
Abstract
Indigenous cattle breeds in Asia are highly adapted to their local environments providing essential commodities such as meat, milk and draught power while also playing a key role in traditional ceremonies, and sports. Despite ongoing efforts to characterize and conserve these breeds, the increasing trend of indiscriminate crossbreeding of Zebu cattle with high-yielding taurine breeds, threatens their genetic diversity. This study investigates the population structure, inbreeding levels, effective population size, gene flow and identification of selection footprints of Asian Zebu (Bos indicus) cattle. Using an Axiom 60 K SNP chip, we analyzed genotypes from 1303 cattle across 36 populations in nine countries, including seven taurine outgroups and 29 Zebu populations from Bangladesh, Cambodia, India, Myanmar, Pakistan, and Sri Lanka. Zebu populations demonstrated moderate genetic diversity, with heterozygosity levels averaging 0.356, inbreeding coefficients ranging from 0.026 to 0.074 and genetic differentiation (FST) varied between 0.01 and 0.11. Breed clusters aligned closely with their geographic locations except for Achai (Pakistan) and Baru Harak (Sri Lanka) breeds that appeared in both Zebu and taurine clusters indicating evidence of taurine admixture. Genomic analyses identified regions under selection using extended haplotype homozygosity (EHH) and fixation index (FST) methods. Candidate genes associated with key biological functions related to environmental responsiveness, including heat tolerance (HSP90AA1), immunity (RIPK3), metabolism and fertility (REC8, CLIC4, TSSK4), were identified, reflecting adaptive traits critical for Zebu survival and utility across diverse environments. These findings provide valuable insights for conservation and management strategies aimed at preserving the unique genetic diversity of Asian Bos indicus breeds.
Similar content being viewed by others


Introduction
Cattle contribute significantly to agricultural economies of many South Asian countries by serving as sources of food products such as milk and meat, draught power for agricultural operations and rural transportation, cultural and recreational traditions of local communities. Bos indicus, also known as Zebu cattle, originated from the Indus Valley, and later spread to Africa and southeast Asia1. More than half of the cattle population worldwide are Zebu cattle, primarily found in Asia due to their economic significance and ability to thrive in different ecological settings and prevailing production systems. Zebu cattle are well adapted to a wide range of climatic conditions ranging from arid and semi-arid to tropical areas of Asia and Africa. Based on their primary utility, Zebu cattle are mainly classified as draught (transport and ploughing), dairy and dual-purpose (milk/meat or draught/meat) cattle. Most of the Zebu breeds in Asia are raised for draught and meat except for a few breeds that are specialized for milk production. Phenotypically Zebu cattle are distinguished by their horns and prominent hump with excess skin around neck and dewlap. They are renowned for their heat tolerance, resistance or lower susceptibility to various diseases, and low nutritional requirements allowing them to survive on coarse feed and crop residues available in tropical and sub-tropical regions2.
Asian Zebu have significant phenotypic variations across breeds ranging from massive animals of Kankrej and Bhagnari to dwarf cattle like Vechur and Punganur3. India holds about 13.1% of the global cattle population, making it the country with highest Zebu count in the world. Out of the total cattle population of 193.17 million, 73.5% are indigenous cattle while the remaining 26.5% are either exotic purebreds or crossbreds. Out of the indigenous cattle population, only 29.5% fall within one of the 40 or more registered breeds4. North and North-West India holds the greater part of dairy and dual-purpose Zebu breeds while the South and South-eastern India mostly holds the draught breeds. Physical characteristics of draught breeds are typically moderate to large size, prominent hump, uniform coat color and phenotypic characteristics such as good load pulling capacity, speed, and endurance (Alambadi, Bargur, Deoni, Hallikar, Kangayam, Ongole, Pulikulam and Umbalacheri). Within the Asian region, aside from India, Pakistan and Bangladesh have the third and fourth largest cattle populations with 51.56 and 24.55 million heads respectively5. Pakistan has approximately 20 registered cattle breeds, some of which are shared with India as transboundary breeds located on both sides of the national border. Many Zebu breeds in Pakistan are dairy type including Red Sindhi, Cholistani, Sahiwal and Tharparkar, which are also known for their resistance to tropical diseases or vectors and tolerance to high temperatures. The draught breeds of cattle from the hot mountainous and dry regions of Pakistan include Bhagnari, Dajal, Dhani and Achai characterized as relatively compact draught breeds6,7.
Myanmar, Mongolia, Cambodia, and Sri Lanka possess 10.3, 5.0, 2.7 and 1.1 million cattle respectively5. The indigenous Zebu cattle breeds represent more than 50% of Sri Lanka’s cattle population, although the dairy industry has experienced high rates of crossbreeding in the past few decades leading to the genetic dilution of local breeds. Zebu breeds in Sri Lanka include the Batu Harak characterized by less prominent hump and primarily raised in the country’s dry region. Thamankaduwa, also known as White Cattle is distinguished by its prominent hump and white coat color. The Thawalam cattle found in the Central and Uva provinces have distinct body conformation and exceptional ability to pull heavy loads in hilly terrains8. Myanmar local cattle breeds are characterized as draught type that includes Pyar Zein, Shwe Ni Gyi and Shwe Ni. These cattle are raised in the central regions of Mandalay, Sagaing and Magway9,10. Cambodia has two native Zebu cattle breeds characterized as red or white cattle namely Kdarm Red or Kdarm White11. Zebu cattle predominantly inhabit South Asian and Southeast Asian countries. In contrast East and Central Asia lack significant populations of Zebu cattle but have their own region-specific taurine breeds alongside the commercial taurine cattle such as Holstein, Jersey, Brown Swiss, and Ayrshire. For instance, Mongolia features local taurine breeds like Dadal and Mongol cattle which are dual purpose type native to the eastern region of the country. The inclusion of the taurine breeds (both European and Asian) in studies of Zebu cattle provides valuable insights into taurine introgressions and admixture levels. This is particularly relevant in the context of ongoing crossbreeding programs aimed at improving dairy production in the region.
