plot of all counts, divided into categories of sample type. The prevalence of bacterial species that could produce collagenase is displayed at each bar.

Bar plots of the prevalence of the bacterial species with the potential to produce collagenase among (a) Short-term storage (+20 °C) samples, and (b) Transport medium (buffer vs. water) samples.
Discussion
This study revealed that enough 1.5 kb of bacterial DNA could be extracted from the FIT sampling tubes to amplify the entire bacterial 16S rRNA region for microbiome analysis. We also found that the characteristics of the individual microbiomes derived from the +1.5 mL residual material of the FIT sampling tubes were stable despite various sampling, transport, and storage conditions. This parallels what has been observed for V3–V4-based microbiomes derived from residual material from FIT sampling bottles13,14,15,16,17,18,19. However, the fraction of bacteria that have the potential to produce collagenase increased with the duration of short-term storage at room temperature in the FIT buffer medium and in the sampling tubes with sterile water, primarily due to the growth of Enterococcus faecalis.
Our analysis revealed that alpha diversity was comparable between surface and core samples across various stool segments, corroborating the findings of an earlier study25. In contrast, we did not find significant differences in the relative abundance compositions of several bacterial families. Another study has shown that replicating the detection of low-abundance taxa in samples taken 1 cm apart in each stool was challenging26 indicating that localized sampling only partially represents the fecal microbiome. Furthermore, stool consistency is correlated with species richness, with liquefied stool having the lowest richness27. Unfortunately, we did not collect information about stool consistency in our study.
There has yet to be a consensus on how to collect, store, sequence, and process data from gut microbiome studies28,29,30,31 nor which FIT sampling tube to use32,33. We detected minimal variation in the gut microbiome collected from different parts of the feces when the OC–Sensor sampling system was used. This contrasts with other reports suggesting that samples taken from similar locations should be homogenized before further processing to prevent bias from sampling variation34. The microbiome composition remained unaffected when comparing aerobe and anaerobe sampling tubes35. Previous research has examined the impact of varying storage durations and temperatures, revealing that storage at room temperature for up to 48 hours25,36,37 or even 7–14 days38,39 did not change the microbiome composition. In contrast, variations were detected as soon as 15 min in other studies40,41. We showed the microbiome was stable after short-term storage at a refrigerator temperature (+4 °C) for up to 48 hours37. After storage at −18 °C (like a household freezer), the microbiome composition was stable for at least three days40. The gold standard for sample storage is −80°C42, and recent studies revealed that the microbiome composition was stable at this temperature after 12 months of storage43,44. In the absence of −80 °C facilities, freeze-drying is a suitable alternative45. Keeping the storage temperature constant is essential to avoid freeze-thawing of the samples, thus damaging the DNA46. Nevertheless, DNA and RNA levels were stable even after nine freeze-thaw cycles in a study of colorectal cancer tissue47. The buffer medium in the sampling tube may allow for postsampling growth of different bacteria, thereby altering the composition of the gut microbiome before DNA extraction. A wide range of buffers is used in microbiome studies. Overall, the composition of the bacteria was stable in these buffers, although variations in bacterial taxonomy can occur depending on the buffer used48,49,50,51. Most importantly, we found that the characteristics of each individual’s gut microbiome were captured irrespective of storage conditions, as the variations originating from differences in storage conditions were minor compared with the interindividual microbiome variation of the donors, which aligns with other studies52.
The HEPES buffer medium of the sampling tubes is a widely used buffering agent in biological and biochemical research and helps maintain a stable pH within the physiological range. We found that the growth of E. faecalis was initially limited by the HEPES buffer compared with sterile water. Nevertheless, storage at room temperature allowed bacterial growth in samples containing buffer medium and stored for 4–10 days. Other types of transport media (Amies and Cary–Blair) resulted in similar growth of E. faecalis after 24–48 h at room temperature, but storing the samples at +4 °C reduced growth53,54.
