
Abstract
The SF-25 antigen (SF-25 Ag), which functions as the amino acid transporter solute carrier family 3 member 2 (SLC3A2), is essential for regulatory T (Treg) cell maintenance and is highly expressed in adult T-cell leukemia/lymphoma (ATL) cells. We analyzed SF-25 Ag expression in CD4+peripheral blood lymphocytes (PBLs) from 28 ATL patients, 52 healthy human T-lymphotropic virus 1 (HTLV-1) carriers, and eight non-infected individuals. Inverse polymerase chain reaction was used to detect monoclonal integration of HTLV-1 proviral DNA. The median (interquartile range) percentages of SF-25 Ag-positive PBLs in acute, chronic, and smoldering ATL were 54.9% (23.9–62.9), 34.9% (23.8–41.9), and 22.1% (8.4–28.5), respectively, which were significantly higher than those in HTLV-1 carriers (0.8% [0.5–1.0]) and non-infected subjects (0.4% [0.3–0.4]) (P < 0.001). Magnetic sorting isolated SF-25 Ag-positive cells, which showed monoclonal HTLV-1 integration, unlike SF-25 Ag-negative cells. Moreover, culture supernatant from healthy donor PBLs stimulated with a murine-human chimeric SF-25 monoclonal antibody (c-SF-25 Mab) induced apoptosis in SF-25 Ag-positive ATL cells. These findings suggest that c-SF-25 Mab, by targeting SLC3A2, may offer a specific therapeutic approach for ATL by eliminating malignant cells while sparing normal tissue.
Similar content being viewed by others


Introduction
Adult T-cell leukemia/lymphoma (ATL) is an aggressive malignancy of mature peripheral T lymphocytes expressing CD3, CD4, CD25, FOXP3, and CCR4 and exhibiting a phenotype similar to that of regulatory T cells (Tregs)1 acquired after long-term infection with human T-lymphotropic virus 1 (HTLV-1). Southwestern Japan, where more than 100 thousand individuals are HTLV-1 carriers2,3, is one of the most highly endemic regions for prevalence of HTLV-1 infection worldwide. ATL is diagnosed by clonal integration of HTLV-1 proviral DNA and classified into four subtypes based on clinical courses: smoldering, chronic, acute (acute leukemic type), and lymphoma (lymphomatosis type)4. Prognosis in patients with acute or lymphoma ATL who are treated with cytotoxic chemotherapy is particularly poor. Although allogeneic hematopoietic cell transplantation (alloHCT) can improve prognosis in younger, transplant-eligible patients with ATL5, more than half of the patients diagnosed with aggressive ATL are older than 65–70 years of age and ineligible for alloHCT6. In 2023, the efficacy of cytotoxic chemotherapy in combination with anti-CCR4 antibody mogamulizumab in older adult patients with ATL was reported7. This report suggested that the humanized monoclonal antibody targeting ATL might be able to eradicate ATL cells by antibody-dependent cellular cytotoxicity (ADCC).
The murine SF-25 monoclonal antibody (m-SF-25 Mab) recognizes a 125-kilodalton glycoprotein, SF-25 antigen (SF-25 Ag). SF-25 Ag is highly expressed in human tumor cells, whereas most normal human tissues are negative for SF-25 Ag by immunohistological staining, with the exception of a very weak staining observed in a subpopulation of proximal tubular cells of the kidney and islet cells of the pancreas8. Pellizzari et al. identified that a murine-human chimeric SF-25 monoclonal antibody (c-SF-25 Mab) recognizes the CD98 heavy chain (CD98hc) encoded by the solute carrier family 3 member 2 (SLC3A2), a cell surface protein that plays key roles in cancer metabolism9. Ikeda et al. found that branched-chain amino acids (BCAAs) are essential for maintaining the expansion and suppressive capacity of Treg cells via SLC3A2 and the mechanistic/mammalian target of rapamycin complex 110.
Magnetic cell sorting is an extremely efficient magnetic separation method. This technique can expedite separating specific cells. Based on their surface antigens, cells of very high purity can be isolated from a cell medium suspension within minutes. This method is highly specific, allowing for isolating rare cells, such as specific T cells, CD133+ hematopoietic stem and progenitor cells, subpopulations of CD34+ hematopoietic progenitor cells, and disseminated carcinoma cells circulating in the peripheral blood11.
The murine-human chimeric SF-25 (c-SF-25) Mab, in which the constant regions of human IgG1 are linked to the variable regions of m-SF-25 Mab, has been constructed using recombinant DNA techniques12. Reportedly, m-SF-25 Mab is not internalized or shed from the tumor cell surface after binding to the antigen and delivered to tumor cells at high density in vivo12,13. In addition, c-SF-25 Mab has been shown to induce the lysis of SF-25 expressing tumor cells via ADCC by human NKs and macrophages12. The variable region genes of murine Mab that encode the antigen-combining sites (Fab) of the antibody, can be spliced into the constant region (Fc) genes of human immunoglobulin (Ig) G1 heavy and light chains using genetic engineering techniques14. The resulting chimeric mouse-human Mabs are substantially less immunogenic to humans than murine antibodies15,16. These chimeric proteins have a longer serum half-life and can be used as therapeutic substances in humans via multiple administration regimens7,17,18. In addition, because human IgG1 binds to type III IgG Fc receptor (FcγR III, CD16) with high affinity19, the mouse-human IgG1 chimeric Mab is able to mediate ADCC, which otherwise is not induced by murine form12. Therefore, chimeric MAbs might serve as more potent cytotoxic reagents compared with their parental murine counterparts20.