The majority of the Asian cattle are kept and used by small-holder farmers, and a significant proportion of the Asian Zebu cattle population are yet to be genetically characterized, remaining classified as non-descript cattle. In the last decades, small-holder farmers have been aiming to increase the milk and meat productivity of their cattle leading to widespread crossbreeding with high-producing taurine breeds. While this approach has improved productivity, it has raised concerns on genetic erosion of the locally adapted indigenous cattle breeds before their unique traits and selection footprints could be studied. In South Asian studies on Zebu cattle, genetic diversity is limited and often rely on small number of microsatellites, mitochondrial DNA, small single nucleotide polymorphisms panels or a restricted set of breeds from specific regions. However, the advent of cost-effective high-throughput genotyping assays over the past decade has enabled large-scale studies on genetic diversity. These studies can provide valuable insights into indigenous populations, shedding light on their breeding systems and informing strategies for their conservation and sustainable use. Additionally, analysis of markers at the genome level can identify associations with local adaptation to diverse climatic conditions and traits such as draughtability, milk production and stature12. The knowledge of the candidate genomic regions gives direction in the design of future breeding strategies tailored to the production objectives of small holder farmers in the region. This study aims to investigate the genetic structure of 29 Asian Zebu breeds from seven countries alongside five taurine outgroup populations and two local taurine populations from Mongolia. The study also explores population relationships, inbreeding levels, effective population size, gene flow and selection signatures using genome-wide SNP data.
Materials and methods
Animal ethics statement
Blood samples were collected by jugular venipuncture into EDTA vacutainer tubes. Blood sampling was performed by qualified veterinarians in the respective native breed tracts of different countries following the standard guidelines and good animal handling practice, after due approval from respective collaborating institutions located in India, Pakistan, Sri Lanka, Bangladesh, Cambodia and Myanmar. The blood samples were collected as part of routine veterinary surveillance and informed consent was obtained from animal owners for the usage of a part of those samples for DNA extraction and analysis in this study. Therefore, no further license/ethical approval was required for the study. However, all efforts were made to ensure ethical and safe handling of the animal subjects during the process of sample collection.
Sample collection and genomic-DNA extraction
A total of 994 Zebu cattle belonging to 29 native breeds located across six countries, India, Pakistan, Sri Lanka, Bangladesh, Myanmar, and Cambodia were utilized for this study. Additionally, 309 cattle from four European (Holstein, Jersey, Ayrshire, and Brown Swiss) and two Mongolian taurine breeds were used for comparison and admixture analysis. The list for Zebu and taurine cattle breeds, breed code and their country of origin are presented in Table 1. The characteristic features and representative pictures of Zebu cattle breeds investigated in the study are presented in Supplementary Table ST1 (S1file). Briefly, a stratified random sampling approach was followed covering representative, true to the type animals located in different regions of the native breed tract. With the absence of pedigree records in most instances, unrelatedness was ensured by interviewing the farmers about the breeding history. Genomic DNA was extracted by (i) standard phenol chloroform method (cattle samples from India) and (ii) MasterPure DNA Purification Kit (Biozym, Illumina Inc, USA) (cattle samples from Bangladesh, Pakistan, Sri Lanka, Myanmar, and Cambodia). The DNA samples were subjected to two step quality control procedures using Nanodrop 2000 spectrophotometer (ThermoScientific, USA) initially to estimate concentration, 260/280 and 260/230 ratios followed by Quant-iT™ PicoGreen™ assay (Invitrogen, ThermoFisher Scientific, USA) to ascertain the quantity of good quality double strand DNA. DNA samples were then stored at -20 °C until further processing.
SNP genotyping and data quality control
All the cattle samples were genotyped on Affymetrix-Axiom platform using Axiom Bovine Genotyping BovMDv3 array that consisted of 63,655 SNPs (single nucleotide polymorphism markers) (Fig. 1). The genotype data was extracted from .cel files following the Best Practices Workflow as implemented in Axiom Analysis Suite version 5.1.1. Downstream quality control was done in PLINK v1.9 and PLINK v2 with all SNPs assigned to mitochondrial chromosomes, X and Y being excluded from the analysis leaving SNPs located in the 29 autosomes for further analysis13,14. In addition, removal of SNPs that were monomorphic, having minor allele frequency (MAF) lower than 0.01, and SNPs that failed the Hardy Weinberg Equilibrium (HWE) test (p < 10–6) was done. The data was filtered for linkage disequilibrium (LD) with –indep-pairwise 50 5 0.2, call which removes SNPs with squared correlation coefficient between two alleles of two SNPs (r2) higher than 0.2 within 50-SNP sliding windows with each window overlapping by 5 SNPs in PLINK v1.9. Individuals with a call rate lower than 0.95 and with relationships > 0.354 were also removed using the –king-cut-off parameter in PLINK v2. Therefore, after performing quality control, a total of 21,737 variants were kept for further analysis. A separate quality control approach was followed for effective population size (Ne) estimation, runs of homozygosity (ROH) and detection of selection signatures. Genotype data was pruned using –geno 0.1, –mind 0.05 and –king-cut-off 0.354 to remove SNPs with genotyping call rate less than 0.90, individuals with genotyping call rate less than 95% and duplicate animals or twins as implemented in PLINK (v1.9, v2). A set of 1304 animals and 43,271 SNPs were retained for use in runs of homozygosity, effective population size, FST and extended haplotype homozygosity estimations.

