
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a highly aggressive cancer with KRAS mutations in ~ 95% of cases. While KRAS inhibitors have shown promise, therapeutic resistance necessitates combination approaches. In particular, it is important to understand how downstream signaling of KRAS supports PDAC growth. For example, DUSP6 has emerged as an important dual-specificity phosphatase regulating KRAS-MAPK signaling. DUSP6 is markedly overexpressed in PDAC tumors compared to normal pancreatic tissue, with transcriptomic and single-cell RNA-seq analyses revealing its enrichment in epithelial tumor cells, especially in metastatic lesions. High DUSP6 expression correlates with the quasi-mesenchymal/squamous molecular subtype and poorer survival outcomes. Gene set enrichment analyses linked DUSP6 to pathways involved in cell migration and metabolism in metastatic samples. Functionally, DUSP6 knockdown in PDAC cells increases ERK/MAPK activation and alters migration. Metabolic profiling revealed enhanced basal glycolysis upon DUSP6 suppression. However, combined glycolysis inhibition and DUSP6 knockdown did not affect migration, suggesting that glycolytic changes are not the driver of altered migratory behavior. These findings reveal that DUSP6 independently regulates migration and metabolism in PDAC, emphasizing its dual role in disease progression. This study underscores the significance of DUSP6 as a potential therapeutic target and provides new insights into its contributions to PDAC progression.
Similar content being viewed by others

Introduction
Pancreatic ductal adenocarcinoma (PDAC) accounts for over 90% of pancreatic cancer cases and is one of the most lethal forms of invasive cancer globally, ranking as the third leading cause of cancer-related mortality1,2,3. Despite significant advancements, the current 5-year survival rate remains low, at 13%3. Late detection of PDAC primarily stems from vague symptoms, lack of early diagnostic markers, and resistance to chemotherapy4,5. Complete resection remains the sole curative option, yet it is unattainable for the majority of patients; 80–90% of patients present with locally advanced or metastatic disease upon diagnosis6,7. Metastasis in PDAC commonly affects the liver, peritoneum, and lungs8,9,10,11,12. Studies indicate that not all circulating tumor cells successfully initiate secondary tumors, suggesting the requirement for genetic and/or epigenetic changes13. Mutations such as those occurring in Kras are prevalent in both primary and metastatic tumors, with certain passenger mutations showing a preference for metastatic sites11,12. For example, loss of DPC4 (SMAD4) is more frequent in metastatic cases, correlating with increased invasiveness14. Gene expression alterations are also important considerations in this context; however, to date, most studies have only documented changes between non-neoplastic pancreatic and tumor tissues15.
Kras mutations are found in approximately 95% of PDAC cases16 and are the first genetic alterations that arise in precancerous lesions. These mutations alter protein conformation and ultimately lead to a prolonged activation state and, consequently, downstream activation of signaling pathways such as the ERK/MAPK pathway17. In tumor cells, ERK phosphorylation drives MYC activation through subsequent phosphorylation, leading to a myriad of effects such as uncontrolled cell proliferation, increased survival, and metabolic rewiring18,19. Although it is intuitive to believe that KRAS constitutive activation is imperative for tumors, its hyperactivation induces oxygen reactive species (ROS) production, and cells are forced to upregulate their antioxidant systems to maintain ROS homeostasis. However, failure to buffer intracellular ROS accumulation can result in oncogene-induced senescence and cytotoxicity20. Therefore, fine-tuned KRAS-MAPK activation is required for tumor cells to survive and concurrently evade the detrimental effects of their metabolic adaptations.
DUSP6 is a dual-specificity phosphatase that targets serine and threonine residues, particularly dephosphorylating ERK1/2 and downregulating the ERK/MAPK pathway21. While DUSP6 is reported to act in a negative feedback loop to downregulate ERK/MAPK activation, it has also been reported that ERK1/2 phosphorylates DUSP6 to target this protein for subsequent proteasomal degradation22. Its role in cancer is context dependent, acting either as an oncogene or as a tumor suppressor23. DUSP6 was previously shown to be upregulated in pancreatic intraepithelial neoplastic (PanIN) lesions (early stage of PDAC tumorigenesis) and downregulated in invasive carcinoma, particularly in poorly differentiated subtypes24. This phenomenon has been reported to occur due to hypermethylation of DUSP6 promoter, leading to abrogation of gene expression and increased activity of the ERK/MAPK pathway25. The complex coordination of the KRAS-ERK-DUSP6 axis and the timely regulation of DUSP6 expression throughout tumorigenesis prompted us to investigate the role of this molecule in PDAC metastasis.
In this study, we demonstrated that in silico gene expression and RNAscope studies in human samples revealed upregulation of DUSP6 in metastatic samples compared to that in primary tumors and non-neoplastic pancreatic tissue. Gene set enrichment analysis (GSEA) of human metastatic PDAC samples revealed enrichment of pathways related to cell migration and metabolism. In vitro assessments demonstrated DUSP6 downregulation in a KRAS mutant cell line, which correlates with increased ERK/MAPK activation, diminished proliferation, and migration capacity; however, this effect was absent in KRAS wild-type cells. Functionally, DUSP6 downregulation led to elevated basal glycolysis, indicating its involvement in glucose metabolism. However, the combined inhibition of DUSP6 and glycolysis did not affect the migratory phenotype of the KRAS mutant cell line. Our findings suggest that, while DUSP6 influences migration capacity and glucose metabolism, these effects may not be directly correlated in this context.
Results
DUSP6 overexpression is correlated with advanced stage PDAC
To investigate the role of DUSP6 in metastasis, we first confirmed its expression in tumor samples. To evaluate DUSP6 expression in PDAC samples, we used three distinct in silico datasets available on public platforms (GSE6245226, GSE2873527 and TCGA/GTEx). The TCGA dataset was selected due to its size and accessibility, making it one of the most comprehensive resources for patient-derived cancer data. In addition, two other datasets containing matched tumor and adjacent non-tumoral tissues from the same patients were included, allowing for direct intra-patient comparisons. This matching reduces inter-patient variability in gene expression analyses and enhances the reliability and interpretability of the findings. Our analysis revealed that DUSP6 was upregulated in primary tumor samples compared to non-tumoral pancreatic tissue across all analyzed datasets (Fig. 1A-C). Based on these findings, we aimed to assess DUSP6 expression in metastatic cases. Utilizing the in silico dataset GSE7172928, we compared DUSP6 expression across a cohort comprising 134 non-tumor tissue samples, 145 primary tumor samples, and 62 metastatic samples derived from various secondary sites. Notably, we observed that DUSP6 was significantly overexpressed in metastatic samples compared to both primary tumor samples (P < 0.0001) and non-tumorous pancreatic tissue (Fig. 1D). Considering that DUSP6 is overexpressed in primary tumors and metastatic samples, we hypothesized that there is a possible correlation between DUSP6 expression and patient prognosis. To answer this question, we first assessed its expression in the PDAC molecular subtypes. We performed in silico analysis utilizing PDAC molecular subtyping29,30. We took advantage of the datasets from Badea et al. (GSE15471) and Bailey et al.30 and analyzed DUSP6 expression in distinct PDAC subtypes. In the Badea et al. cohort (Fig. 1E), we observed that DUSP6 was overexpressed in the quasi-mesenchymal subtype compared to non-tumoral pancreatic tissue (P < 0.0001) and the exocrine-like subtype (P = 0.0211). Likewise, in the Bailey et al. cohort (Fig. 1G), we observed DUSP6 overexpression in the squamous subtype compared to that in the pancreatic progenitor (P = 0.0065) and immunogenic (P = 0.0197) subtypes. Next, we performed the same analysis using another subtyping paradigm28; however, no differences were observed (Fig. 1F). The same subtyping was used to stratify samples in the GSE22456431 (Fig. 1H) dataset, and we observed overexpression of DUSP6 in the basal-like subtype compared to the classical subtype (P = 0.0292). The quasi-mesenchymal, squamous, and basal-like subtypes share similar molecular signatures and worse prognoses than the other PDAC subtypes. Collectively, these findings indicate that DUSP6 is enriched in this tumor phenotype. To further investigate the relationship between DUSP6 expression and ERK1/2 activation in the context of PDAC, we conducted a correlation analysis between MAPK1 (ERK2) and DUSP6 expression in quasi-mesenchymal samples from the GSE15471 dataset32. Consistent with previous observations in lung cancer33, we found a significant positive correlation (R = 0.6787, P = 0.02), supporting the notion that DUSP6 overexpression may be driven by ERK activation as part of a negative feedback mechanism aimed at modulating MAPK signaling (Figure S1A).