The aim of this study was to investigate SF-25 Ag expression in ATL cells and to evaluate the potential of the murine-human chimeric SF-25 monoclonal antibody as a targeted therapeutic agent for ATL.
Results
Detection of the monoclonal integration of HTLV-1 by IPCR
DNA samples from all 28 patients with ATL and 52 healthy HTLV-1 carriers were analyzed by inverse polymerase chain reaction (IPCR). Figure 1 shows the representative results of IPCR (Fig. 1a) and Southern blot analysis (Fig. 1b) of ATL samples and healthy HTLV-1 carriers. All 28 samples from patients with ATL showed monoclonal integration of HTLV-1 proviral DNA, as demonstrated by both IPCR (lanes 2–8 in Fig. 1a) and Southern blot analysis (lanes 2–8 in Fig. 1b). In contrast, seven of the 52 healthy HTLV-1 carriers displayed distinct bands in both assays (lane 12 in Fig. 1a,b), indicating oligoclonal proliferation of HTLV-1-infected cells. The remaining 45 carriers showed no detectable bands of HTLV-1 proviral DNA (lanes 9 and 10 in Fig. 1a,b, Table 1).

IPCR products with samples from patients with ATL and HTLV-1 healthy carriers. (a) Ethidium bromide-stained gel showing unique bands in samples from ATL and HTLV-1 healthy carriers. (b) Hybridization with an internal oligoprobe showed that the amplified bands contained the U5 region. The samples used in each lane were as follows: lane 1, Raji (negative control); lanes 2 and 3, acute ATL; lanes 4 and 5, chronic ATL; lanes 6–8, smoldering ATL; lanes 9 and 10, HTLV-1 healthy carriers; line11, water; and lane 12, HTLV-1 healthy carriers with HTLV-1 oligoclonal integration. ATL adult T-cell leukemia/lymphoma; HTLV-1 human T-lymphotropic virus 1.
SF-25 Ag expression in peripheral blood lymphocytes (PBLs) and HTLV-1 T cell lines
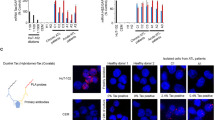
The expression of SF-25 Ag was investigated in ATL cells of CD4-positive PBLs from patients with ATL by two-color flow cytometry using fluorescein isothiocyanate (FITC)-conjugated m-SF-25 Mab and phycoerythrin (PE)-conjugated anti-CD4 Mab. Figure 2a and Supplementary Fig. S1 show the representative results of two-color flow cytometry with smoldering, acute, and chronic ATL samples. SF-25 Ag is specifically expressed in CD4-positive PBLs, which are considered ATL cells. KUT-1 and MT-1 cells also expressed the SF-25 Ag; the levels of expression on their cell surface were higher than those of smoldering ATL samples (Fig. 2b). As shown in Fig. 2C, SF-25 was expressed in the majority of CD4-positive PBLs in patients with acute and chronic ATL. In contrast, the SF-25 Ag was found in only a very small population of CD4-positive PBLs in healthy participants (Fig. 2c). The median (interquartile range) percentages of SF-25 Ag-positive PBL in acute (n = 11), chronic (n = 5), and smoldering type (n = 12) ATL were 54.9% (23.9–62.9), 34.9% (23.8–41.9), and 22.1% (8.4–28.5), respectively. The expression levels of SF-25 Ag in PBLs from patients with ATL were significantly higher than those from HTLV-1 healthy carriers (0.8% [0.5–1.0]) and healthy participants (0.4% [0.3–0.4]) (p < 0.001), as shown in Fig. 2c. The level of SF-25 Ag expression in PBLs from HTLV-1 healthy carriers was similar to that from healthy participants (Fig. 2c). When the cut-off point of SF-25 Ag-positive cells was set to 1.8% according to the 95th percentile of expression in HTLV-1 healthy carriers, all patients with ATL were SF-25 Ag-positive; all patients with non-HTLV-1 carriers were SF-25 Ag-negative. However, among the seven HTLV-1 healthy carriers, two exhibited oligoclonal integration of HTLV-1 proviral DNA using IPCR, with SF-25 Ag-positive cells recorded at 1.9% and 2.3%, respectively (Table 2).

Expression of SF-25 Ag in ATL, HTLV-1 carriers and healthy controls. (a) Representative data of SF-25 Ag and CD4 expression with smoldering ATL. SF-25-expressing cells also expressed CD4. (b) KUT-1 and MT-1 cells were incubated for 30 min at 4 °C with 1 µg/mL of FITC-conjugated murine SF-25 Mab (FITC-m-SF-25 Mab) or FITC-conjugated irrelevant murine IgG1 Mab (FITC-m-IgG1), washed with PBS, and analyzed by flow cytometry. (c) Patients with ATL and healthy controls were categorized as follows: Group A, the healthy controls without HTLV-1 infection; Group B, the healthy HTLV-1 carriers; Group C, the patients with smoldering ATL; Group D, the patients with chronic ATL; and Group E, the patients with acute ATL. Notably, the expression of SF-25 in ATL patients was significantly higher than that in both HTLV-1 healthy carriers and healthy controls without HTLV-1 infection (P < 0.001). ATL adult T-cell leukemia/lymphoma; FITC fluorescein isothiocyanate; HTLV-1, human T-lymphotropic virus 1.