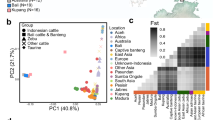
Location of the Asian Zebu samples from Bangladesh, Cambodia, India, Sri Lanka, Pakistan, Myanmar.
Genetic diversity and population structure
The genomic diversity within and between populations was estimated using statistics such as observed heterozygosity (Ho), expected heterozygosity (He) and global fixation index (FST) using the HierFstat R package13,15. Nei (1987) defined the fixation index as a measure of genetic differentiation between populations, that quantifies the differences in allele frequencies between populations. To characterize the genetic differences between the investigated cattle breeds, pairwise FST values (Weir and Cockerham, 1984) were calculated using R. All the results from genetic diversity and differentiation analysis were further visualized using custom R scripts16. The structure of the Asian Zebu cattle and genomic relationship between the populations was investigated using Principal Component Analysis (PCA) as implemented in PLINK v1.9. The –pca function in the software reduces dimension of the data using variance-standardized relationship matrix and classifies samples based on principal components whose eigenvalues represent a fraction of the total genetic variability in the dataset. The results from the function were visualized using the ggplot package in R (https://cran.r-project.org/web/packages/ggplot2/index.html). In addition to the PCA, population structure was investigated using ADMIXTURE v1.3.0 to determine the Asian Zebu ancestries at whole genome level assuming ancestral populations (K values) ranging from 2 to 30 and the optimal K value being the one with the lowest cross validation error (Supplementary Figure SF2 in Supplementary Information S1 File)17. The results of population structure analysis were visualized using R. Population splits and gene flow inference was investigated using the TREEMIX v1.018. The treemix model assumed no linkage among SNPs, parameter K -1000 was set to allow resampling of SNPs. Migrations ranging from 1 to 10 were investigated with root population based on the highest pairwise FST among populations. To further investigate population differentiation Reynolds genetic distance was calculated using a custom script in Ubuntu v20.04 and the pairwise distance matrix was used to construct a Neighbournet tree using SPLITSTREE4 v4.19.119,20.
Effective population size
Two approaches were used to provide effective population size estimates, GONE21 generates recent historical estimates based on LD. The availability of abundant numbers of markers and their location across the genome ensures that Ne analysis using GONE gives a clear picture of historical trends of effective population size. We used the default parameters except for chromosomes which we specified according to species as the software can accommodate up to 200. The other default parameters used were unknown phase, centiMorgans per Megabase with unknown genetic distance between markers, maximum generations for which LD is obtained of 2000, 400 number of bins with 5 generation gaps and maximum recombination rate of 0.05. The other approach was to estimate contemporary effective population size using CurrentNe22. The software has multiple advantages including the ability to include large number of SNPs up to 2 million in one analysis, it does not consider minor allele frequencies, accounts for complex mating systems, small sample sizes and uses less computational power. The CurrentNe estimates using artificial neural networks gives confidence interval for results based on integration over the whole genome as well as LD between chromosomes. The historical trends and contemporary estimation of Ne were visualized using R.
Runs of homozygosity
Runs of homozygosity were detected using cgaTOH Analysis and Clustering Suite v1.0123 identifying genome segments that are homozygous spanning over at least 15 SNPs, classified into several categories of minimum length of genome sequence (1 Mb, 2 Mb, 4 Mb, 8 Mb and 16 Mb). No missing or heterozygous SNPs were allowed within the 1 Mb and 2 Mb length categories, one missing and no heterozygous SNPs were set for ROH with minimum length of 4 Mb, two missing and no heterozygous SNPs were set for ROH with minimum length of 8 Mb, four missing and one heterozygous SNPs were set for ROH with minimum length of 16 Mb. Subsequently all the ROH had to have at least 15 consecutive SNPs and no gaps larger than 1 Mb. The average, minimum and maximum length of ROH was calculated globally for each population. Individual inbreeding coefficients (FROH) values were estimated for each animal as the sum of all ROH divided by the genome length24.
Identification of selection signatures
Asian Zebu cattle breeds belong to three major types based on their utility (i) draught (ii) dairy and (iii) dual purpose types. Draught cattle are used for mostly agricultural operations, transport and for cultural sport whilst other breeds are kept mainly for milk production. Identifying selection signatures within and between the dairy type (PRS, PSH, PTP) and draught type (IHL, IKA, ION) cattle is important to identify genomic regions that were selected for in the populations. In addition, Asia houses some of the world’s smallest cattle breeds which are of Zebu origin. Therefore, identifying selection signatures between large size (IHL, IKA) and small Zebu breeds (IPN, IVE) is of interest to investigate the genomic regions responsible for such a difference in stature. Signatures of selection were investigated using extended haplotype homozygosity statistics (EHH) and FST. The EHH method first identifies a set of SNPs in a small region such that recombination may not occur and later evaluates identified SNPs at increasing distances from the core haplotypes for the decay of LD25. The probability of two chromosomes carrying a specific core homozygous haplotype (EHH) is calculated and compared to all other core haplotypes combined. When one core haplotype has both high relative EHH and higher frequency in the population, it is suggestive of positive selection. Differential selection identification was investigated using cross population extended haplotype homozygosity (XP-EHH) using the rehh package in R with default parameters26. The EHH statistics required haplotype data and SHAPEIT was used to phase the genotyped SNPs into matching haplotypes27. With the classified draught and milk breeds, statistics were calculated for SNPs that indicated a within-population MAF of minimum 0.05. For a region to be classified under selection it had to contain at least three SNPs, not separated by more than 1000 kb that were above − log10 (p-value = 4) significance threshold. The candidate regions of selection were characterized based on ARS-UCD1.2 bovine reference genome using the ENSEMBL genome browser (https://www.ensembl.org/). Inferred functions and phenotypes influenced by genes were drawn from functional enrichment analysis using Database for Annotation, Visualization, Integrated Discovery (DAVID) in conjunction with literature search28. Significant enriched Gene Ontology (GO) biological processes and Kyoto Encyclopedia of Gene and Genomes (KEGG) pathway were defined by a threshold p-value = 0.0529.