DUSP6 overexpression is correlated to tumor metastatic potential and worse prognosis. In silico datasets from GSE6245226 (A), GSE2873527 (B) and GTex/TCGA (C) assessing DUSP6’s differential expression between non-tumoral pancreatic tissue and primary tumor tissue; (D) GSE7172928 assessing DUSP6 expression between non-tumoral pancreatic tissue, primary and metastatic tumor tissue; In silico gene expression analysis utilizing (E) Badea, (F) Moffitt28, (G) Bailey30 and (H) GSE22456431 datasets to assess DUSP6 expression among PDAC molecular subtypes; (I) Kaplan-Meier (Log-rank) plot comparing DUSP6 expression and patients’ overall survival using the TCGA dataset. Padj. = 0.0339, N = 67 for each group; (J) Pearson correlation analysis between DUSP6 expression in CCLE PDAC cell lines and their Metastatic Potential (MetMap 50034). R = 0.6105, P = 0.0003; (K) Distribution of genes negatively and positively correlated with CCLE PDAC cell lines Metastatic Potential (MetMap 50034) highlighting DUSP6 among the Top 10 positively correlated genes. ADEX: aberrantly differentiated endocrine–exocrine; QM: quasi-mesenchymal. (A, H) Mann-Whitney unpaired test. (B, C, F) Welch’s unpaired t-test. (D, G) Brown-Forsythe and Welch unpaired ANOVA test. (E) Kruskal-Wallis unpaired test. * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001.
Supporting the latter, our analysis of TCGA dataset revealed that high DUSP6 expression in PDAC patients correlated with worse overall survival (P = 0.0058; Fig. 1I). Despite the counterintuitive nature of DUSP6 overexpression in metastatic samples, given the prevailing dogma suggesting its negative regulation of the ERK/MAPK pathway, our findings were substantiated by further correlation analysis conducted with 30 PDAC cell lines from the Cancer Cell Line Encyclopedia (CCLE) and the Metastasis Map (MetMap 50034). This analysis revealed a moderate positive correlation between DUSP6 expression and metastatic potential in these cell lines (R = 0.611; P = 0.0003; Fig. 1J). Furthermore, this correlation analysis indicated that DUSP6 gene expression ranked as the eighth strongest positive correlation for metastatic potential within this PDAC cell line cohort among a list of 1000 genes (Fig. 1K). Collectively, these observations suggest that DUSP6 overexpression in PDAC samples is associated with the poorest prognostic outcomes and correlates with progression to metastasis.
To validate these findings, we assessed DUSP6 expression in lesions across different stages of the disease using healthy murine pancreatic tissue, primary tumors, and liver metastasis samples. We observed a trend of temporal control of DUSP6 expression with disease progression, although the results were not statistically significant (Fig. 2A-B). DUSP6 expression is upregulated during the acinar-to-ductal metaplasia (ADM) process and subsequently reduced in pancreatic intraepithelial neoplastic (PanIN) lesions. DUSP6 was again increased in primary tumor lesions and, finally, in liver metastasis samples. A similar trend was observed in lung metastases assessed using the L-iKras mouse model35,36, further supporting the robustness of our findings (Figure S1B). These results suggest that temporal control of DUSP6 expression is differentially utilized during PDAC tumorigenesis and progression, perhaps to fine-tune MAPK signaling.

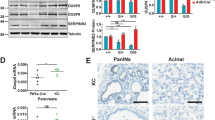
DUSP6 is upregulated in PDAC epithelial cells in human and mouse tissue. (A) DUSP6 immunohistochemistry staining (20x magnification) in wild type (WT) pancreatic tissue (N = 3), KC (N = 3) and KPC (N = 6) mice. Yellow arrows indicate acinar-to-ductal metaplasia (ADM) lesions and red arrows indicate Pancreatic Intraepithelial Neoplasia (PanIN) lesions. Quantification (B) shows DAB OD mean ± SD in DUSP6 positive cells from 10 randomly selected areas in each slide stratified by lesion grade; (C) Multiplex in situ hybridization with DUSP6 (red, RNAscope), panCK (green), αSMA (orange) and DAPI (blue). Images are representative of normal pancreatic tissue/pancreatitis, Intraductal papillary mucinous neoplasms (IPMN), Mucinous Cystic Pancreatic Neoplasms (MCN), Pancreatic Intraepithelial Neoplasia (PanIN), Pancreatic Ductal Adenocarcinoma (PDAC) and PDAC metastasis samples composed from 2 human TMA sections; (D) Quantification of the percentage DUSP6 positive cells normalized by total number of cells in each TMA core; (E) Quantification of the percentage of cells that are positive for both DUSP6 and panCK, normalized by total number of cells in each TMA core. Welch’s unpaired t-test; (F) UMAP plot of all cells captured from single-cell RNA sequencing of eleven human tumor samples35. Cell types are identified by color. Following UMAPs show DUSP6 expression among cell types in healthy pancreatic tissue (G), adjacent normal pancreatic tissue (H) and PDAC samples (I); (J) Percentage of cells co-expressing DUSP6 and KRT19 (epithelial marker) in healthy pancreatic tissue, adjacent normal pancreatic tissue and PDAC samples assessed by single-cell RNA-seq35. * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001.
PDAC has a highly complex microenvironment and is mainly composed of dense fibroinflammatory stroma consisting predominantly of activated stromal fibroblasts and immunosuppressive immune cell types. Our previous analyses were primarily performed on bulk tumors, where gene expression represents a composite of myriad cell types in the tumor microenvironment. Accordingly, we assessed whether DUSP6 overexpression was localized to specific cell types within the tumor. To answer this question, we examined DUSP6 expression in different tumor compartments using GSE9332637. Indeed, our analysis revealed that DUSP6 was significantly upregulated in tumor cells compared to stromal cells (P < 0.0001; Figure S1C). We validated this analysis by assessing DUSP6 protein levels in four mouse PDAC cell lines and two mouse cancer-associated fibroblast (CAF) cell lines. The mouse cell lines exhibited varying levels of the protein, with DT8082 and K8484 demonstrating the highest DUSP6 levels, P4313 showing intermediate levels, and PKT62 displaying the lowest levels (Figure S1D). Subsequently, we investigated Dusp6 expression in mouse tumor tissue using RNA in situ hybridization (RNA-ISH) technology. We examined Dusp6 mRNA levels in tumor sections obtained from LSL-KrasG12D/+, Ptf1aCre/+ (KC), LSL-KrasG12D/+, Tp53R172H/+, Ptf1aCre/+ (KPC) mice, co-stained for either Krt19 (a tumor epithelial cell marker) or Pdgfrb (a stromal marker). As depicted in Figure S1E, Dusp6 (green dots) exhibited strong co-expression with Krt19 (red dots, upper images), but not with Pdgfrb (red dots, lower images), in both genetic backgrounds. Consistent with the western blot analysis of protein levels, both KC and KPC pancreata demonstrated Dusp6 overexpression in malignant epithelial cells, whereas stromal compartment expression remained low (Figure S1E). To further explore the link between DUSP6 expression and MAPK signaling, we leverage GSE9332637 to perform a correlation analysis between DUSP6 and MAPK1 in the epithelial and stromal compartments. Interestingly, we observed a moderate positive correlation in gene expression levels in the epithelial compartment (R = 0.6105, P < 0.0001) but not in the stromal compartment (P = 0.0321, P = 0.789) in this cohort (Figure S1F-G). Altogether, these data indicate that DUSP6 is upregulated in PDAC epithelial cells in the context of hyperactivated MAPK signaling.
To confirm whether these observations could be translated into human disease, we used a panel of human samples from two TMAs containing normal pancreatic tissue/pancreatitis, intraductal papillary mucinous neoplasms (IPMN), mucinous cystic pancreatic neoplasms (MCN), PanIN, PDAC, and PDAC metastasis. We differentially stained tissue compartments with duplex RNA-ISH specific markers using panCK for epithelial cells and αSMA for CAFs (Fig. 2C). A general assessment of DUSP6 positive cells throughout the cores revealed a trend similar to that observed in mouse tissue. Although not statistically significant, we observed a slight decrease in expression in PDAC primary tumors compared to that in normal pancreatic tissue and a subsequent increase in metastatic samples (Fig. 2D). Furthermore, we observed a significant increase in DUSP6 expression in epithelial cells throughout tumor progression, and once again, a notable decrease in DUSP6 expression in PDAC primary tumors, followed by upregulation in metastatic samples (Fig. 2E). These findings suggest that DUSP6 timely upregulation is required for metastatic tumor cells in the foreign environment.