Isolation of SF-25 Ag-positive PBLs and detection of HTLV-1 integration
SF-25 Ag-positive cells were separated from the PBLs from patients with smoldering ATL using the magnetic cell sorting method. More than 90% of the cells in the enriched fraction of SF-25-expressing cells were found to be SF-25 Ag positive. On the other hand, less than 1% of cells in the depleted fraction of SF-25-expressing cells were found to be SF-25 Ag positive.
Figure 3a shows the electrophoretic pattern of the polymerase chain reaction (PCR) products of DNA from cell-enriched fractions of SF-25-expressing and SF-25-negative cells. Figure 3b shows the results of the Southern blot analysis of the PCR products from these cells. The cell fraction enriched in SF-25 Ag-positive cells (> 90%) showed monoclonal integration of HTLV-1 proviral DNA (line 3 in Fig. 3a,b). In contrast, SF-25 Ag-negative cells did not show clonal integration of proviral DNA (line 4 of Fig. 3a,b).

IPCR products from PBLs of one ATL patient with the smoldering type before and after separation using the magnetic cell sorting method are displayed. (a) Shows the electrophoretic pattern of IPCR products from PBLs of the patient with smoldering type ATL before and after separation using the magnetic cell sorting method. (b) Presents the results after Southern blot analysis of IPCR products from SF-25-positive and SF-25-negative cells. The samples used in each lane are as follows: lane 1, Raji (negative control); lane 2, smoldering ATL before separation; lane 3, SF-25 Ag-positive cells after separation; lane 4, SF-25 Ag-negative cells after separation; lane 5, water (negative control). ATL adult T-cell leukemia/lymphoma; IPCR inverse polymerase chain reaction.
Effect of the c-SF-25-PBL-sup on the proliferation of KUT-1 cells
PBLs from healthy participants were co-cultured for 24 h with paraformaldehyde-fixed KUT-1 cells (5 × 105 per 106 PBLs) in the presence of 1 µg/mL of c-SF-25 Mab or m-SF-25 Mab. The effect of the culture supernatants (c-SF-25-PBL-sup, m-SF-25-PBL-sup, or rituximab-PBL-sup) on the growth of KUT-1 cells was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. The representative results are shown in Fig. 2a. c-SF-25-PBL-sup from healthy volunteers strongly inhibits the growth of KUT-1 cells. In contrast, the m-SF-25-PBL-sup and rituximab-PBL-sup from the same healthy volunteers did not inhibit KUT-1 cell growth. Moreover, c-SF-25-PBL-sup in the absence of paraformaldehyde-fixed cells failed to inhibit KUT-1 cell growth (Fig. 4a). These results indicated that the inhibitory effect of the soluble factor(s) on cell growth was not related to direct antigen binding of c-SF-25 Mab to KUT-1 cells and was SF-25 Ag-specific.

The cytotoxic activity of c-SF-25-PBL-sup on KUT-1 cells. (a) Effect of the c-SF-25-PBL-sup on the proliferation of KUT-1 cells. Neutralization of anti-FasL Mab, anti-IFN-γ, TNF-α or the combinations of anti-FasL Mab, anti- IFN-γ, and TNF-α did not neutralize the cytotoxic activity of the c-SF-25-PBL-sup to KUT-1 cells. (b, c) The morphological changes in KUT-1 cells cultured in the presence of c-SF-25-PBL-sup derived from healthy participants. (d) DNA fragmentation was observed in the KUT-1 cells treated with mu-SF-25-PBL-sup. FasL Fas ligand; IFN-γ interferon–γ; TNF-α tumor necrosis factor-α.
Apoptosis of KUT-1 cells induced by c-SF-25-PBL-sup
Figure 4b,c show the morphological changes in KUT-1 cells cultured for 72 h in the presence of c-SF-25-PBL-sup derived from healthy participants. Nuclear condensation and fragmentation were observed in KUT-1 cells cultured with c-SF-25-PBL-sup (Fig. 4b) (Fig. 4c) but not in the control cell culture without c-SF-25-PBL-sup (Fig. 4c) (Fig. 4b). The fragmentation of DNA into oligosomal fragments results in a characteristic DNA ladder on agarose gel electrophoresis, which is often used as a biochemical marker for apoptosis. As shown in Fig. 4d, KUT-1 cells exhibited DNA laddering on agarose gel electrophoresis after treatment with c-SF-25-PBL-sup for 72 h (lane 3 in Fig. 4d). In contrast, no DNA fragmentation was observed in the KUT-1 cells treated with mu-SF-25-PBL-sup (lane 2, Fig. 4d).
Detection of tumor necrosis factor-α (TNF-α), interferon–γ (IFN-γ, and soluble Fas ligand (sFasL)
To investigate the soluble cytotoxic factors in c-SF-25-PBL-sup, the levels of TNF-α, IFN-γ, and sFasL in c-SF-25-PBL-sup were examined. As presented in Table 3, a significant amount of TNF-α, IFN-γ, and sFasL was detected in the c-SF-25-PBL-sup derived from healthy participants. In contrast, the levels of TNF-α, IFN-γ, and sFasL were significantly lower in m-SF-25-PBL-sup, prepared from the same healthy participants, compared with those in c-SF-25-PBL-sup (P < 0.001, t-test) (Table 3) and in Rituximab-PBL-sup (data not shown).