Using the residual buffer material from FIT screening in an OC–Sensor, and possibly other FIT sampling tubes, allows for the accessible collection and analysis of baseline microbiomes, which reflect the adaptation of the microbiomes to the host and vice versa. They, therefore, reveal characteristics of both the gut microbiome and the host. We also found that the standard preanalytical handling of the FIT samples (collection and subsequent transport by mail) had a minimal effect on the composition of the samples, ensuring that the individual characteristics of the microbiomes were preserved. Finally, we demonstrated that microbiomes can be generated from DNA extracted from residual material in FIT sampling tubes, based on the sequence of the full-length V1–V9 hypervariable regions of the 16S rRNA gene. Using the entire V1–V9 sequence, rather than shorter areas, such as the V3–V4 region, has enabled more accurate discrimination between closely related species21.
A strength of this study is that we addressed possible concerns regarding the impact of variations in the preanalytical handling of the types of sampling tubes used in the Danish Colorectal Cancer Screening Program. The results for the fecal microbiomes originating from these types of samples represent the species present, regardless of variations in sampling techniques or storage conditions. A limitation of the study is that we did not use positive controls. During the DNA extraction and PCR steps, we analyzed samples containing sterile water as the sample material, which constitutes negative controls. The results should be interpreted cautiously, especially concerning low-abundance bacteria, as both localized sampling and the choice of primer could impact these findings. Another limitation is the duration of “long-term storage” (6 months). The preanalytical stability of samples stored for several years remains unknown. This study supports the feasibility and reliability of large-scale studies of fecal microbiomes collected from the sampling tubes of a colorectal cancer screening program. Nevertheless, the limitations of microbiome studies should always be taken into account when designing future studies.
We have demonstrated that bacterial DNA can be successfully extracted from OC–Sensor FIT screening sampling tubes and subsequently used to sequence full-length 16S rRNA microbiomes. Furthermore, we found that the collection site (superficial vs. core and front, middle, or tail) of the fecal matter did not significantly impact the composition of the fecal microbiome, nor did the composition change due to short-term (a surrogate for transport time) or long-term storage at various temperatures. Furthermore, despite the limited growth of robust bacteria (e.g., Enterococcus faecalis), the sampling buffer stabilized the bacteria, allowing for minimal growth compared to sterile water. Therefore, the full-length 16S rRNA microbiomes generated from the residual FIT sampling material collected through FIT-based CRC screening programs are representative of the feces at the time of collection. This study lays the groundwork for microbiome research by utilizing sampling tubes from colorectal cancer screening programs.
Materials and methods
This paper was written according to the ”Strengthening The Organization and Reporting of Microbiome Studies” (STORMS) checklist55 (Supplementary Material). The study received ethical approval from the Committee of Research Ethics in Region Zealand (SJ-1069).
Study participants
This study was an observational cohort study. Healthy adult volunteers provided stool samples following specific sampling instructions. None of the participants took daily medication that could modify the gut microbiome composition (e.g., proton pump inhibitors, nonsteroidal anti-inflammatory drugs, opioids, antibiotics, or chemotherapy) within three months of sampling.
Sample collection
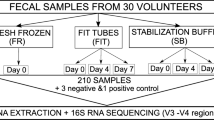
Sampling was performed at the participants’ homes, following written instructions from the Danish Colorectal Cancer Screening Program (DCCSP), which included a cartoon illustrating how to collect the fecal samples. The fecal matter was gathered via the EasySampler Basic paper toilet cover (GP Medical Devices, Holstebro, Denmark). Afterward, samples were collected using an OC–Sensor (OC–Auto Sampling Bottle 3; Eiken Chemical Co., Ltd., Tokyo, Japan) sampling tube. The OC–Sensor is designed to collect fecal matter for a fecal immunochemical test and features a flattened tube with a cap attached to the sampling probe56. The bottom of the sampling tube features a foil seal, allowing access to the sample buffer in which the fecal matter is dissolved. The yield is approximately 10 mg of feces per sample, corresponding to a feces concentration of 5 mg/mL. The transport medium in the sampling tubes was 2 ml of HEPES buffer (N-2-hydroxyethylpiperazine-N’−2-ethane-sulforic acid). When used in the DCCSP, only a few microliters of buffer are used to analyze for the presence of hemoglobin. The remaining buffer medium, therefore, corresponds to the residual material mentioned in the Introduction. For each participant, 20 stool samples were collected from one defecation. The time from sampling in the participants’ homes until the samples were processed was planned to last 1–3 days to mimic the anticipated time for the sample from the patient participating in the DCCSP to laboratory processing in a real-life setting. During this period, the participants were instructed to keep the samples at room temperature to mimic transport by regular mail.