The observation that DUSP6 expression is significantly lower in PDAC primary tumor cells is counterintuitive, considering what we observed in bulk RNA-seq datasets. In view of these previous observations and the complexity of the PDAC microenvironment, we decided to dig further into a publicly available pancreatic tissue single-cell atlas38 to characterize DUSP6 expression patterns in different cell types within the healthy donor pancreas, adjacent normal pancreatic tissue, and primary tumor microenvironment. Figure 2F depicts the UMAP plot of all the cell types identified in the bulk of human patient samples. Interestingly, we observed that regardless of the context, DUSP6 was majorly expressed in myeloid cells, with increased expression in macrophages (Figs. 2G-I, Figure S2A). Nevertheless, when we look specifically at the epithelial cells, in these three contexts, we observe a 2.4-fold increase in the percentage of DUSP6 expressing epithelial cells in PDAC compared to healthy pancreata (Figure S2A). Lastly, we performed co-expression analysis of DUSP6 and KRT19 and found a 1.8-fold increase in DUSP6 positive cells in PDAC KRT19 positive cells compared to healthy pancreata (Fig. 2J), confirming DUSP6 upregulation in tumor epithelial cells. Interestingly, in silico analysis utilizing a bulk RNA-seq dataset (GSE245535) from Bojmar et al.39 reveals that DUSP6 is upregulated in liver biopsy samples from patients who eventually developed PDAC liver metastasis in comparison to those who did not in a 3-year follow up study (Figure S2B). In addition to its presence in epithelial tumor cells, our findings suggest that DUSP6 may play a role in the immune cell compartment, which could explain the variability in the expression observed across different human PDAC samples when analyzed via bulk RNA-seq. Notably, DUSP6 expression may also contribute to the formation of the pre-metastatic niche, further underscoring its multifaceted role in PDAC progression. Together, these results highlight the complex involvement of DUSP6 in both tumor and immune cell populations, as well as its potential influence on metastasis.
DUSP6 expression in metastatic samples correlates with decreased oxidative phosphorylation and increased glycolysis
To gain further insights into the functional significance of DUSP6 overexpression in metastatic samples, we aimed to elucidate the pathways involved. To accomplish this, we revisited the GSE7172928 dataset and conducted gene set enrichment analysis using 62 metastatic samples. Intriguingly, our analyses revealed enrichment of several pathways associated with cell motility, epithelial-to-mesenchymal transition, and metabolism. Notably, these enrichments revealed a positive correlation between DUSP6 expression and cell migration (Fig. 3B-C), suggesting that DUSP6 overexpression in this cohort leads to an upregulation of this process. Moreover, we observed a negative correlation between DUSP6 expression and oxidative phosphorylation (Fig. 3A-C), suggesting a downregulation of this pathway upon DUSP6 overexpression in this cohort. Supporting these observations, Panther Pathway enrichment analyses conducted across two independent datasets (TCGA_PAAD and CPTAC_PDAC) highlighted Glycolysis (P00024) as one of the pathways most strongly correlated with DUSP6 expression in PDAC across all three datasets (Table S1). Moreover, other enrichment analyses utilizing the CPTAC Protein dataset indicated a correlation between DUSP6 protein levels and the Pentose phosphate pathway (P02762), Glucose 6-phosphate metabolic process (GO:0051156), and Carbon metabolism (hsa01200) (Table S1). These findings prompted us to hypothesize that DUSP6 overexpression may play a pivotal role in metabolic reprogramming of tumor cells during metastasis.

DUSP6 downregulation increases glycolysis in PDAC cell lines, but glycolysis inhibition does not rescue migratory phenotype. GSEA analysis of GSE7172928 dataset showing pathways significantly negatively (A & C) and positively (B & C) correlated to DUSP6 expression in metastatic samples; (D) Assessment of migratory capacity in MVPMII upon 24 h treatment with BCI (2µM); (E) Western blot analysis of MVPMII lysates upon short (90 min) and long (24 & 48 h) exposure to BCI treatment (5µM); (F) Representative curves of extracellular acidification rates (ECAR) measured in AsPC-1 and BxPC-3 with sequential injections of glucose (10 mM), oligomycin (1µM) and 2-DG (100mM), respectively in a Glycolysis Stress Test; (G) Basal glycolysis corresponds to glycolytic levels under basal conditions, no stimulation; (H) Glycolytic capacity is measured after inhibition of ATP synthase (complex V of the mitochondrial respiratory chain); (I) Assessment of migratory capacity after 2-DG treatment in AsPC-1 upon DUSP6 knockdown. Seahorse Glycolysis Stress Test was performed in 9–12 replicates in 2 independent experiments. Results represent mean ± SD of one representative experiment. L-Lactate assay was performed in 2 independent experiments with 6 replicates each. Results represent mean ± SEM. Welch’s unpaired t-test; Migration graphs show 3 independent experiments with 4–6 replicates each. Results represent mean ± SEM. Mixed-effects analysis with Šídák’s multiple comparisons test. ns: not significant; * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001.
DUSP6 inhibition reduces migration in PDAC cell lines.
Considering DUSP6 upregulation in epithelial tumor cells derived from metastatic samples (Fig. 2E), we aimed to underscore the functional role of this molecule in tumor epithelial cells. Hence, we performed DUSP6 knockdown in PDAC cell lines exhibiting the highest levels of the protein, one of which was derived from metastatic ascites (AsPC-1, Figure S3A). DUSP6 knockdown was confirmed through gene expression and protein level analyses (Figure S3B). As expected, DUSP6 knockdown led to increased activation of ERK1/2, as confirmed by enhanced phosphorylation on western blotting (Figure S3B). Subsequently, considering that ERK1/2 activation elicits a proliferative phenotype, we evaluated cell proliferation using ethynyldeoxyuridine (EdU) incorporation assays under conditions of full serum (5%) and serum deprivation, designed to either stimulate or inhibit proliferation. Surprisingly, both AsPC-1 and BxPC-3 cell lines exhibited impaired proliferation after DUSP6 knockdown. Notably, AsPC-1 cells displayed a more pronounced decrease, particularly under serum-starved conditions (P < 0.0001; Figure S4A). While BxPC-3 cells also showed significant proliferation impairment (P < 0.0001), they were less affected by serum starvation compared to AsPC-1 cells (Figure S4B).
To confirm our prior observations in human samples, we investigated the impact of DUSP6 knockdown on cell migratory capacity. In vitro migration assays conducted within 24 h of scratching (time 0) revealed distinct phenotypes among the cell lines. AsPC-1 cells exhibited a significant reduction in migratory capacity upon DUSP6 knockdown (P < 0.0001; Figure S4C), whereas BxPC-3 cells showed no significant effect (Figure S4D). To further explore the role of DUSP6 in cell migration, we utilized the pharmacological inhibitor BCI in two murine KRAS-mutant cell lines, MVPMII and AKC. These cells were isolated from KC mice to specifically evaluate the role of DUSP6 in modulating the ERK/MAPK pathway, while minimizing confounding effects from additional tumorigenic mutations. Consistent with our findings in AsPC-1, DUSP6 inhibition significantly impaired migration of both lines (Fig. 3D-S4E). In contrast, BCI treatment did not affect migration in BxPC-3 cells, despite a robust increase in ERK1/2 phosphorylation (Figure S4F). Given the well-established crosstalk between ERK/MAPK and PI3K/AKT signaling pathways – previously reported by our group40 and others, we examined the effect of DUSP6 inhibition on AKT activation. In MVPMII cells, BCI treatment induced an increase in both ERK1/2 and AKT phosphorylation at 90 min, comparable to the response elicited by TGF-α stimulation. While ERK1/2 phosphorylation remained elevated for up to 24 h, AKT activation was not sustained (Fig. 3E). To further investigate the interplay between these pathways, we treated a panel of PDAC cell lines with Trametinib, a MEK1/2 inhibitor, and evaluated DUSP6 levels and AKT activation. Long-term MEK inhibition led to a reduction in DUSP6 levels and a concomitant increase in AKT phosphorylation39 (Figure S4G). Notably, this indirect modulation of DUSP6 resulted in sustained AKT activation (24 h). Finally, a correlation analysis again using the GSE15471 dataset revealed a moderate inverse relationship between DUSP6 and AKT expression in quasi-mesenchymal PDAC samples, further supporting the relevance of our observations (Figure S4H). Collectively, these findings indicate that DUSP6 modulates both proliferation and migration in PDAC cells, potentially through context-dependent regulation of ERK/MAPK and PI3K/AKT signaling pathways.
DUSP6 downregulation increases glycolytic flux in PDAC cell lines
Given the enrichment analysis indicating a consistent correlation between DUSP6 expression and glycolysis in metastatic samples, we sought to determine whether DUSP6 plays a functional role in metabolic reprogramming within epithelial tumor cells. First, we evaluated basal lactate secretion in extracellular media following DUSP6 knockdown using a colorimetric assay. In this context, distinct phenotypes were observed in both cell lines: BxPC-3 cells exhibited a significant increase in lactate secretion compared to the control (P < 0.0001), whereas AsPC-1 cells showed no significant difference compared to the control (Figure S5A). Considering that glucose is the primary source of energy for tumor cells in vitro, we sought to delineate how DUSP6 affects glucose metabolism in each cell line and conducted a metabolic assay to measure glycolysis activity in the absence of DUSP6 (Fig. 3F). Upon DUSP6 knockdown and glucose injection, both AsPC-1 and BxPC-3 cells demonstrated a statistically significant increase in basal glycolysis (Fig. 3G), consistent with observations from the lactate secretion assay. Following treatment with oligomycin, an inhibitor of ATP synthase (complex V), which shuts down mitochondrial respiration and makes cells dependent on glycolysis for ATP production, we observed a significant increase in glycolytic capacity in AsPC-1 cells upon DUSP6 knockdown compared to the control (P < 0.05; Fig. 3H), with no significant differences in glycolytic reserve (Figure S5B). This indicates that the absence of DUSP6 alone leading to pronounced ERK1/2 activation pushes these cells towards their maximum glycolytic potential. Upon the addition of 2-DG, glycolysis was completely abolished under all conditions. These results demonstrate that DUSP6 inhibition affects glycolysis in PDAC under basal metabolic conditions, but subsequent rescue experiments are required to determine the observed changes in glycolytic reserve.
