
Abstract
Integrin subunit alpha V (ITGAV), a subunit of the integrin receptor, is involved in many types of cancers. In order to explore the potential mechanisms of ITGAV in cancers, we carried out a comprehensive pan-cancer analysis using public database. In this study, ITGAV expression in different cancers and the relationship between ITGAV and clinic-pathological features, prognosis, genetic alteration, epigenetic modification, and tumor immune microenvironment were systemically analyzed. Gene enrichment analysis was performed to explore potential functions of ITGAV in gastric cancer (GC). GC tissue microarrays and in vitro cell experiments were used to verify the prediction results in GC. The results revealed that ITGAV was variably expression in different cancers, and ITGAV had a certain prognostic and diagnostic value in most cancers, including GC. ITGAV expression was found to be related to genetic alteration, DNA methylation, immune checkpoint gene, and immune cell infiltration in multiple cancers. Functional analyses revealed that ITGAV was involved in the regulation of EMC remodeling, ferroptosis, and cuproptosis in GC. In vitro experiments verified that ITGAV was correlated with GC cell proliferation, apoptosis, migration, and invasion. Our study demonstrated that ITGAV can be used as an effective prognostic and immunological biomarker for multiple cancers. ITGAV can promote GC malignant progression and could serve as a potential therapeutic target for GC treatment.
Similar content being viewed by others

Introduction
Malignant tumors are the main diseases endangering human health, with nearly 20 million new cases and approximately 9.7 million cancer-related deaths in 2022 1. China is one of the countries with the highest incidence of gastric cancer (GC) cancer, with 478 thousand new cases each year, taking up over 40% of total cases worldwide2. Despite great progress has been gained in the prevention, diagnosis, and treatment of GC, prognosis of advanced stage patients remains dismal3. Tumor recurrence and metastasis are the two main causes of death in patients with advanced GC. Thus, a better understanding of the molecular biological mechanism underlying the pathogenesis of GC is required for discover novel diagnostic biomarkers and therapeutic targets.
Integrins are transmembrane receptors consisting of α and β subunits that mediate cell–cell and cell–extracellular matrix (ECM) interactions4. Recently, a growing number of studies have revealed that the integrin family has played important roles in tumorigenesis and metastasis, suggesting that these molecules may serve as potential biomarkers and therapeutic targets5,6. Integrin subunit alpha V (ITGAV) is a subunit of the integrin receptor, can binds to β1, β3, β5, β6, and β8 subunits to form receptors7. Overexpression of ITGAV was found in various solid tumors including breast cancer, esophageal adenocarcinoma, and neck squamous cell carcinoma, and associated with poor prognosis8,9,10. Moreover, ITGAV has been reported to be involved in the regulation of angiogenesis, tumor invasion, growth, and metastasis11. At present, there are relatively few studies focusing on ITGAV in GC, and its biological function in GC is still unclear. Zhang et al. and Wang et al. reported that ITGAV was overexpression in GC tissues, and high ITGAV expression was related to poorer prognosis12,13. Nevertheless, these studies did not explore the underlying mechanism in GC. Piroozkhah et al., Chen et al., and Zhang et al. explored ITGAV expression and prognostic significance in pan-cancer by bioinformatics analysis14,15,16. However, these studies mainly focus on the role of ITGAV in digest system tumors without a detailed analysis specific to GC and lack support from experimental data.
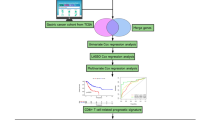
To fill this gap in existing literature, in the present study, we utilized multiple bioinformatic tools to analyze the expression, prognostic and diagnostic value, genetic alteration and methylation status, immune cell infiltration and immunological markers, and gene enrichment analyses of ITGAV in various cancer types. Additionally, tissue microarray-based immunohistochemistry (IHC) was conducted to validate the results in GC tissues specimens in our hospital. Finally, in vitro experiments were performed to confirm the regulatory function of ITGAV in GC cells (Workflow of our study is depicted in Fig. 1). In summary, our study demonstrated that ITGAV might be a diagnostic and prognostic biomarker of GC, and has potential as a therapeutic target for GC.

A workflow of the whole study.
Results
ITGAV expression and localization
The abbreviations of the 33 cancer types in TCGA are provided in Table S1. Based on TCGA datasets and GETx datasets, the ITGAV mRNA expression was extremely upregulated in CHOL, COAD, ESCA, HNSC, LIHC, LUAD, LUSC, and STAD. In contrast, expression of ITGAV was downregulated in KICH, KIRP, and UCEC (Fig. 2A, B). The UALCAN dataset indicated that ITGAV protein expression was elevated in most types of tumors (Fig. S1A). The HPA datasets revealed that ITGAV was significantly high expression in parathyroid gland, heart muscle, and retina tissues (Fig. S1B). The HPA and GeneCards datasets indicated that ITGAV protein mainly localizes in the cytoplasm (Fig. S1C, D). Next, the clinical correlation analysis was carried out to determine the correlation between ITGAV expression and different pathological stages. The results indicated that high ITGAV expression predicted late pathological stage in STAD and KIRP, deeper tumor invasion in STAD and KIRP, and more lymph node metastasis in STAD, CHOL, KIRP, and THCA (Figs. 2C, S2). Analysis of CCLE data revealed that ITGAV expression was elevated in a variety of GC cell lines, GCIY, MKN1, and AGS were the top three expressed (Fig. 2D). Last, single-cell dataset (GSE134520) was used to assess the origin of cells in GC tissues with ITGAV expression. All distinct cell subtypes of GC were included in the cluster analysis. A total of 9 cell types were identified (Fig. 2E). ITGAV was mainly expressed in malignant cells, myofibroblasts, fibroblasts, and mast cells. In addition, ITGAV was also expressed in several immune cell types, such as DC and CD8 T cells. Collectively, ITGAV was upregulate expressed in diverse cancers, and this elevation is associated with disease progression in STAD.