Effect of recombinant TNF-α, IFN-γ, and anti-Fas Mab on KUT-1 cells
The cytotoxicity of TNF-α and IFN-γ on KUT-1 cells was assessed using recombinant cytokines at a final concentration of 1000 pg/mL, a level similar to that found in c-SF-25-PBL-sup (Table 3). As shown in Fig. 4a, neither recombinant TNF-α nor IFN-γ had any effect on the growth of KUT-1 cells. In contrast, KUT-1 cells were sensitive to Fas-mediated apoptosis; their growth was significantly inhibited by anti-Fas Mab treatment (10 ng/mL) (data not shown). However, neutralization of anti-FasL Mab, anti-IFN-γ, or the combinations of anti-FasL Mab, anti- IFN-γ, and TNF-α did not neutralize the cytotoxic activity of the c-SF-25-PBL-sup to KUT-1 cells (Fig. 4a).
Discussion
In this study, the clonal integration of HTLV-1 proviral DNA (Fig. 1) and expression of SF-25 Ag, identified as SLC3A2, an amino acid transporter, on the surface of PBLs from patients with ATL, healthy HTLV-1 carriers (Fig. 2a,c), and an HTLV-1-infected T cell line were evaluated (Fig. 2b). This provides new insight into how SLC3A2 is involved in ATL pathogenesis by supporting cell survival and proliferation.
Because BCAAs are essential for maintaining Treg cell expansion through SLC3A2-mediated transport10, high SF-25 Ag (SLC3A2) expression in ATL cells may explain how these malignant cells sustain their proliferation and immune evasion by mimicking Treg metabolism. This may clarify why targeting SLC3A2 with c-SF-25 Mab could disrupt this metabolic advantage. Furthermore, the findings in this study demonstrated the feasibility of ATL treatment using ADCC activity with c-SF-25 Mab (Fig. 3).
The presence of abnormal lymphocytes is associated with a greater proportion of CD4+/CD25+cells, which is the predominant phenotype in ATL cells21. Moreover, persistent proliferation of HTLV-1-infected clones has been observed primarily in CD4+cells from asymptomatic carriers with a high provirus load22. In our experiments, SF-25-expressing cells also expressed CD4 (Fig. 2a and S1 Figure). The expressions of SF-25 Ag in patients with acute, chronic, and smoldering type ATL were significantly higher than those in HTLV-1 healthy carriers and healthy participants (Fig. 2c). The levels of SF-25 Ag expression in the PBLs of HTLV-1 healthy carriers were similarly low in those of healthy participants (Fig. 2c). If we setup the cutoff point for SF-25 Ag positivity at 1.8%, based on the 95th percentile of expression in HTLV-1 healthy carriers, all patients with acute ATL (11 cases), chronic ATL (five cases), and smoldering ATL (12 cases) were positive for SF-25 Ag (Table 2).
An intermediate state of HTLV-1 infection has also been proposed as a clinical condition between smoldering ATL and HTLV-1 carriers23. The number of HTLV-1-infected cells increased in these patients; however, the integration of HTLV-1 provirus was random, as detected by Southern blot analysis. IPCR facilitates the identification of oligoclonal proliferation of HTLV-1-infected cells in HTLV-1 carriers and intermediate states24. In this study, IPCR detected oligoclonal HTLV-1 integration in some carriers where Southern blot showed polyclonal patterns (Table 2), revealing how low-frequency clones expand before overt ATL develops. This suggests why IPCR combined with SF-25 Ag measurement could serve as an early marker for identifying carriers at risk of progression. These seven individuals were classified as intermediate-state participants. The levels of SF-25 Ag-expressing cells in PBLs from these carriers were 1.2% (0.4–1.9), which was slightly higher than were those in HTLV-1 healthy carriers and healthy participants; however, the difference was not statistically significant (P > 0.05). Nonetheless, two (28.6%) of the seven exhibited SF-25-Ag positive cell rates of 1.9% and 2.3%, respectively, which were higher than 1.8% (Table 2). These findings suggest that the intermediate type may be identified by assessing SF-25 expression in PBLs from HTLV-1 healthy carriers.
ATL is clinically divided into four subtypes: smoldering, chronic, lymphoma, and acute. Diagnosing chronic and acute ATL based on clinical and morphological characteristics is generally straightforward. However, confirming the monoclonal integration of HTLV-1 proviral DNA is the key for diagnosing smoldering-type ATL. Therefore, evaluating the expression of SF-25 Ag in PBLs from HTLV-1 carriers may provide a simple and effective diagnostic tool for ATL, including its smoldering variant. In this study, employing magnetic cell sorting techniques, SF-25 Ag-positive cell fractions in smoldering ATL showed monoclonal integration of HTLV-1 proviral DNA, whereas SF-25 Ag-negative cells did not, using negative selection methods (Fig. 4). These results endorse the expression of SF-25 Ag in ATL cells from patients with smoldering type, as well as those with acute and chronic types; the cell sorting with positive selection did not show effects on the IPCR methods of these sorted cells.