Glycolysis inhibition does not provide an additional impact cell migratory capacity in DUSP6 depleted cells
To better understand how the glycolytic shift induced by DUSP6 knockdown impacts tumor cell behavior, we revisited our previously conducted functional assays and incorporated 2-DG, a well-established inhibitor of glucose metabolism, into the analyses. Firstly, by reducing fetal bovine serum in the media to minimize its metabolic influence, we assessed cell proliferation with and without 2-DG addition. Consistent with our previous findings, both AsPC-1 and BxPC-3 cells exhibited a significant reduction in proliferation upon DUSP6 knockdown compared to their respective controls. Following 2-DG treatment, BxPC-3 cells displayed a notable decrease in proliferation under both conditions, with the reduction being more pronounced in the control cells (P < 0.0001; Figure S5C). In contrast, AsPC-1 showed no discernible effect on proliferation compared to the same conditions without 2-DG (Figure S5D). Subsequently, to establish a link between the metabolic phenotype and metastatic potential of these cells, we conducted a migratory capacity assay combined with 2-DG treatment. DUSP6 knockdown significantly reduced the migratory capacity of AsPC-1 cells, whereas no change was observed in BxPC-3 cells (Figure S4C-D). Unexpectedly, treatment with 2-DG did not significantly alter the migratory phenotype observed in AsPC-1 cells, but significantly reduced BxPC-3 migratory capacity upon DUSP6 knockdown (Fig. 3I-S5E). Interestingly, we noted that in the presence of an external source of pyruvate in the media, the cells maintained their glycolytic potential, even when treated with electron transport chain (ETC) inhibitors (Rotenone and Antimycin A) and 2-DG (Figure S5F). This suggests that pyruvate availability plays a critical role in the decision to sustain aerobic glycolysis or to feed the TCA cycle. These findings suggest that, under nutrient-rich conditions, both AsPC-1 and BxPC-3 utilize different energy sources for migration.
Discussion
DUSP6 is differentially expressed during PDAC tumorigenesis and progression24. However, little is known about the role of phosphatases in the development and establishment of metastasis. Our group observed that DUSP6 was overexpressed in primary tumor samples compared to non-tumoral pancreatic tissues by analyzing in silico datasets and spatial transcript profiling. To further investigate the complex feedback loops involving DUSP6 and the ERK/MAPK signaling pathway, we evaluated in silico data to assess DUSP6 expression in late-stage PDAC samples. We observed that DUSP6 was overexpressed in metastatic samples compared to primary tumor samples by leveraging in silico, immunohistochemistry, and multiplex RNAscope analysis. Translational relevance for DUSP6 expression was observed in survival data; TCGA analysis showed that high DUSP6 expression correlates with lower overall survival in PDAC patients. Finally, when we analyzed DUSP6 expression among different PDAC molecular subtypes29,30, DUSP6 overexpression was pronounced in the quasi-mesenchymal/squamous subtype, which is associated with a worse prognosis41. These data support the hypothesis that DUSP6 overexpression confers a more aggressive phenotype, further impacting metastasis and worsening overall survival in PDAC.
Spatial localization is critical for investigating the functionality. Through in silico analysis of pancreatic tumor samples, we determined that DUSP6 expression was upregulated in the epithelial compartment of the tumor rather than in the stromal compartment. The same pattern was observed in mouse models assessing protein presence by immunohistochemistry and RNA in situ hybridization in mouse pancreatic tumors. In human samples, we observed a significant decrease in DUSP6 expression in PDAC epithelial cells versus normal pancreatic tissue, but increased expression in metastatic epithelial cells. By leveraging a single-cell RNA-seq Human Pancreatic Cancer Atlas, we were able to confirm that DUSP6 expression is increased in PDAC epithelial cells. However, the results also highlighted macrophages as a subset of cells with important changes in DUSP6 expression throughout tumorigenesis. Kidger et al. recently reported similar RNA in situ hybridization results using KC mice, demonstrating that DUSP6 expression is found in ductal cells in PanINs, but not in stromal cells42. Our results confirmed that PDAC tumor cells overexpress DUSP6, suggesting the need for further investigation of its role in tumor development.
Previous studies on multiple tumor types have linked high DUSP6 expression to worse prognosis and metastasis43,44,45. DUSP6 has alternatively been described as a tumor suppressor gene in PDAC. Furukawa et al. reported that downregulation of DUSP6 coordinated PDAC progression via hypermethylation of its promoter24,25. Kidger et al. also proposed a tumor suppressor role for Dusp6, observing that KCD6−/− animals develop a higher number of poorly differentiated tumors and liver metastases42. In their model, Dusp6 was constitutively absent, leading to a permanent state of Erk1/2 activation, which would predictably result in more aggressive tumors and more metastatic foci. Our current hypothesis is that DUSP6 suppression might allow tumor initiation, while its expression is required for metastatic cells to re-establish in a new environment. Reinforcing this idea, DUSP6 overexpression appears to correlate with the more invasive quasi-mesenchymal subtype.
To investigate this, we subjected PDAC cell lines to stable DUSP6 knockdown and evaluated their proliferation and migration capacities. Surprisingly, we did not find consensus regarding the observed phenotypes. All statistically significant changes in proliferation indicated an impairment in this capacity. This observation is counterintuitive because it is widely known as a dogma in the field that ERK1/2 activation leads to increased cell proliferation. However, it is also known that tumor cells require fine-tuning of ERK1/2 activation, in which too much MAPK signaling can bypass the fitness advantage and become detrimental to cell growth46. Similarly, Gutierrez-Prat et al. recently reported that depletion of DUSP4, another ERK1/2 negative regulator, reduces proliferation by driving “oncogene overdose” in BRAF- and NRAS-mutant melanoma cell lines47. The migration phenotype varied among the cell lines, complicating any definitive conclusions on this topic. We hypothesized that this variability might be due to natural compensatory mechanisms within the cells, resulting from dependence on this signaling pathway. AsPC-1 cells carry two alleles of the KRASG12D mutation, which is the most commonly found among PDAC patients and, therefore, more clinically relevant. BxPC-3 cells present with wild-type KRAS which might explain the differences observed between these two cell lines. Reinforcing this observation, under DUSP6 pharmacological inhibition, two murine cell lines harboring the KRASG12D mutation showed a decrease in migration, while BxPC-3 phenotype was not affected by the drug. A recent study by Bulle et al. showed that upon MAPK pharmacological inhibition, DUSP6 is one of the top downregulated genes, demonstrating that DUSP6 expression correlates with ERK/MAPK activation. Supporting this evidence, we observed that DUSP6 was significantly upregulated in tumor cell lines harboring a KRAS mutation compared to tumor cells driven by other mutations in the CCLE dataset (Figure S6). Bulle et al. also demonstrated that upon MAPK inhibition, DUSP6 was targeted for proteasomal degradation, leading to an increase in HER2 activation48. Interestingly, this is the first time that DUSP6 has been described as a negative regulator of HER2, since it was previously thought to be ERK-specific. This discovery shifts the paradigm of DUSP6’s functional role and raises the possibility that it may be involved in the negative feedback for other signaling pathways. Moreover, many studies, including those from our group, have demonstrated that solid tumor cells, such as those in PDAC, exhibit compensatory crosstalk between the ERK1/2 and PI3K/AKT pathways, wherein inhibition of one pathway often leads to the activation of the other40,49,50. DUSP6 has been previously implicated in the regulation of PI3K/AKT signaling in solid tumors, although its role appears to be context-dependent and sometimes contradictory51,52. In this study, we observed a negative correlation between DUSP6 activity and PI3K/AKT signaling. Both direct inhibition of DUSP6 using BCI and indirect suppression via Trametinib treatment resulted in increased phosphorylation of AKT. These findings align with a recent report by Bye et al., who showed that Trametinib-mediated ERK1/2 inhibition in a panel of PDAC cell lines significantly impaired cell migration while concurrently enhancing PI3K/AKT activation. Notably, co-treatment with the dual mTORC1/2 inhibitor Omipalisib further suppressed cell migration in these models40. Additionally, it is important to consider that DUSP6 is not the only phosphatase acting as a regulator of ERK1/2 phosphorylation. Therefore, its downregulation may elicit compensatory responses from other molecules, ultimately offsetting its absence. Ito et al. recently demonstrated a co-dependence of DUSP4 and DUSP6 in MAPK-pathway-driven tumors as a mechanism of drug resistance53. In this regard, we highlight that many DUSPs, among other proteins, are involved in ERK1/2 negative feedback control and have been shown to act synergistically to prevent oncogene-induced senescence23. Similarly, we found a positive correlation between DUSP6 and MAPK1 expression in tumor cells, but not in stromal cells, in a human cohort, suggesting that these cells are also dependent on DUSP6 for ERK1/2 modulation; we cannot, however, rule out the involvement of other DUSPs in the process. Moreover, the BxPC-3 cell line does not harbor a KRAS mutation, and thus, its tumorigenic behavior does not rely on this condition. The observed phenotype in this cell line might be driven by mechanisms different from those initially hypothesized. Conversely, AsPC-1, MVPMII and AKC are all PDAC lines harboring the KRAS mutation, which more closely mimics the genetic background that we aimed to investigate. In these cell lines, we observed a significant impairment in migration and increased ERK1/2 activation upon DUSP6 inhibition.