ITGAV expression in pan-cancer. (A) ITGAV mRNA expression in pan-cancer tissues from TCGA + GTEx. (B) ITGAV mRNA expression in paired tumors and paired adjacent normal tissues from TCGA. (C) Relationship between ITGAV and serosal invasion, lymph node metastasis, and pathologic stage in STAD from TCGA. (D) ITGAV expression in different GC cell lines from CCLE. (E) Single-cell analysis of ITGAV expression in GC from the GSE134520 dataset. P ≥ 0.05, *P < 0.05, **P < 0.01, ***P < 0.001.
Prognostic and diagnostic value of ITGAV
To clarify the prognostic value of ITGAV in pan-can, we systematically analyze the correlation between ITGAV expression and three important survival metrics: Overall Survival (OS), Disease-Specific Survival (DSS), and Progression-Free Interval (PFI). The results indicated that high levels of ITGAV expression was significantly associated with poor OS in CESC, LGG, LIHC, PAAD, and STAD. On the other hand, high ITGAV expression was correlated with good OS in CHOL and KIRC (Fig. 3A, B). To further illustrate the effect of ITGAV on prognosis, we also performed DSS analysis. Higher ITGAV expression was correlated with worse DSS in LIHC, MESO, PAAD, and STAD. In contrast, higher CENPF expression was associated with better DSS in COAD and KIRC. In addition, PFI analysis was also performed. ITGAV expression had a protective influence on PFI in ACC, GBM, KIRP, LGG, and PAAD. Kaplan–Meier curve analysis indicated that ITGAV expression was associated with poor prognosis in LGG (Fig. 3C), PAAD (Fig. 3D) and STAD (Fig. 3E). On the contrary, Kaplan–Meier curve analysis showed that high ITGAV expression was associated with batter prognosis in KIRC (Fig. 3F). ROC curve was used to evaluate the diagnostic efficacy of ITGAV. ITGAV had a high accuracy in predicting CHOL, GBM, LGG, PAAD, THYM, and ESCA (AUC > 0.9) (Fig. 4A–G). Additionally, ITGAV had moderate diagnostic efficiency in STAD (Fig. 4H), ACC, DLBC, HNSC, LAML, LIHC, OV, PCPG, UCEC, and UCS (AUC: 0.7–0.9) (Fig. S3). Taken together, ITGAV might be a potential prognostic and predictive factor in various cancers, including STAD.

Prognostic value of ITGAV expression in pan-cancer. (A) Summary of the correlation of ITGAV expression with OS, DSS, and PFI in pan-cancer. (B) Univariate Cox regression analysis of correlation between pan-cancer ITGAV expression and OS. (C–F) OS survival curves of ITGAV in LGG (C), PAAD (D), STAD (E), and KIRC (F).

Diagnostic value of ITGAV in pan-cancer. (A) Heatmap of diagnostic value of ITGAV. ROC analysis showing predictive efficiency of ITGAV in CHOL (B), ESCA (C), GBM (D), LGG (E), PAAD (F), THYM (G), and STAD (H).
Genetic alteration and methylation status of ITGAV
The genetic alterations of ITGAV in pan-cancer were investigated using the cBioPortal. As shown in Fig. 5A, 2.8% samples harbored ITGAV mutations in all of TCGA samples, and missense mutation, amplification and deep deletion were the main mutation types. The alteration frequency was highest in STAD (10%), and the “deep deletion” type was the most common form of alteration in STAD (Fig. 5B). Furthermore, we analyzed the correlation between ITGAV mRNA expression and mutation count in pan-cancer (Fig. 5C). PCPG exhibited the lowest mutation count, while UCEC had the highest. Additionally, we further explored mutated genes in the high/low expression cohorts of ITGAV in GC. The top five genes were TTN, TP53, MUC16, LRP1B, and SYNE1 in GC (Fig. 5D). Based on GSCA data, we assessed the association between ITGAV expression and methylation levels in pan-cancer. ITGAV expression was negative correlated with promoter DNA methylation in almost all tumor types (Fig. 6A). Results from UALCAN showed that ITGAV was hypermethylated in COAD, HNSC, KIRP, LUAD, LUSC, PRAD, and UCEC. In contrast, it was hypomethylated in BLCA, CHOL, KIRC, and TGCT(Fig. 6B). Many studies have reported that m6A RNA methylation regulators play important roles in cancer incidence and development. Our results revealed that ITGAV was positively correlated with most m6A methylation regulators in pan-cancer (Fig. 6C). In addition, the methylation map of ITGAV2 in STAD was acquired from the MethSurv (Fig. 6D). A total of 14 CpG sites contained. Survival analysis indicated that 5 sites were correlated with prognosis of STAD patients, including cg00422230, cg07177569, cg16866216, cg22208448, and cg23613429 (Fig. S4). All 5 sites showed a protective effect in STAD. These results revealed that the genetic alteration and methylation level of ITGAV may be linked to tumor progression and prognosis for multiple cancers.

Genetic alteration analysis of ITGAV. (A) The genetic alteration of ITGAV in pan-cancer. (B) Alteration frequency and mutation type of ITGAV in different cancers. (C) Correlation between TPM4 mRNA expression and mutation count in pan-cancer. (D). The top 10 genes with the highest frequency of mutations in ITGAV high/low expression groups in GC.

DNA methylation analysis of ITGAV. (A) Correlations between ITGAV expression and Methylation level in pan-cancer. (B) Promoter methylation level in ITGAV between normal and tumor tissues in pan-cancer. (C) The heatmap of correlations between ITGAV expression and m6A methylation regulatory factors in pan-cancer. (D) The heatmap of CpG sites methylation of ITGAV in GC.