The FST analysis was based on two categories of breeds defined according to their type and stature (i) draught vs dairy breeds, and (ii) large-stature vs small stature breeds (Table 2). The pairwise FST was computed, ranked, and used to identify regions under positive selection. The genome-wide significance level was set to 0.005, filtering only the top 0.5% FST values. The identified signatures of selection were characterized based on the ARS-UCD2.0 bovine reference genome using the NCBI ENSEMBL genome browser (https://www.ensembl.org/). Inferred functions and phenotypes influenced by genes were drawn from functional enrichment analysis using DAVID in conjunction with literature search. Significantly enriched GO terms and KEGG pathways were defined by a threshold p-value of 0.05.
Results
Genetic diversity
SNP ascertainment bias arises from the deviation of population genetic statistics, and it can be a result of biased SNP discovery protocols. The SNP ascertainment panel of commercial bovine SNP arrays consisted of predominantly taurine breeds and one or few Zebu breeds. This results in over representation of variability present in taurine populations and under representation of variability in Zebu breeds. In the present study, after initial data quality control measures, the observed heterozygosity of most Zebu cattle breeds was relatively low as compared to commercial European taurine breeds indicating potential bias (Fig. 2). The SNP ascertainment panel used for the design and validation of Axiom BovMD v3 array had 17 taurine breeds but only three belonging to Zebu cattle. The overestimation of heterozygosity statistics for the ascertained SNP set which was higher for populations involved in the discovery process was observed. SNP ascertainment bias can be mitigated by adopting SNP filtering strategies such as LD based pruning. This approach reduces multicollinearity effects by removing SNP markers that are highly correlated with other marker loci in a given window thus reducing the LD among markers left after pruning2,30,31. When the LD threshold for data pruning was set at LD < 0.2, the divergence of heterozygosity statistics between taurine and Zebu cattle breeds narrowed down as illustrated in Fig. 2. The resulting dataset of 21,737 SNPs was used for subsequent estimations of basic diversity statistics, genetic distance, fixation index and principal component analysis. The observed heterozygosity ranged from 0.209 (BGL) to 0.373 (IPN) while the expected heterozygosity varied between 0.316 and 0.349 (IPN) across different Zebu cattle populations (Table 3). These estimates are comparable to diversity measures reported on seven Zebu cattle breeds located in North and North-Western parts of India32. Pairwise FST indicated that most Zebu breeds had low differentiation levels (Fig. 3), with the lowest differentiation of 0.01 found among Cambodian breeds (KDR, KDW) and Myanmar breeds (YSG, YSN, YPZ). The highest pairwise differentiation (0.39) was observed between Jersey and Zebu breeds such as Kangayam, Cholistani and Tharparkar. The highest pairwise difference among the Zebu breeds was 0.11 observed between IKA and PCH- IVE.

Summary statistics on heterozygosity for Asian Zebu and taurine populations (Breed abbreviations Table 1).

Pairwise FST values based on 33,725 SNPs in 36 Zebu and taurine populations from Asia, America, and Europe. (Breed abbreviations Table 1).
Population structure
The first principal component (PC1) of the overall PCA that included both taurine and Zebu breeds explained 23.75% variation while the second principal component (PC2) explained 22.68% of the total variation in the dataset. The results showed the distinct divergence of Zebu and taurine cattle as expected. Within the taurine group, Jersey cattle clustered distinctly while Holstein, Brown Swiss, and Ayrshire formed a separate cluster (Fig. 4a). Interestingly, the East Asian Mongolian taurine cluster was observed to be genetically closer to Brown Swiss cattle among the commercial European taurine breeds investigated in the present study. Apart from this, some individuals belonging to non-descript cattle (PND) of Pakistan, Batu Harak cattle (LBH) of Sri Lanka, and Achai cattle (PAC) of Pakistan were observed to be scattered in between taurine and zebu clusters. These individuals possess significant taurine introgression in them either due to ongoing crossbreeding program or lack of availability of true to the type breeding males. For instance, a section of PND individuals were spread towards Holstein cluster and the remaining towards the Jersey cluster, consistent with the improver breeds used for crossbreeding in their respective regions. These non-descript cattle are generally the subjects of crossbreeding programs for genetic improvement. Significant levels of taurine introgression in these animals are expected, considering the official breeding policy of genetic upgradation using exotic (Jersey/Holstein) semen for improved milk production. Further, to investigate the genetic relationship within the Zebu, PCA was performed with Zebu breeds only. To reduce the confounding effect of recent admixture due to crossbreeding programs, individuals of Zebu cattle with < 99% membership coefficient in the Zebu cluster were filtered out after unsupervised clustering (K = 2) using ADMIXTURE. The PC1 and PC2 accounted for 15.95% and 14.19% of the total variation in the dataset respectively. The results showed distinct clustering of Myanmar and Cambodian cattle from other South Asian Zebu located in India, Pakistan, Sri Lanka, and Bangladesh. Within the major cluster of South Asian Zebu cattle, the Kangayam cattle of South India clustered separately while Cholistani and Sahiwal cattle from Pakistan showed distinctiveness from other breeds located near the center of domestication in the Indus valley region (Fig. 4b). Neighbour-net tree drawn from a pairwise Reynolds genetic distance matrix revealed similar genetic relationships among the investigated cattle breeds. The European and Mongolian taurine cattle clustered together while all the Zebu breeds formed a single large cluster with sub-clusters conforming to their geographic origin. Within the Zebu cattle group, Indian and Pakistani cattle clustered separately while Cambodian and Myanmar cattle clustered distinctly. Interestingly, Zebu breeds that showed significant taurine admixture including PND, IVE, PAC and LBH were placed in between the taurine and Zebu clusters (Fig. 5a).