To understand the mechanisms by which DUSP6 affects the metastatic potential of PDAC cells, we performed gene set enrichment analysis using the metastatic cohort of samples from the GSE7172928 dataset. This analysis indicated a positive correlation with cell migration and a consistent negative correlation with oxidative phosphorylation. Consequently, we conducted a glycolysis stress test and observed that the absence of DUSP6 led to increased glycolytic function in PDAC cell lines. This upregulation of glycolysis was confirmed by the increased extracellular acidification rate upon glucose supplementation (in the absence of pyruvate), which could be completely reversed by the addition of 2-DG. This shift towards a more glycolytic phenotype was expected based on previous literature showing that KRASG12D increases glucose uptake and glycolytic flux through MAPK activation in PDAC cells. Ying et al. demonstrated that increased glycolytic flux induces the non-oxidative pentose phosphate pathway (PPP), which is involved in processes such as nucleic acid synthesis and is closely related to cell proliferative capacity18. However, in this context, we observed that although glycolysis was induced upon DUSP6 knockdown, the cells showed a decreased proliferative capacity, a phenotype that is consistent among the cell lines. This behavior might be linked to inherent differences in cell metabolism among PDAC cell lines, as it has been previously reported that cells can present distinct metabolic profiles despite their growth rates54. Interestingly, Hsu et al. have demonstrated that DUSP6 is required for T-cell receptor-mediated glycolysis, and that in the absence of DUSP6, T cells show higher dependence on glutamine and fatty acids to fuel the TCA cycle55. These findings reinforce DUSP6’s involvement in metabolic reprogramming but highlight the context-dependency of the observed phenotypes.
Finally, we hypothesized that the induction of glycolysis through DUSP6 inhibition would decrease the migratory potential of these cells, which could be rescued by the addition of 2-DG, leading to irreversible glycolysis inhibition. We treated the cells with 2-DG and evaluated their migratory capacity but did not observe significant changes with the combination of DUSP6 and glycolysis inhibition. Previous studies have demonstrated that the induction of glycolysis leads to increased metastatic potential due to the promotion of epithelial-to-mesenchymal transition (EMT)56,57. In the present study, we demonstrated that while cells acquire a more glycolytic phenotype upon DUSP6 inhibition, their migratory capacity is abrogated. However, inhibition of glucose flux failed to rescue the observed phenotype. We hypothesized that, in a context where pyruvate is available in the culture media, cells can bypass 2-DG-mediated glycolysis inhibition. To estimate the effect of glycolysis induction on the metastatic behavior of DUSP6 knockdown cells, it is recommended to closely control nutrient availability in the media. While it is tempting to extrapolate our observations to the decreased metastatic potential of the cells, we must reiterate that metastasis encompasses a variety of complex processes involving both intrinsic and extrinsic signals58. To further understand DUSP6’s role in metastatic potential, it will be necessary to explore other mechanisms involved in metastasis, such as invasive potential and the ability of these cells to adapt to a new environment, which is highly correlated with their metabolic reprogramming capacity.
Conclusions
This study provides new insights into the role of DUSP6 in PDAC metastasis and metabolism. We demonstrated that DUSP6 inhibition has cell-specific effects on migratory potential while simultaneously inducing glycolysis in PDAC cells. However, we were unable to establish a direct correlation between the two processes. Future studies should focus on in vivo metastatic models to explore how tumor epithelial cells adapt their metabolism in the absence of DUSP6 to survive and proliferate in secondary niches. Additionally, as highlighted in this study, DUSP6 may play a role in establishing the pre-metastatic niche, presenting another intriguing avenue for future investigation. Given prior studies implicating DUSP6 in immune cells, its potential role in tumor-associated macrophages also warrants further exploration to deepen our understanding of its contributions to the tumor microenvironment.
Materials & Methods
Cell lines and culture
MIA PaCa-2 (RRID: CVCL_0428), PANC-1 (RRID: CVCL_0480), AsPC-1 (RRID: CVCL_0152), BxPC-3 (RRID: CVCL_0186), Capan-2 (RRID: CVCL_0026), and SW1990 (RRID: CVCL_1723) were purchased from the ATCC (Manassas, VA, USA). K8484 (RRID: CVCL_XD12) and DT8082 (RRID: CVCL_XD11) were acquired from David Tuveson’s laboratory (Cold Spring Harbor Laboratory)20. P4313 was acquired from Andy Lowy (UC San Diego)21. PKT62 is a murine pancreatic tumor cell line derived from a PKT (LSL-KrasG12D/+, TGFBR2fl/fl, Ptf1aCre/+) mouse59 and kindly provided by Michael VanSaun (University of Kansas Medical Center). KPC CAFs and PKT CAFs cell lines were isolated from the respective mouse tumors and validated by western blotting. MVPMII and AKC were isolated from KC (LSL-KrasG12D/+, Ptf1aCre/+) mice at the VanSaun laboratory and validated by western blotting. Briefly, tumors were collected, digested, minced and cells were sorted with EpCAM beads (BioLegend Inc., cat #741444). EpCAM− cells were kept in culture and until full confluence, protein samples were isolated and fibroblastic phenotype was confirmed by the presence of α-SMA and absence of CK19 and RasG12D. Capan- 2, AsPC-1 and SW1990 were cultured in high glucose (4.5 g/L) RPMI-1640 medium (ThermoFisher Scientific, Waltham, MA USA cat #A1049101) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Atlanta Biologicals/RD Systems, Atlanta, GA USA) and antibiotic-antimycotic (Thermo Fisher Scientific cat# 15240062). The K8484 cell line was cultured in high-glucose (4.5 g/L) Dulbecco’s Modified Eagle Media (Thermo Fisher Scientific, cat# 11-995-073) supplemented with 5% heat-inactivated FBS (R&D Systems-Atlanta Biologicals) and antibiotic-antimycotic (Thermo Fisher Scientific). All other cell lines were maintained in high-glucose DMEM supplemented with 10% FBS and antibiotic-antimycotic. Cells were maintained in culture conditions (37 °C, 5% CO2 and humid atmosphere) as recommended by the ATCC, utilized within 15 passages from purchase and regularly tested for mycoplasma.
Plasmids and gene knockdown
To generate DUSP6 knockdown cells, short hairpin RNA (shRNA) constructs targeting three independent regions of DUSP6 mRNA (#1 5’-CTGTGGTGTCTTGGTACATTG-3’; #2 5’- TCTAATCCAAAGGGTATATTT-3,’ #3 5’-ATTCGGCATCAAGTACATCTT-3’) were obtained from Millipore (Burlington, MA, USA). The empty vector pLKO.1 puro (RRID: Addgene_8453) was used as a control. Packaged lentiviruses were generated by transfecting HEK293T (RRID: CVCL_0063) cells with the respective plasmids and the Trans-Lentiviral™ Packaging Mix (Open Biosystems) to produce viral supernatants, which were subsequently concentrated with Lenti-X™ concentrator (TakaraBio, cat #631231). Next, 2.5 × 105 pancreatic cancer cells were seeded in 6-well plates and incubated for 24 h with lentiviral suspension. Successfully transduced cells were selected with puromycin treatment (1–2.5 µg/mL) until complete cell death was achieved in the non-transduced cells.
RNA isolation and RT-qPCR
Briefly, 106 cells were seeded in 6-well plates and incubated overnight. The next day, cells were washed with 1X PBS and harvested using QIAzol Lysis Reagent (Qiagen, GmbH, cat #79306). Samples were stored at -80 °C until total RNA was isolated using the Direct-zol RNA Miniprep kit (Zymo Research Corporation), according to the manufacturer’s protocols. RNA quantification and quality analyses were performed using NanoDrop 2000 (Thermo Fisher Scientific Inc.). For cDNA, 1.5–2 µg of RNA was processed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, cat #4368814) according to the manufacturer’s instructions. The product was further diluted 1:10 before storage at -20 °C. Real-time PCR analysis was carried out with the 2X SYBR Green qPCR Master Mix (ApexBio Technology, cat #K1070) on a ViiA 7 Real-Time PCR System (Applied Biosystems) using the following primers: human DUSP6 (Fwd: ATGGTAGTCCGCTGTCCAAC/Rev: ACGTCCAAGTTGGTGGAGTC), mouse Dusp6 (Fwd: TGTTTGAGAATGCGGGCGAGTT / Rev: ACAGTTTTTGCCTCGGGCTTCA), human ACTB (Fwd: TCGTGATGGACTCCGGTGAC/Rev: CGTGGTGGTGAAGCTGTAG), and mouse Actb (Fwd: GTGACGTTGACATCCGTAAAGA / Rev: GCCGGACTCATCGTACTCC). Each sample was run in triplicate using the 2−ΔΔCT method (Livak & Schmittgen, 2001) and the fold-change was evaluated relative to normal samples and determined using ACTB/Actb levels as a reference.