The present study demonstrated that SF-25 Ag was highly expressed in ATL cells and that c-SF-25-PBL-sup derived from healthy participants inhibited the growth of KUT-1, an HTLV-1-infected T-cell line (Fig. 4a), by activating apoptosis (Fig. 4b,c). The c-SF-25 Mab can induce ADCC in target cells that express the SF-25 Ag12. It is possible that c-SF-25 Mab activates PBLs in the presence of KUT-1 target cells and produces soluble factors that induce apoptosis in HTLV-1-infected T cells.
The cytotoxicity of c-SF-25-PBL-sup is not an experimental artifact, as stringent experimental steps were taken to negate the influence of lymphocyte stimulation by endotoxins, degradation products from target cells, or immune complexes formed with soluble antigens. First, all reagents were treated with an immobilized endotoxin affinity ligand (END-X endotoxin neutralization resin) to eliminate any potential endotoxin contamination. Second, immune complexes and aggregates were removed from the antibodies by ultracentrifugation in our experiments to prevent binding to the Fc receptor and subsequent cytokine production stimulation25. In addition, the monomeric nature of c-SF-25 Mab after ultracentrifugation was verified by autoradiography after iodination of the antibody with [125I] and sodium dodecyl sulfate–polyacrylamide gel electrophoresis under non-reducing conditions (data not shown). Third, the KUT-1 target cells were fixed to prevent degradation. Fixation of the tumor cells with 2% paraformaldehyde at 4 °C for 1 h effectively immobilized the cells but did not decrease the expression of SF-25 Ag on the tumor cell surface when they were examined by flow cytometry (data not shown). KUT-1 target cells were fixed under these conditions and cultured with PBLs (106 PBLs per 5 × 105 fixed KUT-1 cells) with 1 µg/mL of c-SF-25 Mab or m-SF-25 Mab that had been treated with END-X and ultracentrifugation. Furthermore, TNF-α, sFasL, or IFN-γ was not detected in m-SF-25-PBL-sup (Table 3). Our experimental system excluded the possibility that an activated complement was present in the supernatant. Moreover, the lack of cytotoxicity to KUT-1 cells in rituximab-PBL-sup is evidence that the killing is antigen-specific (Fig. 4a). The c-SF-25-PBL-sup contained at least three soluble cytotoxic molecules, including TNF-α, IFN-γ, and sFasL (Table 3). However, the cytotoxicity mediated by c-SF-25-PBL-sup was not blocked by anti-TNF-α, IFN-γ, or sFasL neutralizing antibodies or their combination (Fig. 4a). Therefore, the c-SF-25-PBL-sup may contain cytotoxic factor(s) that mediate apoptosis of ATL cells in non-TNF-α, non-IFN-γ, and non-Fas-receptor-mediated pathways. FasL exists in two forms: membrane-bound (mFasL) and soluble (sFasL)26. Furthermore, the apoptosis-inducing capacity of naturally processed sFasL reportedly reduces by more than 1000-fold compared with mFasL27. Notably, our finding that the cytotoxic activity of c-SF-25-PBL-sup containing sFasL was not inhibited by the neutralizing anti-FasL antibody (Fig. 4b) was consistent with the concept that sFasL is not an apoptosis-producing ligand under physiological conditions. The perforin-granzyme system is reportedly critical for the elimination of tumor cells, whereas the Fas pathway seems less important in tumor-bearing mouse models28,29. We must clarify some potential limitations in this study. The expression of SF-25 Ag is not tumor-specific; in this experiment, lymphocytes derived from healthy individuals are used as effector cells. The effects of in vivo when tumor immune tolerance is acquired and side effects of SF-25 Ag being expressed in normal Treg cells have not been fully tested.
The Fc domain of the mouse-human IgG1 c-SF-25 Mab may bind to FcγRI (CD64) on antigen-presenting cells, promoting cross-presentation of tumor antigens and enhancing Th1 immune responses30. This mechanism could be more effective than ADCC alone in eliminating malignant cells, suggesting that c-SF-25 Mab may exert anti-tumor effects through both direct cytotoxicity and activation of Th1-mediated immunity.
Despite these promising results, this study has limitations. The sample size was relatively small, which may affect generalizability, and the functional effects of c-SF-25 Mab were only evaluated in vitro. Further in vivo validation is essential to clarify how this approach would perform under immune tolerance and why off-target effects, especially on normal Treg cells that express SLC3A2, must be carefully assessed.
In summary, this study demonstrates how strong expression of SLC3A2 (SF-25 Ag) in ATL cells provides a rationale for targeted therapy. It shows why c-SF-25 Mab could be a novel strategy for eliminating ATL cells through multiple cytotoxic mechanisms, including ADCC, bystander apoptosis, and potentially Th1 activation. These findings offer a promising perspective for future therapies targeting the amino acid transporter SLC3A2 in ATL.
Methods
Patients
Between April 2000 and April 2003, 28 patients with ATL and 52 healthy HTLV-1 carriers were evaluated. ATL diagnosis was based on clinical and hematological data; presence of anti-HTLV-1 antibody, and monoclonal integration of HTLV-1 proviral DNA detected by the Southern blot method. Eight healthy participants without HTLV-1 infection were included as controls. The characteristics of the patients and healthy participants are presented in Table 1.