Principal component analysis for (a) Asian Zebu populations with outgroup taurine breeds from America, Europe, and Northen Asia (b) Asian Zebu populations with more than 0.9375 membership coefficient in the Zebu cluster. (Breed abbreviations Table 1).


(a) Neighbour-net tree plotted using Reynolds distance (b) Admixture plot of Asian Zebu cattle (c) Proportion of sampled cattle (in%) with different levels of taurine admixture. (Breed abbreviations Table 1).
The population structure and genetic admixture of Zebu cattle were further assessed using ADMIXTURE analysis. Through the analysis of ancestry components at varying levels of K (the number of inferred populations), ADMIXTURE allows the identification of genetic clusters and the extent of admixture between populations that have complex evolutionary history due to domestication and subsequent migration events. Analysis was performed at K = 2 to K = 30 to elucidate historical gene flow, and crossbreeding events among Zebu and taurine cattle populations. At K = 2 there was a distinct separation between taurine and Zebu cattle. Non-descript Zebu populations such as BGL and PND showed significant taurine admixture while well-defined breeds such as Vechur (IVE), Batu Harak (LBH) and Achai (PAK) showed moderate levels of taurine introgression in them (Fig. 5b). The level of taurine admixture in Zebu cattle was quantified based on their proportion of membership coefficient assigned to taurine clusters at K = 2. The proportion of sampled individuals with admixture levels of at least 6.25%, 12.5% and 25% was calculated for each of the investigated Zebu breeds (Fig. 5c). The level of taurine admixture was negligible in all the purebred Zebu cattle breeds of Myanmar (YPZ, YSG,YSN) and Cambodia (KDR, KDW), White cattle (LWC) of Sri Lanka, Alambadi (IAL), Bargur (IBR), Kangayam (IKA), Ongole (ION) of South India, Bhagnari (PBN), Cholistani (PCH), Dhani (PDH), Dajal (PDJ) and Tharparkar (PTP) from Pakistan. However, more than 75% of Pakistani Non-Descript (PND), Sri Lankan Batu Harak (LBH), Pakistani Achai (PAC) and South Indian Vechur (IVE) cattle had at least 6.25% taurine admixture. In the same breeds, 62%, 18%, 15% and 1% of the sampled cattle showed at least 25% taurine admixture respectively. At K = 3, distinct ancestries of Southeast Asian (Myanmar and Cambodia) and South Asian (Pakistan, India, Sri Lanka, and Bangladesh) cattle was observed, consistent with the results of PCA. Within the South Asian group, beginning K = 4, more than 95% of Kangayam (IKA), Cholistani (PCH) and Sahiwal PSH cattle were assigned to a single inferred cluster whilst most individuals of other breeds were assigned to more than one inferred cluster. Significant differences were also observed in the ancestry composition of Pakistani cattle (located close to the Zebu domestication center) and South Indian or Sri Lankan cattle. Beginning K = 7, Kdarm White (KDW) cattle of Cambodia showed significant admixture of Brahman ancestry reflecting the ongoing crossbreeding program to improve local Cambodian cattle for beef production.
Effective population size, gene flow and migrations
Historical trends in effective population size were estimated up to 100 generations in the past using GONE. The method provides relatively stable estimates of Ne against deviations from assumptions such as non-random mating, overlapping generations, population sub-structuring and genetic admixtures21. Except for a few breeds, the effective population size estimated up to 100 generations in the past, showed a non-linear decline from generation 5–15 before present until the most recent generation as shown in Fig. 6a. The Punganur (IPN) cattle breed in South India experienced consistent and significant decline in Ne until 35 generations before present after which the population size stabilized. Similarly, the Pulikulam (IPM) cattle from the same region experienced a steep decline beginning 70 generations and up to 60 generations before present. In case of Kdarm Red (KDR) cattle from Cambodia and Shwe Ni Gyi (YSG) cattle from Myanmar, estimates of effective population size fluctuated until 40 generations before present after which there was a significant decline. In contrast the estimates of effective population size in Tharparkar (PTP) cattle from Pakistan remained stable for more than 80 generations while there was a slight increase in Sri Lankan White (LWC) cattle during the last 25 generations or more. We further estimated the contemporary Ne of investigated cattle populations using CurrentNe22. The method provides relatively accurate estimates of Ne in problematic scenarios of large population sizes or small sample sizes and provides confidence intervals that are more consistent than resampling methods33. The contemporary Ne estimates (Fig. 6b) were the lowest among South Indian dwarf types (Punganur (IPN), Vechur (IVE)), and Pakistani (Cholistani (PCH) and Tharparkar (PTP)) cattle. In contrast, the highest contemporary Ne estimates were observed in Myanmar (Pyar Zein (YPZ), Shwe Ni (YSN) and Shwe Ni Gyi (YSG)) cattle breeds. Overall, variations in Ne trends shown by breeds in different countries were comparable to demographic patterns. For instance, the census population size of Vechur (IVE) and Punganur (IPN) cattle from South India counts few hundreds and they are classified under endangered category34.