Western blotting
Briefly, 106 cells were plated in 6-well plates, incubated overnight, and collected the next day for protein isolation. Cells were washed with 1X PBS, lysed in ice-cold RIPA buffer (1X RIPA [Cell Signaling Technology]; 10mM NaF; 1mM PMSF), sonicated, and incubated on ice for 10 min. The cells were then centrifuged at 16,000 × g for 10 min at 4 °C. Protein quantification was performed using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific), following the manufacturer’s instructions. For western blotting assays, 10 µg of total protein was loaded in each lane of a 10% polyacrylamide gel and subjected to electrophoresis. Proteins were transferred to nitrocellulose membranes using the semi-dry method on a Trans-Blot Turbo Transfer System (Bio-Rad). Specific proteins were detected using the following primary antibodies: Anti-DUSP6 (Abcam cat# ab76310, RRID: AB_1523517, link), P-ERK1/2 (Cell Signaling Technology, cat #4370, RRID: AB_2315112, link), ERK1/2 (Cell Signaling Technology, cat #4695, RRID: AB_390779, link), phospho-AKT (Cell Signaling Technology, cat #4060, RRID: AB_2315049, link), total AKT (Cell Signaling Technology, cat #4298, RRID: AB_10693940, link), β-actin (Cell Signaling Technology, cat #12262, RRID: AB_2566811, link), and Vinculin (Santa Cruz Biotechnology, cat #sc-25336, RRID: AB_628438, link). Secondary antibodies used were Peroxidase AffiniPure F(ab’)₂ Fragment Donkey Anti-Mouse IgG (H + L) (cat #715-036-151, RRID: AB_2340774, link) and Donkey Anti-Rabbit IgG (H + L) (cat #711-036-152, RRID: AB_2340590, link) from Jackson ImmunoResearch Laboratories Inc. Images were obtained using FluorChem M (Bio-Techne) or a Sapphire FL Biomolecular Imager (Azure), and analyzed using ImageJ 1.54f software (NIH, Bethesda, MD, USA).
Immunohistochemistry staining
Healthy pancreata, primary tumor, and metastatic samples were collected from wild-type C57BL/6J (RRID: IMSR_JAX:000664) mice, LSL-KrasG12D/+, Ptf1aCre/+ (KC), LSL-KrasG12D/+, Tp53R172H/+, Ptf1aCre/+ mouse pancreatic tissue (KPC) maintained in a C57BL/6J background. Mice were obtained from The Jackson Laboratory. Male and female mice were used for experiments and mice ages varied from 5.9 to 86.6 weeks. Samples were collected, embedded in paraffin, and sectioned into 4 μm sections. Primary tumor and lung metastasis samples from L-iKras (Ccsp-rtTa; TetO-KrasG12D; Trp53LSL − R172H/+) mice were gently donated by the Pasca di Magliano Lab, at the University of Michigan, and are described in a previously published study36. Before staining, the paraffin sections were processed as follows: 15 min in xylene (2x); 5 min in 100% ethanol (2x) and 5 min in 75% ethanol. Finally, the sections were washed 3x in PBS at room temperature (RT). Antigen retrieval was performed by boiling the sections in 10mmol/L sodium citrate solution for 10 min and waiting for the sections to cool down and achieve RT. Next, the sections were washed 3x with PBS, quenched with 3% H2O2 for 15 min, and blocked with 5% BSA for 20 min. Whole-slide scanning was performed using a PANNORAMIC Scan II scanner (3DHISTECH). Immunohistochemical staining was performed by iHisto Inc. (Salem MA; iHisto.io) using an anti-DUSP6 antibody (Abcam, cat #ab76310, RRID: AB_1523517, link). Ten fields per independent section from each condition were randomly selected for quantification using the QuPath software60. The study was performed under protocols 2005N000148 and 2019N000116 approved by the IACUC at the Massachusetts General Hospital, and at Pharmaron Inc. and Champions Oncology under approved protocols.
RNA in situ hybridization
RNA in situ hybridization was performed using the RNAscope® technology (Advanced Cell Diagnostics, Inc.). Briefly, tumors were collected from the mice (The Jackson Laboratory), sectioned, and immediately fixed in 10% buffered formalin for 24 h. Tumor sections were preserved in 70% ethanol, embedded in paraffin blocks, cut into 5 μm sections, and placed in pre-treated slides. The sections were freshly cut for the assay and dried at RT to preserve RNA integrity. Sections were baked for 1 h at 60 °C and then deparaffinized using Histo-Clear (Electron Microscopy Science, cat #6411004) and 100% ethanol. Next, sections were treated with RNAscope® Hydrogen Peroxide for 10 min at RT, followed by a target retrieval step with RNAscope® 1X Target Retrieval Reagent for 15 min, and finally, RNAscope® Protease Plus treatment for 30 min at 40 °C. Finally, we used the RNAscope® 2.5 HD Duplex Reagent Kit, following the manufacturer’s protocol. Specific probes used in this assay were Mm-Dusp6 (cat #429321-C1), Mm-Krt19-C2 (cat #402941- C2) and Mm-Pdgfrb-C2 (cat #411381-C2). The sections were imaged at 20x magnification using an EVOS™ M5000 Imaging System (Thermo Fisher Scientific). Experiments were performed in accordance with the approved IACUC Protocols (22-02-226) at the University of Kansas Medical Center.
Clinical samples
The human patient samples used in this study were derived from previously published tissue microarrays (TMAs)61,62. These samples were obtained from pancreas resections performed between 2002 and 2015, at the University of Michigan Health System, due to pancreatitis, cystic neoplasms, or pancreatic ductal adenocarcinoma (PDAC). All studies were carried out in accordance with an Institutional Review Board (IRB)-approved protocol (IRB number: HUM00098128), with written informed consent from patients, and in compliance with the Declaration of Helsinki.
RNA in situ hybridization combined with immunofluorescence multiplex staining
Tissue microarray (TMA) sections were obtained, set up and processed as described in62. Fluorometric in situ hybridization was performed according to the manufacturer’s (Advanced Cell Diagnostics, Inc.) instructions. Briefly, FFPE preserved TMAs were cut in 5 μm sections, and placed in pre-treated slides. Sections were freshly cut for the assay and dried at RT to preserve RNA integrity. Sections were baked for 1 h at 60 °C, deparaffinized in xylene three times for 15 min, washed in 100% ethanol twice for 3 min and air dried. Slides were treated with pretreatment 1, 2 and 3 buffers, rinsed in double-distilled water (ddH2O) and incubated with the DUSP6 (cat #1032061-C1) or control probes for 2 h at 40 °C in a humidity chamber. Slides were then treated with Amp solutions 1–6 before the fluorogenic substrate Opal 690 TSA (Akoya Biosciences, Inc.) was applied to the slide. After a 10 min incubation, the slides were washed and then subjected to dual immunofluorescent staining on a Ventana Discovery Ultra stainer (Roche Diagnostics, North America) with smooth muscle actin (BioCare Medical, cat #001, RRID: AB_2910606, link, 1:100, 60 min, Opal 570) and pan-cytokeratin (Abcam, cat #ab9377, RRID: AB_307222, link, 1:200, 20 min, Opal 520). Slides were mounted using Prolong Gold with DAPI and scanned using a Vectra Polaris™ (Akoya Biosciences, Inc.) whole slide scanner. Images were analyzed using the de-array TMA tool in QuPath and DAPI was used to identify individual cells in each core.
Proliferation assay
Cell proliferation was assessed by ethynyldeoxyuridine (EdU) incorporation. Briefly, 2.5-5 × 104 cells were seeded in 24-well plates in complete media and incubated overnight. The next day, the media was replaced with the appropriate treatments and incubated for 18 h. At this point, EdU was added to the wells at a final concentration of 10µM and cells were incubated for an additional 6 h. At the end of this period, the cells were trypsinized, harvested, and fixed with 10% buffered formalin overnight at 4°C with gentle agitation. The next day, the cells were washed with PEB-T (1x PBS, 2mM EDTA, 1% BSA, 0.5% Triton-X) and subsequently with PB (1x PBS, 1% BSA). Next, the cells were resuspended in Click-It Reaction (H2O, 100mM Tris pH 8.5, 1mM CuSO4, 1µM azide-dye) and 100mM ascorbic acid. Cells were incubated with 0.5 µg/mL propidium iodide (PI), washed twice, and resuspended in PEB (1x PBS, 2mM EDTA, 1% BSA) for flow cytometry analysis. The cells were analyzed for 530 nm and 695 nm emission using an Attune NxT Flow Cytometer (Thermo Fisher Scientific). FCS files were analyzed with FlowJo™ v10.8 Software (BD Biosciences) and the percentage of EdU-positive cells was taken from the PI-positive single cells.