Sample handling
The participants were instructed to sample superficially from the stool’s proximal, middle, and distal parts to investigate the differences in bacterial distribution throughout the feces. Additionally, two samples from the core of the stool were collected by introducing the tip of the sampling device into the center of the stool at two locations and then twisting it a couple of times. To mimic the transport conditions for patient samples before they reach the laboratory performing the FIT, six localized superficial samples were pooled and stored at room temperature for up to 10 days before being stored at −80 °C until further processing.
Furthermore, seven localized superficial samples were pooled and stored at either −18 °C or −80 °C for 1–6 months before DNA extraction to examine the effects of long-term storage. Finally, we investigated the impact of the transport medium on sample stability. The participants collected two localized samples, one of which was the transport medium that had been replaced with sterile water before collection. An overview of the four substudies is provided in Fig. 7.

Overview of the four substudies. (a) Superficial sampling vs. sampling from the core of the stool. (b) Localized, pooled samples were stored at room temperature (+20 °C) until analysis. (c) Localized, pooled samples were stored in a freezer (–18 °C or −80 °C) until analysis. (d) Localized samples were stored in either transport medium or sterile water.
DNA extraction
DNA extraction from the fecal samples was performed via the AllPrep PowerFecal Pro DNA/RNA Kit (Cat. No. 80244; Qiagen, Hilden, Germany). Briefly, 200 µL of FIT sample buffer was transferred to a lysis tube provided by the manufacturer and eluted in 650 µL of buffer. Additionally, 25 µL of dithiothreitol was added. Both bead beating and chemical lysis were used in the DNA extraction process. DNA extraction was performed according to the manufacturer’s protocol. The concentrations of the extracted DNA were measured via a Qubit 1X dsDNA HS Assay Kit (Q33231, Thermo Fisher Scientific, Waltham, USA) and a Qubit™ 4 fluorometer (Q33226, Invitrogen, Carlsbad, USA).
Polymerase chain reaction
After the DNA concentration in the extracts was quantified, polymerase chain reaction (PCR) was performed via the 16S Barcoding Kit SQK–RAB204 (Oxford Nanopore Technologies, Oxford, UK) according to the manufacturer’s instructions. The 16S primers 27F and 1492R were used to amplify the entire 16S region, including the nine hypervariable regions V1–V9, resulting in a PCR product of approximately 1,5 kBs. The primers also had rapid attachment chemistry and contained 24 barcodes to distinguish the samples during sequencing. For each PCR, 10 ng or 15 µL of extracted DNA was added, along with 10 µL of primer and 25 µL of LongAmp Taq 2X master mix (M0287L, New England Biolabs, Ipswich, USA). The 60-second initial denaturation at 95 °C was followed by 40 cycles of denaturation at 95 °C for 20 s, annealing at 55 °C for 30 s, extension at 65 °C for 120 s, and a final extension at 65 °C for 300 s.