Correlation between immunological characteristics and ITGAV
First, we evaluated the relationship between ITGAV expression and immune cell infiltration in pan-cancer. The ssGSEA algorithm results indicated that high ITGAV expression was significantly associated with increased infiltration of numerous immune cells, including macrophages, neutrophils, T-helper cells, Tcm, and Tem in most cancer types (Fig. 7A). Then, the ESTIMATE algorithm was used to determine tumor purity and immune infiltration in pan-cancer. The results revealed that ITGAV expression was positively related to ESTIMATE scores in 20 cancer types, including STAD, whereas it was negatively related within 2 cancer types, namely, TGCT and UCEC (Fig. 7B, S5). Last, we investigated the association between ITGAV expression and immune infiltration in GC. The results of Violin and Laplace plots indicated that ITGAV was positively correlated with most immune cell infiltration in GC, with the exception of NK CD56 bright cells and Th17 cells (Fig. 7C, D). A chord diagram showed similar results (Fig. 7E). Our results indicated that ITGAV expression was correlated with multiple immune cell infiltration in GC.

Immunoinfiltration analysis of ITGAV. (A) ITGAV expression and immune cell infiltration in pan-cancer. (B) Correlation between ITGAV expression and ESTIMATE scores in pan-cancer. (C) Differences of enrichment scores in different immune cells between low and high ITGAV expression groups in STAD. (D) Correlation between ITGAV expression and infiltration scores of different immune cell types in STAD. (E) Correlation between ITGAV expression and infiltration scores of six common immune cell type in STAD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Previous studies have discovered that immune checkpoint (ICP) genes, mismatch repair (MMR) genes, tumor mutation burden (TMB), and microsatellite instability (MSI) were closely related to the efficacy of immunotherapy17. In this study, the correlation between ITGAV expression and common immunotherapy biomarkers was explored. Our results indicated that ITGAV expression was positively associated with eight ICP genes in most cancer types, including COAD, OV, PAAD, STAD, and UVM. In contrast, ITGAV was negatively correlated with these genes in TGCT (Fig. 8A, Table S2). Next, we verified the associations between ITGAV and 4 ICP genes using TIMER 2.0 database. The similar results were observed, and a positive correlation was found between ITGAV and PD-1(rho values = 0.146, P = 0.0096), PD-L1(rho values = 0.310, P < 0.001), CTLA-4(rho values = 0.221, P < 0.001), and LAG-3(rho values = 0.176, P < 0.001) in STAD (Fig. 8B). Then, we evaluated the co < 0.001rrelation between ITGAV and MMR genes. ITGAV expression was positively correlated with five MMR genes (MLH1, MSH2, MSH6, PMS2, and EPCAN) in most cancers, especially in COAD, LIHC, PRAD, READ, STAD, THYM, and UCEC (Fig. 8C). Moreover, ITGAV expression was positively correlated with TMB in LAML, LGG, and THYM and negatively correlated with BRCA, HNSC, LUSC, and UVM (Fig. 8D). ITGAV expression was positively correlated with MSI in COAD, MESO, READ, TGCT, and UCEC and negatively correlated with LUAD and PRAD (Fig. 8E). However, ITGAV had no significant association with TMB or MSI in GC. The tumor immune dysfunction and exclusion (TIDE) score was a superior predictor of immunotherapy, and high TIDE score indicated a lower immunotherapy response. ITGAV was positively correlated with TIDE score in most cancers, including GC (Figs. 8F, S6). These findings suggest that the relationship between ITGAV and immunotherapy efficacy in GC remains incompletely characterized, further researches will be required to clarify the relationship between ITGAV and response to immunotherapy in GC.

Relationship between ITGAV expression and immunotherapy response. (A) Relationship between ITGAV expression and immune checkpoints in pan-cancer. (B) Relationship between ITGAV and PD-1, PD-L1, CTLA-4 and LAG-3 in pan-cancer. (C) Relationship between ITGAV expression and MMR markers. (D) Relationship between ITGAV expression and TMB. (E) Relationship between ITGAV expression and MSI. (F) Correlation between ITGAV and TIDE scores in STAD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Functional analysis of ITGAV in GC
To better understand the potential function of ITGAV in GC, we identified the differentially expressed genes (DEGs) between high and low ITGAV expression groups in TCGA-STAD (n = 375). A total of 744 DEGs were identified, and 273 genes were considered as high‐expression genes, and 471 genes were considered as low‐expression genes (Fig. 9A, Table S3). The top 30 genes positively and negatively related to ITGAV expression were presented in Fig. 9B, C. Subsequently, the top 100 genes with positives correlated with ITGAV were displayed in protein–protein interaction (PPI) network (Fig. S7, Table S4). the 10 hub genes were CSRP3, MYH7, ACTC1, MYL3, TNNI3, TNNT2, TPM1, TCAP, LDB3, and SCN5A (Fig. 9D, Table S5). Additionally, GO/KEGG and GSEA analyses of the top 100 positive and negative DEGs were performed. GO analysis suggested that most of DEGs were mainly negative enriched in “keratinization”, “epidermal cell differentiation”, and “enzyme inhibitor activity” (z-scores < − 1), and was positive enriched in “collagen-containing ECM” and “ECM structural constituent” (z-scores > 0) (Fig. 9E, Table S6). Moreover, KEGG analysis revealed that ITGAV was positively enriched in “Calcium signaling pathway” and “Vascular smooth muscle contraction” (z-scores > 1), and was negatively enriched in the “Pancreatic secretion”, “protein digestion and absorption”, and “Fat digestion and absorption” (z-scores < -1). Through GSEA analysis, we found that ITGAV was positively associated with categories such as “Basement membranes”, “ECM organization”, and “focal adhesion”, while it was negative correlated with “keratinization” and “formation of the cornified envelope” (Fig. 9F, Table S7). Ferroptosis and Cuproptosis are the two novel identified forms of programmed cell death and are promising therapeutic targets for GC18. Finally, the co-expression analysis indicated that ITGAV was positively correlated with two ferroptosis related genes and seven cuproptosis related genes as previously reported in in TCGA-STAD (N = 375) (Fig. 9G). These findings suggested that ITGAV may contribute to various biological processes to affect GC development and progression.

Functional analysis of ITGAV in GC. (A) A volcano map of DEGs between high and low ITGAV expression groups in TCGA-STAD. (B and C) Heat maps showing the top 30 genes positively (B) and negatively (C) correlated with ITGAV expression in GC. (D) The top 10 hub genes of the network were identified. (E) GO and KEGG enrichment analysis. (F) GSEA enrichment analysis. (G) Correlation analyses of ITGAV expression with ferroptosis- and cuproptosis-related genes in GC.