Clinical sample collection
PBLs were isolated by density gradient centrifugation using sodium diatrizoate polysaccharide (Axis-Sheld PoC AS, Oslo, Norway), and genomic DNAs were extracted from the PBLs from patients with ATL, HTLV-1 healthy carriers, and healthy participants using DNAzol (Molecular Research Center, Inc., Montgomery Rd., Cincinnati, OH., U.S.A.). This study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee and Institutional Review Board of Kagoshima University (240100EKI) and of National Hospital Organization Kagoshima Medical Center (2024-COI-8). All patients provided informed consent.
IPCR of HTLV-1
IPCR methods were used for detecting the integration of HTLV-1 proviral DNA into PBLs from HTLV-1 carriers. The details of these methods have been previously reported21. Briefly, one microgram of genomic DNA was digested with SauAI (Takara, Kyoto, Japan) and then ligated with T4 DNA ligase (Takara) to cause self-ligation. The ligated DNA was then digested with Sac II (TOYOBO CO, LTD, Osaka, Japan) to eliminate circular DNA derived from the 5′ proviral DNA. DNA was used as a template. In the first step of the PCR, primer 1:5′-AAGCCGGCAGTCAGTCGTGA-3′ and primer 2:5′-AAGTACCGGCAACTCTGCTG-3′ were used at a final concentration of 200 nmol/L. Five microliters of PCR product were then performed with the nested primer 3:5′-GAAAGGGAAAGGGGTGGAAC-3′ and primer 4:5′-CCAGCGACAGCCCATTCTAT-3′. After PCR amplification, the products were analyzed using Southern blotting. Subsequently, they were electrophoresed on a 2% agarose gel and transferred onto nylon membrane. Dig-labeled oligonucleotide (5′-CTCCAGGAGAGAAATTTAGTACAC-3′; residues 9012-9035 of HTLV-1, 50μL (100 pmol/μL) was used as a probe for hybridization at 42 °C for 12 h to confirm specific band. The hybridized probe was immunodetected with anti-digoxigenin-AP and Fab fragments (Roche Diagnostics GmbH, Mannheim, Germany) and visualized using the chemiluminescent substrate CDP-Star (Applied Biosystems, Bedford, MA., U.S.A.). The blots were imaged by placing them in contact with a standard X-ray film.
Flow cytometric analysis
The expression of both SF-25 Ag and CD4 in PBLs was determined by flow cytometry using FITC)-conjugated m-SF-25 mAb and PE-conjugated anti-CD4 mAb (Coulter, Marseille, France). FITC- and PE-conjugated murine IgG1 were used as isotype-matched nonspecific control antibodies (Coulter). NK cells were detected by PE-conjugated anti-CD16 Mab (Coulter, Hialeah, FL., U.S.A.). Regarding the flow cytometric analysis, PBLs were stained with a saturated concentration of the Mabs for 30 min at 4 °C using saturated concentrations of the Mabs. The stained cells were washed with cold phosphate-buffered saline (PBS) and immediately examined using a flow cytometer (EPICS XL™, Coulter, Miami, FL, U.S.A.). The results are expressed as a percentage of the positive cells in the whole cell population.
Magnetic cell sorting method
A total of 107 PBLs were suspended in 60 μL of PBS containing 2 mM EDTA and 0.5% bovine serum albumin (pH 7.2). Subsequently, 20 μL of FcR Blocking Reagent (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) was added to these PBLs to block non-specific binding to monocytes. The cells were stained with 10 μL of FITC-conjugated m-SF-25 Mab and 10 μL of PE-conjugated anti-CD4 Mab for 5 min at 4 °C. The cells were then washed twice, resuspended in 90 μL, and 10 μL of Anti-FITC Microbeads (Miltenyi Biotec GmbH) were added. The cells were incubated for 15 min at 6 °C, followed by another wash. Positive selection columns MS + (Miltenyi Biotec GmbH) and a Mini & MidiMACS Starting Kit (Miltenyi Biotec GmbH) were used to separate the SF-25 Ag-expressing cells from non-expressing cells. Subsequently, HTLV-1 integration into these two cell populations was investigated using IPCR.
Cell line
KUT-1 (JCRB Cell Bank, NIHS1729)31 and MT-1 (JCRB Cell Bank, JCRB0610), which are HTLV-1-infected T cell lines, were established from T cells in the peripheral blood of a patient with ATL. Raji (JCRB Cell Bank, JCRB0072), an HTLV-1 non-infected B-lymphoblast-like cell line established from Burkitt’s lymphoma32, was used as a negative control. The cell line was maintained in RPMI-1640 (Life Technologies, Rockville, MD., U.S.A.) supplemented with 10% heat-inactivated fetal calf serum (FCS) (Advanced Biotechnologies, Silver Spring, MD., U.S.A.), 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin at 37° C in 95% air, 5% carbon dioxide, and 90% humidity.
Preparation of culture supernatant
KUT-1 cells were first fixed with 2.0% paraformaldehyde for 2 h at 4 °C and washed three times with endotoxin-free saline. Fixation of KUT-1 cells under these conditions effectively immobilized the cells but did not decrease the expression of SF-25 Ag on the tumor cell surface when examined by radioimmunometric binding assay and flow cytometry (data not shown). PBLs from two healthy volunteers were separated by density gradient centrifugation using Ficoll-Paque Plus. PBLs (1 × 106 cells/mL) were co-cultured in 24-well plates (Falcon, Warren, N.J., U.S.A.) with 1 mL of RPMI-1640 supplemented with 10% heat-inactivated FCS. This culture was maintained for 24 h alongside paraformaldehyde-fixed KUT-1 cells (5 × 106 cells/mL) in the presence of 1 µg/mL of c-SF-25 Mab and m-SF-25 Mab, which had been treated overnight at 4 °C with an immobilized endotoxin affinity ligand (END-X; Associates of Cape Cod INC. Woods Hole, MA., U.S.A.).