The PCR products were purified via AMPure XP beads (A63881, Beckman Coulter, Indianapolis, USA) according to the 16S barcoding kit SQK–RAB204 protocol. In brief, the PCR products were transferred to a 1.5 mL centrifuge tube along with 30 µL AMPure XP beads. The samples were incubated on a Hula mixer (V.3A01, Life Technologies AS, Oslo, Norway). After 5 min, the samples were spun down and placed on a DynaMag™–2 (12321D, Invitrogen, Oslo, Norway) for 1 min until the beads had gathered on one side. The supernatant was discarded, and the beads were washed twice with 70% ethanol. After the second wash, the samples were spun down and placed on a magnet. The remaining liquid ethanol was pipetted away, and the samples were left to air-dry for 30 s to remove the last traces of ethanol. The samples were resuspended in 10 µL of 10 mM Tris–HCl (pH 8.0) with 50 mM NaCl and incubated at room temperature for 5 min. The samples were then placed back on the magnet and left for 1 min for the beads to gather on one side. Finally, the supernatant was transferred to a new 1.5 mL centrifuge tube, and the DNA concentration was measured via the Qubit 1X dsDNA HS Assay Kit, as described above. The amplification size was assessed during sequencing rather than by gel electrophoresis. Negative controls were used in the PCR.
Library preparation and sequencing
Six purified samples were pooled in equal amounts to achieve a concentration of 5 fmol/µL, and rapid adapter protein (OXNTEXP–RAP001, Oxford Nanopore Technologies, Oxford, England) was added, along with sequencing buffer and loading beads, according to the manufacturer’s protocol. Each prepared library was loaded onto a new flow cell (version R9.4.1) and sequenced on a GridION Mk1 × 5 (ONT), with a sequencing process duration of 16 hours. We did not use quantitative microbiome profiling. The samples were sequenced in random order.
Software and data processing
The MinKNOW software version 5.7.5 (ONT) performs base calling via Dorado version 7.1.4 in the “super accurate” setting. The threshold for the Phred score was set to 7, corresponding to a base call accuracy of greater than 80%. The full-length 16S rRNA sequences were demultiplexed using the EPI2ME™ (ONT) desktop agent fast 16S workflow. The nucleotide sequences were then identified using the BLAST from the United States National Library of Medicine, National Institutes of Health. We used the National Center for Biotechnology Information (NCBI) 16S + 18S rRNA database with default parameters (minimum length filter = 0, maximum length filter = 0, BLAST E-value filter = default [e = 0.01], minimum coverage % = 30, minimum identity % = 77, maximum target sequences = 3). Coverage information per read was calculated as the number of identical matches/query lengths. All read classifications were filtered for > 77% min. identity and > 30% coverage to remove spurious alignments, which are the default settings. Further analyses were performed using RStudio version 2023.09.1 (PBC, Boston, USA), running R version 4.3.1 (R Core Team). We analyzed the microbiome data using the phyloseq version 1.44.0 package57.
Statistical calculations
Rarefaction curves were visualized for each sample to assess sequencing depth. To correct for differences in sequencing depth, we used normalization (i.e., the number of reads for each species divided by the total number of reads per sample) on all samples. We used Deming regression to fit a straight line to the data in the scatter plot, presented with the R2 value. Richness, a fundamental component of alpha diversity measures, was defined as the number of unique species in each sample. Alpha diversity was calculated via the Shannon diversity index. Differences in the alpha diversity between the two groups were calculated via the Wilcoxon rank-sum test. We used the Kruskal–Wallis test to determine the differences between three or more groups. Differences in beta diversity between the participants were calculated using the permutational analysis of variance (PERMANOVA) test. The relative abundances of the samples were visualized as stacked bar plots (detection = 0.5%; prevalence = 50%), and differences in proportions were calculated using the Wilcoxon rank-sum test, with the Benjamini‒Hochberg correction to adjust for the false discovery rate. We used the Canberra distance in principal coordinate analysis (PCoA). All values are presented as medians with interquartile ranges in parentheses. Adjusted P values < 0.05 were considered statistically significant.
Data availability
Owing to Danish legislation, sharing or storing raw biological information data outside approved Danish institutions is prohibited. You can contact the corresponding author if you have questions about the dataset.