Migration assay
First, 2-4 × 105 cells were seeded into 24-well plates and cultured until they reached full confluence. The next morning, cells were incubated with Mitomycin C (5µg/mL) (Thermo Fisher Scientific, cat #AAJ67460XF) for 2 h to arrest proliferation. Then, a scratch was made at the center of the plate using a 200µL pipette tip. Wells were washed twice with 1X PBS to discard all debris, and media with appropriate treatments were added. The plates were photographed at 0 and 24 h using an EVOS™ M5000 Imaging System (Thermo Fisher Scientific). Scratch area was calculated using the Wound Healing Coherency Tool (Montpellier Ressources Imagerie) on ImageJ (NIH, Bethesda, MD, RRID: SCR_003070, link) and results are expressed as healing percentage – the difference in scratch area between time 0 and time 24.
In silico analysis
DUSP6 expression analysis was performed using publicly available datasets obtained from The Cancer Genome Atlas (TGCA_PAAD), Genotype-Tissue Expression (GTEx, normal pancreatic tissue) and Gene Expression Omnibus (GEO, RRID: SCR_005012, link; GSE6245226, GSE2873527, GSE1547132, GSE7172928 and GSE22456431). Overall survival analysis was performed by Gene Expression Profiling Interactive Analysis (GEPIA, RRID: SCR_018294, link) using the UCSC Xena project (http://xena.ucsc.edu) datasets. Differential expression of DUSP6 in the epithelial and stromal compartments was performed using GSE9332637 with the R2 Genomics Analysis and Visualization Platform. The latter was also utilized for accession of Bailey et al.30 dataset since ICGC Data Portal Repository was discontinued. Finally, DUSP6 expression correlation to Metastatic Potential (MetMap 50034) was assessed through Pearson correlation analysis at the DepMap Portal using the Cancer Cell Line Encyclopedia (CCLE). We specifically filtered the cell lines for Exocrine Adenocarcinoma, which corresponds to PDAC cell lines. Gene set enrichment analysis was conducted in a cohort of metastatic samples from the publicly available dataset GSE7172928 using the hallmark gene set collection (version 2024) available at GSEA 4.3.3. software (RRID: SCR_003199, link)63. Single-cell analysis was performed using a previously published38 pancreatic tissue single-cell atlas (link).
Glycolysis stress test
Glycolysis rates were determined using the Seahorse XF Glycolysis Stress Test Kit (Agilent, Santa Clara, CA, USA), following the manufacturer’s protocol. Briefly, 2-4 × 104 cells were seeded into a 96-well plate and incubated overnight. The next day, cells were washed twice with Seahorse XF DMEM (pH 7.4 (2mM glutamine) and a final volume of 180µL of the same was added to each well. The extracellular acidification rate (ECAR) was measured (mpH/min) at 11 time points with sequential injections of 10mM Glucose, 1µM Oligomycin and 100mM 2-Deoxy-D-glucose (Thermo Scientific, cat #AC111980010). For nuclear staining, 5 µg/mL Hoechst was added to the oligomycin solution. The ECAR values were calculated and normalized to the cell count number using an automated imaging and cell counting workflow. For normalization, the ECAR value per 103 cells was set, and the Agilent Seahorse XF system automatically calculated and generated normalized results.
Lactate assay
For lactate measurement, 2.5 × 104 cells were seeded in 96-well plates and allowed to sit overnight in the incubator. The next day, the complete media in the plate was replaced with new media containing 0.5% FBS, and the cells were incubated for 24 h. At the end of the experiment, culture plates were centrifuged at 450 × g for 5 min and 10µL of media was collected from each well for the subsequent assay. For lactate quantification, we utilized the Glycolysis Cell-Based Assay Kit (Cayman Chemical Company, Ann Arbor, MI, USA, cat #600450), following the manufacturer’s protocol. The absorbance was read at 490 nm using a Synergy H1 Multimode microplate reader (Bio-Tek/Agilent Technologies).
Statistical analysis
Sample sizes were chosen to achieve a minimum of triplicate samples for all the experiments. For the assessment of statistical significance between two groups, Welch’s t-test was used for parametric data, while Mann-Whitne test was used for non-parametric data. For comparison across multiple groups, Brown-Forsythe and Welch ANOVA test or mixed-effects analysis with Šídák’s multiple comparisons test were used when appropriate for parametric data; for non-parametric data, Kruskal-Wallis test was performed. Data normality was determined based on Q-Q plots distribution. All statistical tests were performed using GraphPad Prism 9 software (RRID: SCR_002798, link), and the results were considered significant at α = 0.05. * P < 0.05, ** P < 0.01, *** P < 0.001, and **** P < 0.0001.
Data availability
Data supporting the herein conclusions are provided within the manuscript or supplementary information files.
References
Wood, L. D. & Hruban, R. H. Pathology and molecular genetics of pancreatic neoplasms. Cancer journal (Sudbury, Mass) 18 (6). (2012).
Muñoz, A. R. et al. Pancreatic cancer: current status and challenges. Curr. Pharmacol. Rep. 12. 3(6). (2017).
Siegel, R. L., Giaquinto, A. N. & Jemal, A. Cancer statistics, 202474 (A Cancer Journal for Clinicians, 2024).
Bardeesy, N. et al. Pancreatic cancer biology and genetics. Nat. Rev. Cancer. 2 (12), 2002–2212 (2002).
McGuigan, A. et al. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 11/11 (43), 24 (2018).
Vincent, A. et al. Pancreatic cancer. Lancet (London, England), 2011/08/08. 378 (9791).
Rawla, P., Sunkara, T. & Gaduputi, V. Epidemiology of pancreatic cancer: global trends, etiology and risk factors. World J. Oncol.,02. 10 (1). (2019).
Disibio, G. & French, S. W. Metastatic patterns of cancers: results from a large autopsy study. Arch. Pathol. Lab. Med. 132 (6), 931–939 (2008).
Kamisawa, T. et al. Hematogenous metastases of pancreatic ductal carcinoma. Pancreas 11 (4), 345–349 (1995).
Mao, C. et al. Observations on the developmental patterns and the consequences of pancreatic exocrine adenocarcinoma. Archives of Surgery 130 (2).
Embuscado, E. E. et al. Immortalizing the complexity of cancer metastasis: genetic features of lethal metastatic pancreatic cancer obtained from rapid autopsy. Cancer Biol. Ther. 4 (5), 548–554 (2005).
Yachida, S. & Iacobuzio-Donahue, C. A. The pathology and genetics of metastatic pancreatic cancer. Arch. Pathol. Lab. Med. 133 (3), 413–422 (2009).
Haeno, H. et al. Computational modeling of pancreatic cancer reveals kinetics of metastasis suggesting optimum treatment strategies. Cell 148 (1–2), 362–375 (2012).
Wilentz, R. E. et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 60 (7), 2002–2006 (2000).
CA, I. D. Genetic evolution of pancreatic cancer: lessons learnt from the pancreatic cancer genome sequencing project Gut 61 (7). (2012).
Hayashi, A. et al. The pancreatic cancer genome revisited. Nat. Rev. Gastroenterol. Hepatol. 18 (7), 2021–2006 (2021).
EA, C. et al. A central role for RAF→MEK→ERK signaling in the genesis of pancreatic ductal adenocarcinoma Cancer Discov., (2012)
Ying, H. et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 04 (3), 149 (2012).
Than, M. T. et al. KRAS-Driven tumorigenesis and KRAS-Driven therapy in pancreatic adenocarcinoma. Mol. Cancer Ther., 23 (10). (2024).
N, D. and S. P, Targeting reactive oxygen species in development and progression of pancreatic cancer Expert Rev. Anticancer Therapy 17(1). 2017.
Alonso, A. et al. Protein tyrosine phosphatases in the human genome. Cell 117 (6), 699–711 (2004).
S, M., et al., Extracellular signal-regulated kinases phosphorylate mitogen-activated protein kinase phosphatase 3/DUSP6 at serines 159 and 197, two sites critical for its proteasomal degradation. Mol. Cell. Biol. 25 (2). (2005).
Ruckert, M. T. et al. Protein tyrosine phosphatases: promising targets in pancreatic ductal adenocarcinoma. Cell. Mol. Life Sci. 76 (13), 2571–2592 (2019).
Furukawa, T. et al. Potential tumor suppressive pathway involving DUSP6/MKP-3 in pancreatic cancer. Am. J. Pathol. 162 (6), 1807–1815 (2003).
Xu, S. et al. Abrogation of DUSP6 by hypermethylation in human pancreatic cancer. J. Hum. Genet. 50 (4), 159–167 (2005).