(a) Historical effective population size estimated using GONE (Ne) and (b) Contemporary effective population size estimated using CurrentNe considering integration over the whole genome showing point estimates, lower and upper bound at 90% confidence interval. (Breed abbreviations Table 1).
Population history relationships and gene flow were identified by constructing a maximum likelihood tree and residual matrix using Treemix software. To focus on historical migration patterns, analysis was performed using (a) Zebu cattle only (Fig. 7) and (b) Zebu cattle with taurine outgroups (Supplementary Figure SF3 in Supplementary Information S1 File). Migration events were modeled sequentially from 1 to 10, and the model with 5 migration edges had at least maximum likelihood errors indicating the optimal fit to data. The results revealed notable gene flow and migration patterns among the cattle breeds studied. Gene flow from Ongole (ION) to Brahman (KBR) supports the known development of the Brahman as a composite breed. Among the Cambodian cattle, Kdarm White (KDW) showed gene flow from Kdarm Red (KDR) and Brahman (KBR) cattle. The gene flow observed between the Pakistani Tharparkar (PTP), and Indian Deoni (IDN) cattle is likely due to shared ancestry influenced by both geographic proximity as well as physical and functional traits. Another interesting evidence of gene flow was between India’s Kangayam (IKA) and Sri Lankan White cattle (LWC), considering the geographic proximity, historical and cultural connections among the livestock keepers in the native tract of these two breeds. When taurine outgroups were included, the most prominent migration edge indicated Holstein introgression into Zebu breeds like Umbalachery (IUM) and Thawalam (LTM). This likely reflects historical crossbreeding efforts aimed at improving milk productivity in local cattle. Another migration edge indicated gene flow from Jersey (LJR) into Punganur (IPN), the miniature Zebu breed from India with unique phenotypes.

Treemix analysis of Zebu populations (a) model assuming k = 1000 and up to 5 migrations (b) Residual plot for 5 migrations model.
Runs of homozygosity
The Runs of Homozygosity analysis provided insights into inbreeding levels in the studied cattle breeds by examining continuous homozygous regions along their genomes. The ROH-based inbreeding coefficient, or FROH, was estimated by analyzing the proportion of ROH segments within the total length of autosomes. Inbreeding levels were classified according to five ROH length categories (1, 2, 4, 8, and 16 Mb), reflecting both historical and recent inbreeding events. A total of 75,468, 15,913, 5284, 1912 and 626 ROH were observed for 1 Mb, 2 Mb, 4 Mb, 8 Mb and 16 Mb length categories respectively. The FROH values varied across breeds, indicating different degrees of inbreeding across populations. For most Zebu cattle breeds with the exception of Tharparkar, the inbreeding levels remained below 5%, suggesting low to moderate inbreeding. Tharparkar (PTP) had the highest inbreeding levels across all categories, with FROH of up to 7.4% in the shortest ROH segments (1 Mb), suggesting a history of recent inbreeding likely due to population bottlenecks or selective breeding practices. Such high levels of genomic inbreeding estimates have also been reported previously in Tharparkar cattle population from the Indian side of the native breed tract. Other Pakistani breeds such as Cholistani (PCH) and Red Sindhi (PRS) showed moderate inbreeding levels.
Short ROH Segments (1–4 Mb) are indicative of historical inbreeding events. Breeds with high FROH in these categories, such as Punganur (IPN) and Vechur (IVE) from India, have small, isolated populations where historical inbreeding occurred over generations. Breeds from Myanmar, such as Shwe Ni Gyi (YSG) and Pyar Zein (YPZ), had among the lowest inbreeding levels across all ROH categories, suggesting more diverse genetic backgrounds and lack of selective breeding practices historically. Further, considering the similarity, the Cambodian cattle breeds, Kdarm Red (KDR) and Kdarm White (KDW) exhibited relatively low levels of inbreeding across all ROH lengths (Fig. 8), consistent with a more diverse gene pool, possibly due to historical gene flow or recent crossbreeding with Brahman (KBR) cattle.

Genomic inbreeding (FROH) estimated from the proportion of runs of homozygosity (ROH) in the total length of autosomes. Inbreeding levels are categorised based on four ROH lengths (1, 2, 4, 8, 16 Mb).