Change history
09 October 2025
The original online version of this Article was revised: In the original version of this Article Anders Bech Jørgensen, Louise Almer, Birgitte Brandstrup and Lennart Friis-Hansen were omitted as equally contributing authors. The statement has now been added and reads: “Anders Bech Jørgensen, Louise Almer, Birgitte Brandstrup and Lennart Friis-Hansen: These authors contributed equally to this work.”
References
Lynch, S. V. & Pedersen, O. The human intestinal Microbiome in health and disease. N Engl. J. Med. 375, 2369–2379 (2016).
Littman, D. R. & Pamer, E. G. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell. Host Microbe. 10, 311–323 (2011).
Hasan, N. & Yang, H. Factors affecting the composition of the gut microbiota, and its modulation. PeerJ 7, e7502 (2019).
Turjeman, S. & Koren, O. Using the Microbiome in clinical practice. Microb. Biotechnol. 15, 129–134 (2022).
Baker, M. 1,500 scientists lift the lid on reproducibility. Nature 533, 452–454 (2016).
Lippi, G. et al. Preanalytical quality improvement: in quality we trust. Clin. Chem. Lab. Med. 51, 229–241 (2013).
Human Microbiome Project Consortium. Structure, function and diversity of the healthy human Microbiome. Nature 486, 207–214 (2012).
Stumptner, C. et al. The Pre-Analytical CEN/TS standard for Microbiome Diagnostics—How can research and development benefit?? Nutrients 14, 1976 (2022).
Tang, Q. et al. Current sampling methods for gut microbiota: A call for more precise devices. Front. Cell. Infect. Microbiol. 10, 151 (2020).
Green, B. B. et al. Reasons for never and intermittent completion of colorectal cancer screening after receiving multiple rounds of mailed fecal tests. BMC Public. Health. 17, 531 (2017).
Kalla, R. et al. Patients’ perceptions of faecal calprotectin testing in inflammatory bowel disease: results from a prospective multicentre patient-based survey. Scand. J. Gastroenterol. 53, 1437–1442 (2018).
Njor, S. H. et al. Three years of colorectal cancer screening in Denmark. Cancer Epidemiol. 57, 39–44 (2018).
Baxter, N. T., Koumpouras, C. C., Rogers, M. A. M., Ruffin, M. T. & Schloss, P. D. DNA from fecal immunochemical test can replace stool for detection of colonic lesions using a microbiota-based model. Microbiome 4, 59 (2016).
Grion, B. A. R. et al. Identification of taxonomic changes in the fecal bacteriome associated with colorectal polyps and cancer: potential biomarkers for early diagnosis. Front. Microbiol. 14, 1292490 (2023).
Grobbee, E. J. et al. First steps towards combining faecal immunochemical testing with the gut Microbiome in colorectal cancer screening. United Eur. Gastroenterol. J. 8, 293–302 (2020).
Gudra, D. et al. A widely used sampling device in colorectal cancer screening programmes allows for large-scale Microbiome studies. Gut 68, 1723–1725 (2019).
Istvan, P. et al. Exploring the gut DNA Virome in fecal immunochemical test stool samples reveals associations with lifestyle in a large population-based study. Nat. Commun. 15, 1791 (2024).
Krigul, K. L., Aasmets, O., Lüll, K., Org, T. & Org, E. Using fecal immunochemical tubes for the analysis of the gut Microbiome has the potential to improve colorectal cancer screening. Sci. Rep. 11, 19603 (2021).
Zouiouich, S. et al. Stability of the fecal and oral Microbiome over 2 years at -80°C for multiple collection methods. Cancer Epidemiol. Biomarkers Prev. 32, 444–451 (2023).
Vogtmann, E. et al. Comparison of collection methods for fecal samples in Microbiome studies. Am. J. Epidemiol. 185, 115–123 (2017).
Matsuo, Y. et al. Full-length 16S rRNA gene amplicon analysis of human gut microbiota using minion™ nanopore sequencing confers species-level resolution. BMC Microbiol. 21, 35 (2021).
Johnson, J. S. et al. Evaluation of 16S rRNA gene sequencing for species and strain-level Microbiome analysis. Nat. Commun. 10, 5029 (2019).