Expression of ITGAV and clinicopathologic characteristics of GC patients
To validate the relationship between ITGAV expression and clinical characteristics in GC, we utilized a GC tissue microarray from our center to analyze its expression by immunohistochemistry. Immunohistochemistry revealed that ITGAV was mainly expressed in the cytoplasm (Fig. 10A). 62.7% (69/110) of GC tissues stained positively for ITGAV, which were notably higher than the positive staining in paracancerous tissues (46.4%, 51/110) (P = 0.015) (Fig. 10B). As shown in Table 1, over-expressions of ITGAV were closely related with serosal invasion, lymph node metastasis and pTNM stage (P < 0.05). The Kaplan–Meier survival analysis revealed that positive expression of ITGAV was associated with the poor OS of patients with GC (Fig. 10C). Univariate and multivariate analyses showed that ITGAV expression were significantly correlated with OS in GC (Fig. 10D, E). Overall, these results demonstrated that ITGAV was overexpression in GC tissues and correlated with disease advancement. ITGAV may serve as a potential prognostic marker for GC.

Expression and prognostic value of ITGAV in GC. (A) Representative IHC image of ITGAV expression in GC. (B) Comparisons of ITGAV expression in GC tissues and paracancerous tissues. (C) Survival analysis of ITGAV expression in 110 GC patients after curative resection. (D) Univariate analysis of OS in GC patients. (E) Multivariate analysis of OS in GC patients.
The malignant behaviors of ITGAV in GC
To further validate the ITGAV function, we chose two independent GC cell lines BGC-823 and AGS to perform the cellular function assays. As shown in Fig. 11A, ITGAV was knocked down successfully in both cell lines. CCK-8 assay indicated that the proliferation of BGC-823 and AGS cells were significantly decreased after knockdown of ITGAV (Fig. 11B). Consistently, clone formation experiments revealed that knockdown of ITGAV can effectively inhibit the cloning ability of BGC-823 and AGS cells (Fig. 11C). In addition, cell apoptosis assays indicated that knockdown of ITGAV remarkably increased apoptosis of GC cells (Fig. 11D). Scratch wound assay and Transwell assay showed that the cell migration and invasion capacities after ITGAV knockdown (Fig. 11E–F). To further explore the molecular mechanisms of ITGAV in migration and invasion of GC, we examined the protein change of E-cadherin (E-cad) and Vimentin (VIM) by Western blot. The results demonstrated that the levels of E-cad were significantly upregulated after ITGAV knockdown; however, the VIM were levels downregulated in GC cells (Fig. 11G). These fundings indicated that ITGAV facilitated malignant behaviors of GC cells and may serve as a potential therapeutic target for GC.

Function of ITGAV in GC. (A) Knockdown efficiency of Si-ITGAV was verified in AGS and BGC-823 cells by qRT-PCR analysis. (B) CCK-8 assays indicated ITGAV knockdown suppress the proliferation of GC cells in vitro. (C) Colony formation assay showed knockdown of ITGAV inhibit the cloning ability of GC cells in vitro. (D) Cell apoptosis assays indicated that ITGAV knockdown could increase apoptosis of GC cells in vitro. (E) Cell scratch assays showed the knockdown of ITGAV could inhibit the migration of GC cells in vitro. (F) Transwell migration and invasion assays indicated ITGAV knockdown could restrain the migration and invasion of GC cells in vitro. (G) Western blot analysis indicated that the protein expression levels of ITGAV, E-cad, and VIM were changed after ITGAV knockdown.
Discussion
ITGAV is a member of the integrin family and is encoded by the ITGAV gene on the long arm of chromosome 2, plays a critical role in cell-surface signal transmission7. Previous studies have revealed that ITGAV is aberrantly expressed in various tumors and is involved in multiple malignant biological behaviors8,9,10. However, the molecular mechanisms underlying the effects of ITGAV remains unclear. Although several previous studies have investigated the function of ITGAV in pan-cancer14,15,16, the integrated bioinformatics analysis specifically focusing on GC remain relatively limited. In this study, we conducted a pan-cancer comprehensive bioinformatics analysis of ITGAV using multiple databases, with a particular focus on its role in GC. In addition, we performed several experiments in GC tissues and cells to validate the results. Our findings will contribute to reveal the probable biological function of ITGAV in the development and advancement of GC.
In our study, the results indicated that ITGAV expression was heterogeneous in different cancers. ITGAV expression was upregulated in 8 cancer types, including GC. ITGAV expression was found to be lower express in KICH, KIRP, and UCEC. Survival analysis unveiled that highly expressed ITGAV was frequently correlated with unfavorable prognosis in various tumor types, particularly LGG, MESO, PAAD, and STAD. Conversely, ITGAV was identified as a protective factor in CHOL and KIRC in term of OS. Those heterogeneities may imply that ITGAV plays divergent roles in different cancers. Besides, the results of ROC analysis indicated that ITGAV expression level has excellent diagnostic value for multiple tumors, including GC. Consistent with bioinformatics results, our cohort study also showed that ITGAV expression was up-regulated in GC tissues. High ITGAV expression was correlated with malignant phenotypes and was an independent adverse prognostic factor for GC. These results were aligned with Zhang et al.12 studies. However, our research used an expanded GC cohort of patients who underwent radical resection to overcome sample limitations of prior studies and strengthen the reliability of our results. Taken together, ITGAV could be used as a potential biomarker for diagnosis and prognosis evaluation in different types of cancer, including GC.