To determine whether the production of apoptotic factors was antigen-specific, we used another isotype-matched humanized antibody, rituximab (chimeric anti-CD20 Mab containing human IgG1 and constant regions with murine variable regions) (Zenyaku Kogyo, Tokyo, Japan), as a control because KUT-1 cells do not express CD20 (data not shown). The culture supernatants (c-SF-25-PBL-sup, m-SF-25-PBL-sup, and Rituximab-PBL-sup) were collected by centrifugation at 1600 rpm for 10 min and stored at − 85 °C so that the supernatants were cell-free and activated complement-free.
Enzyme-linked immunosorbent assay (ELISA)
TNF-α, IFN-γ, and sFasL in the supernatant were determined by commercially available ELISA kits (TNF- α, IFN-γ; Biosource, Camarillo, CA., USA. and sFasL, MBL, Nagoya, Japan).
MTT assay
The cytotoxic activity of c-SF-25-PBL-sup on KUT-1 cells was quantitatively determined using the MTT assay33. KUT-1 cells (2 × 104) were cultured for 72 h in a 96-flat bottom-well plate (Falcon) with 100 µL of c-SF-25-PBL-sup containing 10% fresh FCS. At the end of the culture period, MTT stock solution (2.5 mg/mL in H2O) was added to 96-well culture plates (10 µL per well) and incubated at 37 °C for 4 h. After removing the culture supernatant by centrifugation, the blue formazan product formed in the living cells by oxidation was solubilized by acid-isopropanol (0.04N HCl in isopropanol) and the absorbance of each well was measured using a microplate reader at 590 nm.
The MTT assay was also performed in the presence of neutralizing anti-human TNF-α rabbit polyclonal antibody (Genzyme, Cambridge, MA., U.S.A.), anti-human IFN-γ polyclonal antibody (PeproTech, Rocky Hill, N.J., U.S.A.), or anti-FasL Mab (clone 4H9; MBL) to assess the roles of TNF-α, IFN-γ, and sFasL in the c-SF-25-PBL-sup. The final concentrations of anti-TNF-α, anti-IFN-γ, and anti-FasL antibodies were 1:1,000 fold dilution, 5 µg/mL, and 1 µg/mL, respectively.
Detection of apoptosis
After incubating KUT-1 cells with c-SF-25-PBL-sup, DNA fragmentation was examined by agarose gel electrophoresis. First, DNA was extracted from KUT-1 using a commercially available DNA extraction kit (Wako, Osaka, Japan), and the extracted DNA was subjected to 2.0% agarose gel electrophoresis. Fragmented DNA was visualized by staining the gel with 1.0% ethidium bromide. KUT-1 cell apoptosis was examined morphologically after May-Giemsa staining.
Statistical analysis
Statistical significance was determined using the Mann–Whitney U test. Results were considered significant at P < 0.05.
Data availability
Data is provided within the manuscript files.
Change history
01 October 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41598-025-21843-4
References
Yano, H. et al. Regulatory T-cell function of adult T-cell leukemia/lymphoma cells. Int. J. Cancer 120, 2052–2057 (2007).
Proietti, F. A., Carneiro-Proietti, A. B. F., Catalan-Soares, B. C. & Murphy, E. L. Global epidemiology of HTLV-I infection and associated diseases. Oncogene 24, 6058–6068 (2005).
Hanada, S. et al. The prevalence of human T-cell leukemia virus type I infection in patients with hematologic and nonhematologic diseases in an adult T-cell leukemia-endemic area of Japan. Cancer 64, 1290–1295 (1989).
Shimoyama, M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. A report from the Lymphoma Study Group (1984–87). Br. J. Haematol. 79, 428–437 (1991).
Iqbal, M. et al. Efficacy of allogeneic hematopoietic cell transplantation in human T cell lymphotropic virus type 1-associated adult T cell leukemia/lymphoma: Results of a systematic review/meta-analysis. Biol. Blood Marrow Transplant. 25, 1695–1700 (2019).
Ishitsuka, K. & Tamura, K. Human T-cell leukaemia virus type I and adult T-cell leukaemia-lymphoma. Lancet Oncol. 15, e517-526 (2014).
Choi, I. et al. A phase 2 trial of CHOP with anti-CCR4 antibody mogamulizumab for elderly patients with CCR4-positive adult T-cell leukemia/lymphoma. J. Clin. Oncol. 41, 7504–7504 (2023).
Takahashi, H. et al. In vivo localization of human colon adenocarcinoma by monoclonal antibody binding to a highly expressed cell surface antigen. Cancer Res. 48, 6573–6579 (1988).
Pellizzari, G. et al. Immunotherapy using IgE or CAR T cells for cancers expressing the tumor antigen SLC3A2. J. Immunother. Cancer 9, e002140 (2021).