Welham, Z., Li, J., Engel, A. F. & Molloy, M. P. Mucosal Microbiome in patients with early bowel polyps: inferences from Short-Read and Long-Read 16S rRNA sequencing. Cancers (Basel) 15, (2023).
Jørgensen, A. B., Jonsson, I., Friis-Hansen, L. & Brandstrup, B. Collagenase-producing bacteria are common in anastomotic leakage after colorectal surgery: a systematic review. Int. J. Colorectal Dis. 38, 275 (2023).
Liang, Y. et al. Systematic Analysis of Impact of Sampling Regions and Storage Methods on Fecal Gut Microbiome and Metabolome Profiles. mSphere 5, (2020).
Wu, G. D. et al. Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC Microbiol. 10, 206 (2010).
Vandeputte, D. et al. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 65, 57–62 (2016).
Pérez-Burillo, S. et al. Effect of Freezing on Gut Microbiota Composition and Functionality for In Vitro Fermentation Experiments. Nutrients 13, (2021).
Xu, Z. et al. Variation in the metagenomic analysis of fecal Microbiome composition calls for a standardized operating approach. Microbiol. Spectr. 12, e0151624 (2024).
Poulsen, C. S., Kaas, R. S., Aarestrup, F. M. & Pamp, S. J. Standard sample storage conditions have an impact on inferred Microbiome composition and antimicrobial resistance patterns. Microbiol. Spectr. 9, e0138721 (2021).
Isokääntä, H. et al. High-throughput DNA extraction strategy for fecal Microbiome studies. Microbiol Spectr 12, (2024).
Zouiouich, S. et al. Comparison of fecal sample collection methods for microbial analysis embedded within colorectal Cancer screening programs. Cancer Epidemiol. Biomarkers Prev. 31, 305–314 (2022).
Brezina, S. et al. Using fecal immmunochemical cartridges for gut Microbiome analysis within a colorectal cancer screening program. Gut Microbes. 15, 2176119 (2023).
Jones, J., Reinke, S. N., Ali, A., Palmer, D. J. & Christophersen, C. T. Fecal sample collection methods and time of day impact Microbiome composition and short chain fatty acid concentrations. Sci. Rep. 11, 13964 (2021).
Tunsakul, N., Wongsaroj, L., Janchot, K., Pongpirul, K. & Somboonna, N. Non-significant influence between aerobic and anaerobic sample transport materials on gut (fecal) microbiota in healthy and fat-metabolic disorder Thai adults. PeerJ 12, e17270 (2024).
Carroll, I. M., Ringel-Kulka, T., Siddle, J. P., Klaenhammer, T. R. & Ringel, Y. Characterization of the fecal microbiota using high-throughput sequencing reveals a stable microbial community during storage. PLoS One. 7, e46953 (2012).
Cunningham, J. L. et al. Impact of time and temperature on gut microbiota and SCFA composition in stool samples. PLoS One. 15, e0236944 (2020).
Lauber, C. L., Zhou, N., Gordon, J. I., Knight, R. & Fierer, N. Effect of storage conditions on the assessment of bacterial community structure in soil and human-associated samples. FEMS Microbiol. Lett. 307, 80–86 (2010).
Tedjo, D. I. et al. The effect of sampling and storage on the fecal microbiota composition in healthy and diseased subjects. PLoS One 10, (2015).
Gorzelak, M. A. et al. Methods for improving human gut Microbiome data by reducing variability through sample processing and storage of stool. PLoS One. 10, e0134802 (2015).
Nagata, N. et al. Effects of storage temperature, storage time, and Cary-Blair transport medium on the stability of the gut microbiota. Drug Discov Ther. 13, 256–260 (2019).
Guan, H. et al. Comparison of fecal collection methods on variation in gut metagenomics and untargeted metabolomics. mSphere 6, e0063621 (2021).