Growing evidence has shown that genetic mutations and epigenetic alterations significantly contribute to tumorigenesis and development19. Our results demonstrated that the genetic alteration of ITGAV was observed in a variety of tumors, and missense mutation was the most common alteration type. These results were consistent with previous study by Shi et al.16. Moreover, we specifically investigated the mutational of ITGAV in GC. ITGAV exhibits a high mutation frequency in GC, and deep deletion was the most prevalent alteration type. Previous studies have revealed that the “deep deletion” mutation can lead to loss of gene function, which might result in tumorigenesis and tumor progression in certain tumors20. Notably, the ITGAV overexpression was associated with TP53 mutations in GC. Previous articles have reported that TP53 mutations could promote GC progression and metastasis by facilitating proliferation and EMT21. Furthermore, the present study revealed that ITGAV expression was negatively correlated with methylation levels across various tumor types. Evidence suggests that m6A modification may contribute to progression of oncogenesis, especially in GC22. Our findings showed that ITGAV was positively associated with multiple m6A methylation regulators in most cancers, including GC. In addition, we also found that ITGAV methylation at certain CpG sites was correlated with good prognosis in GC patients, indicating that methylation levels of ITGAV may act as an effective prognostic biomarker for GC. Here, these findings indicated that ITGAV was a prospective biomarker for detecting mutations and epigenetic changes in pan-cancer. Further experiments are needed to confirm these results.
Recently, immunotherapy has been wildly used in tumor therapy and achieved remarkable success. Previous studies have shown that the tumor immune microenvironment is closely associated with the efficacy of immunotherapy23. Muir et al. reported that ITGAV can activates autophagy and regulates B cell activation24. In our study, we observed that ITGAV had strong positive correlation with infiltration levels of multiple immune cells in multiple cancers. In addition, the expression of ITGAV was correlated with a variety of ICP genes as well as MSI, TMB, MMR markers, and TIDE score in different cancers. These results suggested that ITGAV has the potential to modulate the tumor immune microenvironment and effect immunotherapy efficacy. Furthermore, we also analyzed the relationship between ITGAV and immune cell infiltration in GC. Single-cell enrichment analyses showed that ITGAV was mostly concentrated in myofibroblasts, fibroblasts, and mast cells in GC. ITGAV was positively correlated with various immune cells, immune checkpoint markers, and immunostimulatory factors in GC. Thus, we hypothesized that ITGAV may participate in the regulation of immune infiltrating cell in GC and influence the efficacy of immunotherapy. Further studies are required to investigate the exact role of ITGAV in the immune microenvironment.
Integrins are matrix receptors that mediate cell adhesion and transduce signals to the cell interior. ITGAV belongs to the integrin family. To investigate the function of ITGAV in GC, we analyzed DEGs and co-expression genes from TCGA-STAD database. We identified ten hub genes of ITGAV, such as ACTC1, TNNT2, TPM1, and SCN5A et al. Previous studies have demonstrated that these molecules were abnormally expressed in many tumors and were closely associated with tumorigenesis and development of multiple type of cancers. Based on the GO, KEGG, and GSEA analysis, ECM-related pathway may be one of important pathways in GC patients with ITGAV overexpression. ECM remodeling can affect cell migratory ability, and ECM destruction was considered as critical role in initiating cancer invasion and metastasis5. Further studies to elucidate the relationship between ITGAV and ECM proteins may help to reveal the role of ITGAV in GC and its related mechanism. The dysregulation of cell death is critical for cancer initiation and development. Recent studies reported that ferroptosis and cuproptosis play vital roles in GC progression and tumor immune evasion18. We found that ITGAV was positive correlated with multiple ferroptosis and cuproptosis related genes in GC. ITGAV may be involved in the regulation of ferroptosis and cuproptosis in GC. In vitro experiment results shown that IVGAV knockdown significantly inhibited proliferation and promote apoptosis of GC, thus suggesting that ITGAV may participate in the pathogenesis of GC by promoting proliferation and inhibiting apoptosis. EMT is a critical process in regulating migration and invasion, which is often correlated with a decrease of E-cad and an increase of VIM. Our study results demonstrated that ITGAV knockdown can reverse the EMT phenotypic change and decreased migration and invasion capacity of GC cells. Taken together, our results suggested that ITGAV can affects the multiple biological behavior of GC cells, and maybe be correlated with cell cycle regulation, cell death modulating, and EMT. More experiments are required to explore the specific mechanism of ITGAV in GC.
In this study, we systematically analyzed multiple public databases, adopted various prediction algorithms, and found that ITGAV was aberrant expression in in various human cancers and may play disparate roles in pan-cancers. In addition, we particularly focus on the functions of ITGAV in GC. However, this study had some limitations. First, the pan-cancer analysis was based on publicly available datasets, further basic experiments are needed to validate the role of ITGAV. Second, we predicted the potential function of ITGAV in GC, further experiments are required to validate the findings of bioinformatics analysis. Finally, this study only conducted in vitro experiments in GC cells, further in vivo experiments are required.
Conclusion
This comprehensive pan-cancer analysis revealed that ITGAV was aberrantly expressed in a variety of cancers and can be used as a potential biomarker for prognosis, diagnosis, and immunotherapy efficacy of different types of cancer. ITGAV expression was correlated with genetic alteration, epigenetic modification, and immune cell infiltration in multiple malignancies. In GC, ITGAV was upregulated in cancer tissues, and this elevation was associated with tumor invasiveness and poor prognosis. ITGAV can promote GC cell proliferation, invasion, and migration abilities in vitro. In a word, our study demonstrated that ITGAV can promote GC progression and may serve as a potential diagnostic biomarker and therapeutic target in GC.
Materials and methods
Analysis of ITGAV expression
The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov/) and Genotype-Tissue Expression (GTEx, http://gtexportal.org) databases from UCSC XENA (https://xenabrowser.net/datapages/) uniformly processed by the Toil were used to assess ITGAV mRNA expression. We excluded the cancer species with less than three samples in a single cancer species, and finally obtained the expression data of 33 cancer species. A total of 8295 normal and 9807 tumor samples were included. The Wilcoxon rank sum test was used to investigate the relationship between ITGAV levels and clinicopathological features in GC. The human protein atlas (HPA, https://www.proteinatlas.org/)25 was utilized to analyze ITGAV distribution characteristics in different human organs. The Ualcan database (http://ualcan.path.uab.edu/index.html)26 was used for analyzing ITGAV protein expression in pan-cancer. The Cancer Cell Line Encyclopedia (CCLE, https://sites.broadinstitute.org/ccle)27 was used to assess ITGAV expression in GC cell lines. The GeneCards (https://www.genecards.org/)28 and HPA databases was used to visualize the subcellular locations of ITGAV. The single-cell data of ITGAV expression in GC (GSE134520) were download from the Tumor Immune Single-cell Hub (TISCH, https://tisch.comp-genomics.org)29. The R software MAESTRO and Seurat were used to process and analyze the single-cell data, and the t-SNE method was used to re-cluster the cells.