Signatures of selection based on EHH
Draught versus Dairy Zebu cattle: To perform selection signature analysis, we identified zebu breeds that are well known for their draught and dairy characteristics. The Mysore type cattle breeds from Southern India have been traditionally selected and bred for their draught performance and hence three breeds Kangayam, Hallikar and Ongole were utilized. A pair of Kangayam bullocks can pull a load of 3787 ± 51.4 kg over a distance of 10–20 kms without rest35,36. Hallikar bullocks can work in the field for an average duration of 8.18 h/day during busy days of agricultural operations. Their average load pulling capacity was estimated as 2.75 tonnes with an average speed of 5 km/h37. Similarly, Ongole bullocks can draw heavier loads of more than 2000 pounds on a fair road. However, the milk performance of these draught type breeds are significantly low37,38,39. The cattle breeds from North-Western India have traditionally been selected and bred for milk production and hence three breeds viz, Sahiwal, Red Sindhi and Tharparkar were utilized. The 305 days milk yield, total milk yield, lactation length, wet average and peak yield of Sahiwal cattle varied from 1822.59 to 2166.36 kg, 1822.59 to 2259.86 kg, 268.95 to 303.02 days, 6.62 to 7.93 kg/day and 10.64 to 14.09 kg respectively40. In case of Red Sindhi cattle, the mean first lactation milk yield, milk yield per day, lactation length and lifetime milk yield were reported to be 1448 ± 23.2 kg, 6.6 ± 0.1 kg, 277.3 ± 5.6 days and 5203.4 ± 178.4 kg respectively41. With respect to Tharparkar cattle, the mean total milk yield (TMY), lactation length (LL), peak yield (PY) were reported to be 1633.40 ± 45.79 kg, 272.55 ± 4.64 days and 10.83 ± 0.17 kg respectively42. The comparative analysis of draught and dairy type Zebu cattle using XP-EHH approach identified six genomic regions under selection. These regions encompassed a total of 418 SNPs that surpassed the significant threshold (-log10 ≥ 4) strongly indicating the presence of selection pressures that might have shaped distinct phenotypic traits in these populations (Fig. 9). Functional annotation revealed these regions were significantly enriched for biological processes associated with immune regulation, including the cytokine-mediated signaling pathway, negative regulation of endopeptidase activity, and type I interferon signaling pathway. These processes are critical for modulating innate immune responses and protection against infectious diseases, a vital adaptation for cattle in tropical conditions. Enrichment analysis further highlighted four significant KEGG pathways such as necroptosis, JAK-STAT signaling pathway, toll-like receptor signaling pathway and natural killer cell mediated cytotoxicity pathway. These pathways underscore the role of adaptive and innate immune mechanisms in the resilience and productivity of Zebu cattle. Key candidate genes driving these biological processes and KEGG pathways (Table 4) included IFNAR1, IFNAR2, IFNGR2, IL10RB, SERPINB1, SERPINB9, SERPINB6, KCNE1, KCNE2. These genes play pivotal roles in mediating antiviral responses, cytokine signaling, and immune modulation. In particular, the serpin family genes are involved in the regulation of proteolysis during immune responses, as well as wound healing (Table 4)43. Additional genes linked to economically important traits, such as reproduction (REC8, CLIC4, TSSK4, HTR3A, HTR3B), homeostasis (DRD2, MYH6, MYH7), and milk production, were also identified within these regions. Identification of MYH6 and MYH7 suggests selection for muscle function and contraction, traits critical for draught animals. Together, these findings reveal a complex network of genetic factors shaped by selection pressures, reflecting the distinct functional and adaptive requirements of dairy and draught type zebu cattle populations.

Genome-wide distribution of standardised cross population extended haplotype homozygosity (XP-EHH) (a) Draught vs Dairy (b) Miniature vs Large size Zebu cattle. The dashed horizontal line shows the cut off value to call SNPs outliers.
Large versus small statured Zebu cattle: To perform selection signature analysis on stature, dwarf breeds viz. Vechur and Punganur and large sized breeds viz. Hallikar and Kangayam were utilized. The breeds were selected based on their morphometric data. The mean height at withers, body length and heart girth of Vechur and Punganur cattle were reported to be 91.8 cm and 91.01 cm, 96.0 cm and 103.77 cm, 113.2 cm and 124.46 cm, respectively44,45. In contrast, the mean body length, height at withers, and heart girth in adult Hallikar males and cows were 138.94 cm and 130.17 cm, 134.55 cm and 124.75 cm, and 163.15 cm and 148.45 cm respectively. Similarly, the mean values of these traits in adult Kangayam males and cows were 139.5 cm and 124.56 cm, 144.33 cm and 130.72 cm, and 169.92 cm and 155.32 cm respectively36,37. The comparative analysis of large and small statured Zebu cattle following XP-EHH approach detected three genomic regions under selection, comprising 199 SNPs above the significance threshold. Functional annotation and enrichment analysis of these genes revealed 11 significant GO terms (Table 4) predominantly associated with biological processes regulating heart rate, visceral muscle development, cardiac growth and muscle filament sliding. The myosin heavy chain genes such as MYH6 and MYH7 were recurrent among these terms, indicating their central role in determining muscle strength, contractility, and overall physiological performance in response to selection for body size and stature. Further, the KEGG analysis identified necroptosis (p-value = 0.002) and proteosome pathways (p-value = 0.005) as significantly enriched pathways that are intricately linked to immune responses, including antigen processing, stress signaling, and inflammation. Genes such as CHMP4A and RIPK3 are critical mediators of these pathways, emphasizing their roles in maintaining cellular homeostasis and responding to viral infections. Reproductive genes such as REC8, CLIC4, and TSSK4 were also enriched, alongside genes associated with fertility-related processes, such as sperm capacitation. The identified genes and pathways elucidate the genetic basis underlying the key physiological and reproductive traits selected for in large versus small sized Zebu cattle populations.