Soukupova, H., Rehorova, V., Cibulkova, I. & Duska, F. Assessment of faecal microbiota transplant stability in Deep-Freeze conditions: A 12-Month ex vivo viability analysis. J. Clin. Lab. Anal. 38, e25023 (2024).
Barko, P., Nguyen-Edquilang, J., Williams, D. A. & Gal, A. Fecal Microbiome composition and diversity of cryopreserved canine stool at different duration and storage conditions. PLoS One. 19, e0294730 (2024).
Bensch, H. M., Tolf, C., Waldenström, J., Lundin, D. & Zöttl, M. Freeze-drying can replace cold-chains for transport and storage of fecal Microbiome samples. PeerJ 10, e13095 (2022).
Cardona, S. et al. Storage conditions of intestinal microbiota matter in metagenomic analysis. BMC Microbiol. 12, 158 (2012).
Fan, X. J. et al. Impact of cold ischemic time and Freeze-Thaw cycles on RNA, DNA and protein quality in colorectal Cancer tissues biobanking. J. Cancer. 10, 4978–4988 (2019).
Wang, Z. et al. Comparison of fecal collection methods for Microbiome and metabolomics studies. Front. Cell. Infect. Microbiol. 8, 301 (2018).
Bartolomaeus, T. U. P. et al. Quantifying technical confounders in Microbiome studies. Cardiovasc. Res. 117, 863–875 (2021).
Choo, J. M., Leong, L. E. X. & Rogers, G. B. Sample storage conditions significantly influence faecal Microbiome profiles. Sci Rep 5, (2015).
Isokääntä, H. et al. Comparative metabolomics and Microbiome analysis of ethanol versus omnimet/gene•gut fecal stabilization. Anal. Chem. 96, 8893–8904 (2024).
Bundgaard-Nielsen, C., Hagstrøm, S. & Sørensen, S. Interpersonal variations in gut microbiota profiles supersedes the effects of differing fecal storage conditions. Sci. Rep. 8, 17367 (2018).
Tano, E. & Melhus, A. Evaluation of three swab transport systems for the maintenance of clinically important bacteria in simulated mono- and polymicrobial samples. APMIS 119, 198–203 (2011).
DeMarco, A. L. et al. Survival of vaginal microorganisms in three commercially available transport systems. Anaerobe 45, 44–49 (2017).
Mirzayi, C. et al. Reporting guidelines for human Microbiome research: the STORMS checklist. Nat. Med. 27, 1885–1892 (2021).
Schreuders, E. H., Grobbee, E. J., Spaander, M. C. W. & Kuipers, E. J. Advances in fecal tests for colorectal Cancer screening. Curr. Treat. Options Gastroenterol. 14, 152–162 (2016).
McMurdie, P. J. & Holmes, S. Phyloseq: an R package for reproducible interactive analysis and graphics of Microbiome census data. PLoS One. 8, e61217 (2013).
Funding
This original article was written as part of a PhD, supported by the Danish Cancer Society, The Vissing Foundation, The Health Science Research Foundation of Region Zealand; House of Research, Holbæk Hospital; Department of Surgery, Holbæk Hospital; and Dagmar Marshalls Foundation.
Author information
Authors and Affiliations
Contributions
Conceptualisation (A.B.J., B.B., L.F-H.). Methodology (A.B.J., L.A., B.B., L.F-H.). Formal analysis (A.B.J., L.A., B.B., L.F-H.). Visualization (A.B.J., B.B., L.F-H.). Writing – original draft (A.B.J., L.A., B.B., L.F-H.). Writing – review and editing (A.B.J., L.A., B.B., L.F-H.). Funding acquisition (A.B.J., B.B.). Supervision (B.B., L.F-H.).
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study was conducted following the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all participants included in the study.
The authors have no competing interests to declare.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jørgensen, A.B., Almer, L., Brandstrup, B. et al. Fecal microbiomes from screening sampling tubes are stable despite varying sampling and storage conditions. Sci Rep 15, 26951 (2025). https://doi.org/10.1038/s41598-025-12506-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-12506-5