Prognostic and diagnostic value analysis
The univariate Cox regression analysis and Kaplan–Meier curve were utilized to assess the association between ITGAV expression and overall survival (OS), disease-specific survival (DSS), and progression-free interval (PFI) in pan-cancer. The median expression value was used as the cutoff. The results were visualized in forest maps. The diagnostic value of ITGAV was analyzed using receiver operating characteristic (ROC) curve and area under the ROC curve (AUC). Diagnostic value was considered high accuracy (AUC > 0.9), moderate accuracy (AUC: 0.7–0.9), and low accuracy (AUC < 0.7).
Genetic alteration and DNA methylation analysis
The cBioPortal database V6.3.1 (https://www.cbioportal.org/)30 was used to assess ITGAV genetic alteration with 10,967 samples which obtained from 10,953 patients in 32 types of cancer from the 32 most recent TCGA studies. The alteration frequency, mutation types and mutation counts across various tumors were analyzed using “OncoPrint”, “Cancer Type Summary” and “Plots” modules. The maftools package was used to visualize the somatic mutations of 372 GC patients from TGCA-STAD cohort. The top ten genes with the highest alteration frequency based on ITGAV expression in STAD were exhibited in a waterfall map. The correlation between ITGAV expression and DNA methylation levels were calculated using the Gene Set Cancer Analysis database (GSCA) (https://guolab.wchscu.cn/GSCA/)31. The UALCAN database was used to explore methylation levels in tumor and normal tissues. We excluded the tumor species with normal cases less than three. Correlations between ITGAV expression and N6- methyladenosine (m6A) methylation regulators involved were analyzed using Spearman correlation analysis. The MethSurv database (https://biit.cs.ut.ee/methsurv/)32 was applied to determine the methylation level of each CpG site in GC.
Association between ITGAV and immune infiltration
Immune infiltration analysis was performed using the single-sample Gene Set Enrichment Analysis (ssGSEA) algorithm from the GSVA package. The relationships between ITGAV and each cell immune infiltrate in GC were analyzed using TCGA-STAD dataset. The ESTIMATE algorithm was performed to evaluate the ESTIMATE scores in pan-cancer. Spearman’s analysis was used to assess the relationship between ITGAV expression and ESTIMATE scores. Next, the correlation between ITGAV and common immunotherapy biomarkers in pan-cancer were evaluated using Spearman correlation analysis from TCGA datasets. TMB is derived from the Thorsson et al. article33. MSI is derived from the Bonneville et al. article34. The TIMER V2.0 database (http://timer.cistrome.org/)35 was used to verify the results. Potential ICB response was predicted with TIDE algorithm.
Genes correlation and functional analysis of ITGAV in GC
The “DESeq2” package was used to identify differentially expressed genes (DEGs) between ITGAV high or low expression groups in TCGA-STAD (N = 375) with|Log2 (Fold Change)|≥ 1 and adjusted P value < 0.05. DEGs are presented in volcano plots and heat maps. The PPI network for the top 100 genes positively correlated with ITGAV expression was obtained using STRING V12.0 (https://string-db.org/)36, with interaction score: medium confidence (0.400) and number of interactors (100). The top 10 hub genes were identified using “CytoHubba” in Cytoscape and ranked by the MCC score. To further explore the function of ITGAV, the “clusterProfiler” package was utilized for Gene Ontology (GO) analysis (biological process, cellular component, and molecular function), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis37, and GSEA enrichment analysis for the top 100 DEGs. Furthermore, the correlation between ITGAV and ferroptosis- and cuproptosis-related genes as previously reported in TGCA-STAD database (N = 375) was evaluated using Spearman correlation analysis38.
Tissue specimens
A total of 110 pairs of paraffin-embedded GC and paracancerous tissue samples of patients underwent radical gastrectomy were collected at Tianjin medical university cancer institute and hospital from 2015 to 2017. All cases were without any preoperative therapy and underwent radical (R0) resection with D2 lymph node dissection. All patients were followed through December 2023.
Cell culture and transfection
The human GC cell lines (AGS, BGC-823) were ordered from the Chinese Academy of Sciences in Shanghai, China. All cell lines were cultured in DMEM supplemented with 10% fetal bovine serum and 1% antibiotics (100 U/ml penicillin and 100 mg/ml Streptomycin) at 37˚C with 5% CO2. ITGAV small-interfering RNA (siRNA) (sequence: forward 5′-CCCUCUGACAUUGAUUGUUTT-3′, reverse 5′-AACAAUCAAUGUCAGAGGGTT-3′) and scrambled siRNA (NC-siRNA) were purchased from Genepharma (Shanghai, China). Cell transfection was performed as previously described39.
Tissue microarrays (TMAs) and immunohistochemistry (IHC)
The GC and paracancerous tissues were fixed by paraffin and punched cylindrical cores (1.5 mm) to create TMAs. For IHC, the TMAs were dewaxed in xylene and hydrated in gradient ethanol. Primary anti-ITGAV antibody (Abcam, ab179475, 1:500) was applied. The sections were then incubated with HRP-conjugated second antibody and incubated in DAB. The IHC results were evaluated by two experienced pathologists according to the percentage of positive cells and the intensity of staining, as previously described40.