Ikeda, K. et al. SLC3A2 mediates branched-chain amino-acid-dependent maintenance of regulatory T cells. Cell Rep. 21, 1824–1838 (2017).
Stanciu, L. A. & Djukanović, R. T Cell Protocols: Development and Activation (ed. Kearse, K. P.) 133–141 (Humana Press, 2000).
Takahashi, H., Nakada, T., Nakaki, M. & Wands, J. R. Inhibition of hepatic metastases of human colon cancer in nude mice by a chimeric SF-25 monoclonal antibody. Gastroenterology 108, 172–182 (1995).
Takahashi, H. et al. Radioimmunolocation of hepatic and pulmonary metastasis of human colon adenocarcinoma. Gastroenterology 96, 1317–1329 (1989).
Yarnold, S. & Fell, H. P. Chimerization of antitumor antibodies via homologous recombination conversion vectors. Cancer Res. 54, 506–512 (1994).
LoBuglio, A. F. et al. Mouse/human chimeric monoclonal antibody in man: kinetics and immune response. Proc. Natl. Acad. Sci. U S A 86, 4220–4224 (1989).
Khazaeli, M. B. et al. Pharmacokinetics and immune response of 131I-chimeric mouse/human B72.3 (human gamma 4) monoclonal antibody in humans. Cancer Res. 51, 5461–5466 (1991).
Hale, G. et al. Remission induction in non-Hodgkin lymphoma with reshaped human monoclonal antibody CAMPATH-1H. Lancet 2, 1394–1399 (1988).
Coiffier, B. et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med. 346, 235–242 (2002).
Ravetch, J. V. & Kinet, J. P. Fc receptors. Annu. Rev. Immunol. 9, 457–492 (1991).
Takahashi, H., Nakada, T. & Puisieux, I. Inhibition of human colon cancer growth by antibody-directed human LAK cells in SCID mice. Science 259, 1460–1463 (1993).
Welles, S. L. et al. The distribution of T-cell subsets among HTLV-I carriers in Japan. J. Acquir. Immune. Defic. Syndr. 1988(7), 509–516 (1994).
Etoh, K. et al. Persistent clonal proliferation of human T-lymphotropic virus type I-infected cells in vivo. Cancer Res. 57, 4862–4867 (1997).
Yamaguchi, K. et al. Polyclonal integration of HTLV-I proviral DNA in lymphocytes from HTLV-I seropositive individuals: An intermediate state between the healthy carrier state and smouldering ATL. Br. J. Haematol. 68, 169–174 (1988).
Takemoto, S., Matsuoka, M., Yamaguchi, K. & Takatsuki, K. A novel diagnostic method of adult T-cell leukemia: Monoclonal integration of human T-cell lymphotropic virus type I provirus DNA detected by inverse polymerase chain reaction. Blood 84, 3080–3085 (1994).
Debets, J. M., Ruers, T. J., van der Linden, M. P., van der Linden, C. J. & Buurman, W. A. Inhibitory effect of corticosteroids on the secretion of tumour necrosis factor (TNF) by monocytes is dependent on the stimulus inducing TNF synthesis. Clin. Exp. Immunol. 78, 224–229 (1989).
Tanaka, M., Suda, T., Takahashi, T. & Nagata, S. Expression of the functional soluble form of human fas ligand in activated lymphocytes. Embo. J. 14, 1129–1135 (1995).
Schneider, P. et al. Conversion of membrane-bound Fas(CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J. Exp. Med. 187, 1205–1213 (1998).
Sayers, T. J. et al. Molecular mechanisms of immune-mediated lysis of murine renal cancer: Differential contributions of perforin-dependent versus Fas-mediated pathways in lysis by NK and T cells. J. Immunol. 161, 3957–3965 (1998).
Smyth, M. J. et al. Perforin is a major contributor to NK cell control of tumor metastasis. J. Immunol. 162, 6658–6662 (1999).
Soleimanpour, S., Hassannia, T., Motiee, M., Amini, A. A. & Rezaee, S. A. R. Fcγ1 fragment of IgG1 as a powerful affinity tag in recombinant Fc-fusion proteins: Immunological, biochemical and therapeutic properties. Crit. Rev. Biotechnol. 37, 371–392 (2016).
Hanada, S., Tsubai, F. & Namba, Y. The characteristics of T-cell lines derived from peripheral blood of patients with adult T-cell leukemia-lymphoma. Recent Adv. Res. 25, 124–135 (1985).
Pulvertaft, J. V. Cytology of burkitt’s tumour (AFRICAN LYMPHOMA). Lancet 1, 238–240 (1964).
Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63 (1983).
Acknowledgements
This work was supported by a JSPS KAKENHI grant (Grant number: 19K08870 to S.S.) for Clinical Research (Grant-in-Aid for Scientific Research) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Author information
Authors and Affiliations
Contributions
SS, XL, and SH designed and performed the experiments, analyzed the data, and wrote and revised the manuscript. KU, MO, SO, KH, MY, KI, and HT analyzed the data and wrote and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained errors in Figure 2. Full information regarding the corrections made can be found in the correction for this Article.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Suzuki, S., Lin, XY., Hanada, S. et al. Chimeric SF-25 monoclonal antibody as a targeted therapy for SLC3A2 in adult T-cell leukemia/lymphoma. Sci Rep 15, 29379 (2025). https://doi.org/10.1038/s41598-025-14572-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-14572-1