S, Y. et al. A novel MIF signaling pathway drives the malignant character of pancreatic Cancer by targeting NR3C2.Cancer Res., 07 76(13). (2016)
G, Z. et al. DPEP1 inhibits tumor cell invasiveness, enhances chemosensitivity and predicts clinical outcome in pancreatic ductal adenocarcinoma.PloS One, 7(2). (2012).
Moffitt, R. A. et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 47 (10), 2015–2009 (2015).
Collisson, E. A. et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011 17 4, 2011-04-03. 17(4).
Bailey, P. et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531 (7592), 47–52 (2016).
Y, O. et al. SERPINB3-MYC axis induces the basal-like/squamous subtype and enhances disease progression in pancreatic cancer. Cell Rep., 42(12). (2023)
Badea, L. et al. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology, 55 11. (2008).
Zhang, Z. et al. Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK signaling in lung cancer cells. Carcinogenesis, 31(4). (2010).
Jin, X. et al. A metastasis map of human cancer cell lines. Nature, 9. 588 (7837). (2020).
MA, C. et al. Metastatic pancreatic cancer is dependent on oncogenic Kras in micePloS One, 7(12). (2012).
EL, L. O. et al. KRASG12D drives immunosuppression in lung adenocarcinoma through paracrine signaling JCI Insight, 10(1). (2025).
C, M. et al. Experimental microdissection enables functional harmonisation of pancreatic cancer subtypes Gut, 68(6). (2019)
Carpenter, E. S. et al. Analysis of donor pancreata defines the transcriptomic signature and microenvironment of early neoplastic lesions. Cancer Discov., 13(6). (2023).
Bojmar, L. et al. Multi-parametric atlas of the pre-metastatic liver for prediction of metastatic outcome in early-stage pancreatic cancer. Nat. Med. 30 (8), 2024–2006 (2024).
Bye, B. A. et al. Combined omipalisib and MAPK Inhibition suppress PDAC growth. Cancers, 29. 17 (7). (2025).
Birnbaum, D. J. et al. Validation and comparison of the molecular classifications of pancreatic carcinomas. Mol. Cancer. 16 (1), 168 (2017).
Kidger, A. M. et al. Suppression of mutant Kirsten-RAS (KRAS(G12D))-driven pancreatic carcinogenesis by dual-specificity MAP kinase phosphatases 5 and 6. Oncogene 41 (20), 2811–2823 (2022).
Kato, M. et al. Dual-specificity phosphatase 6 plays a critical role in the maintenance of a cancer stem-like cell phenotype in human endometrial cancer. Int. J. Cancer. 147 (7), 1987–1999 (2020).
Wu, F. et al. Nuclear-biased DUSP6 expression is associated with Cancer spreading including brain metastasis in triple-negative breast Cancer. Int. J. Mol. Sci. 06. 20 (12). (2019).
Wu, Q. N. et al. Pharmacological Inhibition of DUSP6 suppresses gastric cancer growth and metastasis and overcomes cisplatin resistance. Cancer Lett. 412, 243–255 (2018).
Timofeev, O. et al. ERK pathway agonism for cancer therapy: evidence, insights, and a target discovery framework. Npj Precision Oncol. 8 (1), 2024–2003 (2024).
Gutierrez-Prat, N. et al. DUSP4 protects BRAF- and NRAS-mutant melanoma from oncogene overdose through modulation of MITF. Life Sci. Alliance, 17. 5(9). (2022).
Bulle, A. et al. Combined KRAS-MAPK pathway inhibitors and HER2-directed drug conjugate is efficacious in pancreatic cancer. Nat. Commun. 15 (1), 2024–2003 (2024).
Carracedo, A. et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Invest. 21. 118 (9). (2008)
Brown, W. S. et al. Overcoming adaptive resistance to KRAS and MEK inhibitors by Co-targeting mTORC1/2 complexes in pancreatic Cancer. Cell. Rep. Med. 17.1 (8). (2020).
H, L. et al. Calcium saccharate/DUSP6 suppresses renal cell carcinoma glycolytic metabolism and boosts sunitinib efficacy via the ERK-AKT pathway. Biochem. Pharmacol., 224. (2024).
Duan, S. et al. Loss of FBXO31-mediated degradation of DUSP6 dysregulates ERK and PI3K-AKT signaling and promotes prostate tumorigenesis. Cell. Rep. 19. 37(3). (2021).
T, I. et al. Paralog knockout profiling identifies DUSP4 and DUSP6 as a digenic dependence in MAPK pathway-driven cancersNat. Genet. 53(12). (2021).
Daemen, A. et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc. Natl. Acad. Sci. U.S.A., 112 (32) (2015)
Hsu, W. C. et al. DUSP6 mediates T cell receptor-engaged glycolysis and restrains TFH cell differentiation. Proceedings of the National Academy of Sciences, 2018-8-21. 115 (34).
Li, M. et al. Biological role of metabolic reprogramming of cancer cells during epithelial–mesenchymal transition (Review). Oncol. Rep.. 41(2) (2019)
Kang, H. et al. Role of metabolic reprogramming in Epithelial–Mesenchymal transition (EMT). Int. J. Mol. Sci., 04. 20(8). (2019).
Joshi, V. B., Ruiz, O. L. G. & Razidlo, G. L. The cell. biology metastatic invasion pancreat. cancer: Updates Mechanistic Insights Cancers.15 (7). (2023).
Nagathihalli, N. S. et al. STAT3 mediated remodeling of the tumor microenvironment results in enhanced tumor drug delivery in a mouse model of pancreatic Cancer. Gastroenterology, 7. 149(7). (2015).
Bankhead, P. et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 1, 2017-12-04. 7(1).
Nwosu, Z. C. et al. Uridine-derived ribose fuels glucose-restricted pancreatic cancer. Nature 618 (7963), 2023–2005 (2023).
Yi, Z. et al. KDM6A regulates cell plasticity and pancreatic Cancer progression by noncanonical activin pathway. Cell. Mol. Gastroenterol. Hepatol. 13(2) (2022)
A, S. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102(43) (2005).
Acknowledgements
We would like to acknowledge Dr. David Tuveson (CRUK) for supplying the murine K8484 cell line used in these studies; Dr. Andrew Lowy (UCSD) for supplying the P4313 cell line; Dr. Dafydd Thomas, at the Research Histology and Immunohistochemistry Core (University of Michigan), for conducting the RNA-ISH multiplex assay; the Merchant Lab (University of Miami) for use of the PKT62 cell line; and the Flow Cytometry Core Laboratory at KUMC, which is sponsored, in part, by the NIH/NIGMS COBRE grant P30 GM103326 and the NIH/NCI Cancer Center Grant P30 CA168524.
Author information
Authors and Affiliations
Contributions
MTR: methodology, data collection, formal analysis, visualization, paper writing, and editing; RMW: data collection, formal analysis, and editing; BBB, AEE, WY, FB, JS, ELLO, MPM: data collection and analysis; CAL: supervision, methodology, data analysis, editing; MNV: supervision, methodology, data analysis, paper writing, and editing; VSS: funding acquisition, project administration, conceptualization, supervision, methodology, data analysis, paper writing, and editing.
Corresponding authors
Ethics declarations
Competing interests
In the past three years, CAL has consulted for Astellas Pharmaceuticals, Odyssey Therapeutics, Third Rock Ventures, and T-Knife Therapeutics, and is an inventor on patents pertaining to Kras regulated metabolic pathways, redox control pathways in pancreatic cancer, and targeting the GOT1-ME1 pathway as a therapeutic approach (US Patent No: 2015126580-A1, 05/07/2015; US Patent No: 20190136238, 05/09/2019; International Patent No: WO2013177426-A2, 04/23/2015). All the remaining authors declare no conflict of interest.
Compliance with ethical standards
Experiments involving mice were approved by the Ethics Committee on Animal Research at Ribeirao Preto Medical School, University of São Paulo (protocol# 016/2018), and conducted in accordance with the Guidelines of the Brazilian College of Animal Experimentation. Experiments performed at the Massachusetts General Hospital and the University of Kansas Medical Center were subjected to IACUC protocols (2005N000148 and 2019N000116; and 22-02-226, respectively) and were conducted according to the ARRIVE guidelines.
Financial support
This study was supported by São Paulo Research Foundation (FAPESP 2015/10694-5) granted to VSS. MTR was supported by CAPES Foundation (Finance Code 001) and Fulbright Association. JS is supported in part by the National Cancer Institute of the National Institutes of Health under award number R37CA262209. MVS received funds from the University of Kansas Cancer Center. CAL was supported by the NCI (R37CA237421, R01CA248160, and R01CA244931).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ruckert, M.T., Walsh, R.M., Bye, B.A. et al. DUSP6 is upregulated in metastasis and influences migration and metabolism in pancreatic cancer cells. Sci Rep 15, 33996 (2025). https://doi.org/10.1038/s41598-025-12967-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-12967-8