Western blot analysis
The GC cells were lysised in RIPA lysis buffer (Beyotime, China). Samples were loaded onto SDS-PAGE gel and electrotransferred to PVDF membranes(Millipore, USA). Membranes were incubated in primary anti-ITGAV antibody (Abcam, ab179475, 1:2000), anti-E-cad antibody (Abcam, ab40772, 1:2000), anti-VIM antibody (Abcam, ab92547, 1:2000), and treated with HRP-lsbeled secondary antibody (Zhongshan Jinqiao Biotechnology, China). Protein bands were detected using ECL reagent and semi-quantified using ImageJ software.
RNA extraction and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from cells using TRIzol reagent (Invitrogen, USA). RNA was reverse transcribed into cDNA using PrimeScript Kit (Takara, Japan). qRT-PCR was performed on a ABI 7900 System (Life Tech, USA). The relative gene expression was calculated by the 2−△△Ct method using GAPDH as the internal control. The sequences of primers were as follows: ITGAV forward: ATCTGTGAGGTCGAAACAGGA, ITGAV reverse: TGGAGCATACTCAACAGTCTTTG,GAPDH forward: GCACCGTCAAGGCTGAGAAC, GAPDH reverse: TGGTGAAGACGCCAGTGGA.
Cell counting kit-8 (CCK-8) and colony formation assay
Cells were cultured in 96-well plates (5 × 103 per well, 100 ul), and proliferation was assessed using CCK-8 (Abcam, USA) following the manufacturer’s protocol. Cell proliferation was detected on hours 0, 24, 48, 72, and 96. For colony formation assay, 500 cells were seeded in a six-well plate and cultured for 10 days. The colonies were fixed with methanol and stained with crystal violet. The colonies were then photographed and counted.
Cell apoptosis assay
Cell apoptosis was assessed through the utilization of the annexin V/PI double-staining assay.The treated cells were gathered and stained with the Annexin V/PI apoptosis kit (BD, USA). Subsequently, the flow cytometry technique (BD, USA) was employed to analyze the rate of apoptosis.
Cell scratch assay
Cells were seeded in 6-well plates (7 × 105 per well) and cultured for 24 h, then a line was drawn using a 10 ul pipette tip. The wounds were photographed at 0 and 48 h. The distances were measured using ImageJ.
Transwell migration and invasion assay
Transwell assay was used to evaluate cell migration and invasion ability. For migration assay, cells cultured in FBS-free DMEM were plated in the upper chambers of transwell (Corning, USA), and DMEM with 10% FBS was added to the lower chamber. Upper chamber coated with Matrigel (BD, USA) for invasion assay. After incubated for 24 h, chamber was fixed in methanol and stained using crystal violet.The stained cells on the bottom surface were examined.
Statistical analyses
The SPSS 26.0(IBM, USA) and GraphPad Prism 8.0(GraphPad, USA) were adopted to analyze statistics. The differences between groups were analyzed using Student’s t-test for continuous or one-way ANOVA for normality variables.. The association between categorical data was assessed using the Pearson Chi-square test for independent samples.. Survival analysis was performed using Kaplan–Meier method, and the log-rank test was used to compare the differences. Representative data were expressed as mean ± standard deviation (SD) based on 3 independent trials. A P value < 0.05 was considered statistically signifcant.
Data availability
The datasets presented in current study are available from the corresponding authors.
References
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J. Clini. 74, 229–263. https://doi.org/10.3322/caac.21834 (2024).
Han, B. et al. Cancer incidence and mortality in China, 2022. JNCC 4, 47–53. https://doi.org/10.1016/j.jncc.2024.01.006 (2024).
Wu, M., Yuan, S., Liu, K., Wang, C. & Wen, F. Gastric cancer signaling pathways and therapeutic applications. Technol. Cancer Res. Treat. 23, 15330338241271936. https://doi.org/10.1177/15330338241271935 (2024).
Hynes, R. O. Integrins: Bidirectional, allosteric signaling machines. Cell 110, 673–687. https://doi.org/10.1016/s0092-8674(02)00971-6 (2002).
Anderson, L. R., Owens, T. W. & Naylor, M. J. Integrins in development and cancer. Biophys. Rev. 6, 191–202. https://doi.org/10.1007/s12551-013-0123-1 (2014).
Huang, Q., Wang, J., Ning, H., Liu, W. & Han, X. Integrin β1 in breast cancer: mechanisms of progression and therapy. Breast Cancer (Tokyo, Japan) https://doi.org/10.1007/s12282-024-01635-w (2024).
Weis, S. M. & Cheresh, D. A. αV integrins in angiogenesis and cancer. Cold Spring Harb. Perspect. Med. 1, a006478. https://doi.org/10.1101/cshperspect.a006478 (2011).
Cheuk, I. W. et al. ITGAV targeting as a therapeutic approach for treatment of metastatic breast cancer. Am. J. Cancer Res. 10, 211–223 (2020).
Loeser, H. et al. Integrin alpha V (ITGAV) expression in esophageal adenocarcinoma is associated with shortened overall-survival. Sci. Rep. 10, 18411. https://doi.org/10.1038/s41598-020-75085-7 (2020).
Xu, L. et al. ITGAV promotes the progression of head and neck squamous cell carcinoma. Curr. Oncol. 31, 1311–1322. https://doi.org/10.3390/curroncol31030099 (2024).
Brzozowska, E. & Deshmukh, S. Integrin alpha v beta 6 (αvβ6) and its implications in cancer treatment. Int. J. Mol. Sci. https://doi.org/10.3390/ijms232012346 (2022).
Zhang, W., Chen, Y., Qiao, Z. & Liu, Y. Overexpression of Integrin alpha V (ITGAV) in gastric cancer and its prognostic significance. Asian J. Surg. 46, 5863–5864. https://doi.org/10.1016/j.asjsur.2023.08.171 (2023).
Wang, H. et al. Integrin subunit alpha V promotes growth, migration, and invasion of gastric cancer cells. Pathol. Res. Pract. 215, 152531. https://doi.org/10.1016/j.prp.2019.152531 (2019).
Chen, Z. Z., Wang, W. P., Xue, H. M. & Liang, Y. The lncRNA-miRNA-integrin alpha V ceRNA network can affect the occurrence and prognosis of gastric cancer. Int. J. Clin. Exp. Pathol. 15, 388–402 (2022).
Piroozkhah, M., Zabihi, M., Jalali, P. & Salehi, Z. Comprehensive multi-omics analysis reveals NPC2 and ITGAV genes as potential prognostic biomarkers in gastrointestinal cancers. Cancer Rep. (Hoboken, NJ) 7, e70087. https://doi.org/10.1002/cnr2.70087 (2024).
Shi, X. et al. Comprehensive analysis of the multifaceted role of ITGAV in digestive system cancer progression and immune infiltration. Front. Immunol. 16, 1480771. https://doi.org/10.3389/fimmu.2025.1480771 (2025).
Ke, B. et al. Oncogenic and immunological role of EDIL3 in human tumours: From pan-cancer analysis to validation in gastric cancer. Heliyon 10, e32291. https://doi.org/10.1016/j.heliyon.2024.e32291 (2024).
Wang, S. et al. Cell death pathways: Molecular mechanisms and therapeutic targets for cancer. MedComm 5, e693. https://doi.org/10.1002/mco2.693 (2024).
Esteller, M. et al. The Epigenetic hallmarks of cancer. Cancer Discov. 14, 1783–1809. https://doi.org/10.1158/2159-8290.Cd-24-0296 (2024).
Hieronymus, H. et al. Deletion of 3p13-14 locus spanning FOXP1 to SHQ1 cooperates with PTEN loss in prostate oncogenesis. Nat. Commun. 8, 1081. https://doi.org/10.1038/s41467-017-01198-9 (2017).
Cai, H. Q., Zhang, L. Y., Fu, L. M., Xu, B. & Jiao, Y. Mutational landscape of TP53 and CDH1 in gastric cancer. World J. Gastrointest. Surg. 16, 276–283. https://doi.org/10.4240/wjgs.v16.i2.276 (2024).
Xu, S. et al. N6-methyladenosine-related lncRNAs identified as potential biomarkers for predicting the overall survival of Asian gastric cancer patients. BMC Cancer 22, 721. https://doi.org/10.1186/s12885-022-09801-z (2022).
Tang, T. et al. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal Transduct. Target. Ther. 6, 72. https://doi.org/10.1038/s41392-020-00449-4 (2021).
Muir, V. et al. Transcriptomic analysis of pathways associated with ITGAV/alpha(v) integrin-dependent autophagy in human B cells. Autophagy 19, 926–942. https://doi.org/10.1080/15548627.2022.2113296 (2023).
Sjöstedt, E. et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 367, 5947. https://doi.org/10.1126/science.aay5947 (2020).
Chandrashekar, D. S. et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 25, 18–27. https://doi.org/10.1016/j.neo.2022.01.001 (2022).
Ghandi, M. et al. Next-generation characterization of the cancer cell line encyclopedia. Nature 569, 503–508. https://doi.org/10.1038/s41586-019-1186-3 (2019).
Stelzer, G. et al. The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. https://doi.org/10.1002/cpbi.5 (2016).
Han, Y. et al. TISCH2: expanded datasets and new tools for single-cell transcriptome analyses of the tumor microenvironment. Nucleic Acids Res. 51, D1425-d1431. https://doi.org/10.1093/nar/gkac959 (2023).
de Bruijn, I. et al. Analysis and visualization of longitudinal genomic and clinical data from the AACR project GENIE biopharma collaborative in cBioPortal. Can. Res. 83, 3861–3867. https://doi.org/10.1158/0008-5472.Can-23-0816 (2023).
Liu, C. J. et al. GSCA: an integrated platform for gene set cancer analysis at genomic, pharmacogenomic and immunogenomic levels. Brief. Bioinform. https://doi.org/10.1093/bib/bbac558 (2023).
Modhukur, V. et al. MethSurv: A web tool to perform multivariable survival analysis using DNA methylation data. Epigenomics 10, 277–288. https://doi.org/10.2217/epi-2017-0118 (2018).
Thorsson, V. et al. The immune landscape of cancer. Immunity 48, 812-830.e814. https://doi.org/10.1016/j.immuni.2018.03.023 (2018).
Bonneville, R. et al. Landscape of microsatellite instability across 39 cancer types. JCO Precis. Oncol. https://doi.org/10.1200/po.17.00073 (2017).
Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48, 509–514. https://doi.org/10.1093/nar/gkaa407 (2020).
Szklarczyk, D. et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638-d646. https://doi.org/10.1093/nar/gkac1000 (2023).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Zhou, X., Xiao, B., Jiang, M. & Rui, J. Pan-cancer analysis identifies EMC6 as a potential target for lung adenocarcinoma. iScience 27, 108648. https://doi.org/10.1016/j.isci.2023.108648 (2024).
Ke, B. et al. Sonic Hedgehog/Gli1 signaling pathway regulates cell migration and invasion via induction of epithelial-to-mesenchymal transition in gastric cancer. J. Cancer 11, 3932–3943. https://doi.org/10.7150/jca.42900 (2020).
Ke, B. et al. EDIL3 is a potential prognostic biomarker that correlates with immune infiltrates in gastric cancer. PeerJ 11, e15559. https://doi.org/10.7717/peerj.15559 (2023).
Funding
This research was funded by National Natural Science Foundation of China (81401952), Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-009A).
Author information
Authors and Affiliations
Contributions
KB designed the research and guided work. KB and JP performed the experiments. WXJ and ZRP analyzed data. KB, LN and MG finalized the manuscript. All authors contributed to the article and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study was approved by the Ethics Committee of Tianjin cancer hospital (No.bc2023054) and written informed consent was obtained from all patients. All methods in this study were performed in accordance with the principles of the Declaration of Helsinki and current ethical guidelines.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ke, B., Jin, P., Wang, XJ. et al. Pan-cancer landscape of ITGAV and its potential role in gastric cancer. Sci Rep 15, 28934 (2025). https://doi.org/10.1038/s41598-025-14342-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-14342-z