Signatures of selection based on FST
The breeds under dairy vs draught type and large vs small sized zebu cattle groups used for the extended homozygosity haplotypes statistics were utilized for the fixation index-based approach as well. Genomic regions containing highly differentiated SNPs (top 0.5% of FST) were considered as under selection (Fig. 10).

Genome-wide Manhattan plots of fixation index (FST) analysis for comparison between (a) Draught vs Dairy (b) Miniature vs Large size Zebu cattle. Chromosomes are differentiated by colour, each SNP is represented by a dot, the red line represents the cutoff value of 0.5% per category and the blue line represents the mean FST. The SNPs above the red line were considered to map significant regions of interest.
Draught versus dairy: A total of 21 significant (p < 0.05) GO terms for biological processes were identified, many of which were immune-related. These included antigen processing and presentation, MHC class Ib molecules, antimicrobial humoral immune response, and exogenous lipid antigen presentation. Positive regulation of lactation was amongst the significant processes directly related to milk production. Processes related to embryonic skeletal system morphogenesis were also enriched highlighting their contribution to the morphological and physiological differences observed between the two groups. Growth and energy related processes such as insulin receptor signaling pathway and other biological processes are presented in Supplementary Table ST2 in S1 file along with the list of genes. The KEGG pathway analysis identified 18 significant pathways including neutrophils signaling pathway (p = 0.02), that modulates intracellular signaling cascades such as MAPK, Ras, and PI3K-Akt. These pathways influence cell differentiation, growth, and survival, all essential for animal productivity. Other noteworthy pathways included the GnRH signaling pathway, which regulates reproductive functions, growth hormone synthesis and action, which directly impact physical growth. Genes associated with bone homeostasis and muscle function, such as those involved in the parathyroid hormone and relaxin signaling pathways, were also enriched, emphasizing their relevance in draught animals.
Large versus small statured: The FST analysis identified 38 significant GO terms related to growth, reproduction, immune response, and physiological adaptations (Supplementary Table ST3 in S1 file). Biological processes such as embryonic skeletal system morphogenesis and canonical Wnt signaling were noteworthy, emphasizing their roles in bone and skeletal muscle development. Embryonic skeletal system morphogenesis is a biological process that causes embryonic tissue or organ to develop its shape by controlling the spatial distribution of cells during embryonic development. The involvement of WNT3, CTNNB1, and FZD1 in osteoblast differentiation underscores the genetic basis of stature differences between large and small Zebu cattle. Additionally, processes linked to kidney function (metanephros morphogenesis), homeostasis, and reproduction (BSP1, BSP3, BSP5) were also enriched significantly. KEGG analysis revealed 17 significant pathways, with growth hormone and relaxin signaling pathways emerging as key regulators of growth and reproductive traits. Pathways associated with energy metabolism, such as glycolysis/gluconeogenesis, and immune regulation (e.g., Th1/Th2/Th17 cell differentiation) were also enriched, reflecting the diverse selection pressures shaping these populations. Milk production traits were supported by enrichment in the prolactin signaling pathway, while reproductive and physiological adaptations were evident from pathways such as estrogen and progesterone-mediated oocyte maturation.
Discussion
In South Asia, countries such as India, Pakistan, Sri Lanka, Bangladesh, Myanmar, and Cambodia are home to a diverse array of indigenous Zebu cattle breeds. These breeds are highly valued for their remarkable adaptability to challenging production environments, including reliance on low-quality feeds and forages, tolerance to tropical heat, and resilience against disease-causing vectors. During the past few decades, crossbreeding with commercial breeds has been extensively promoted to enhance productivity and ensure food and nutritional security in the region. While this approach holds promise, it also poses a significant risk of eroding the genetic diversity that underpins the resilience of these indigenous breeds. To conserve and sustainably manage these genetic resources, characterization of genomic diversity and assessment of admixture levels with exotic taurine breeds are essential steps. Genomic characterization of Zebu cattle using single nucleotide polymorphic arrays involves challenges such as SNP ascertainment bias. Most commercial SNP arrays were developed using taurine cattle genomes, with limited representation of Zebu breeds. For instance, the SNP discovery panel for the development of Axiom BovMDv3 array, utilized in this study, relies on data from only three Zebu breeds—Nelore, Brahman, and Gir21,30,32. While this helps address certain gaps in Zebu representation, it fails to capture the extensive genetic diversity present across geographically distinct Zebu cattle populations. Studies indicate that approximately 40–50% of SNPs on commercial arrays are informative for Zebu cattle, resulting in biased estimates skewed toward taurine genetic diversity. This bias is particularly concerning given the genetic variability of intensively selected taurine breeds compared to more diverse Zebu populations, as also observed in this and other studies33,46.
To mitigate SNP ascertainment bias, we employed linkage disequilibrium (LD)-based pruning (Supplementary Figure SF1 in S1 file) and identified an LD threshold of 0.2 as optimal for minimizing its effects. However, this process resulted in the loss of information at 11,988 SNP loci, underscoring the need to develop a comprehensive SNP array tailored specifically to Zebu breeds. An alternative approach is to perform whole-genome resequencing (WGR), which provides an unbiased and exhaustive representation of genetic variation. Despite its potential, the high costs and technical expertise required for WGR remain significant challenges, particularly for researchers in developing countries. A cost-effective and affordable solution would be the design and development of a Zebu-specific SNP chip, which would improve the accuracy of genetic diversity assessments and facilitate genome-wide studies targeting economically important traits. Such advancements would allow researchers to explore and obtain better genomic insights in Zebu cattle, aiding in both conservation and sustainable utilization of these critical genetic resources.
The observed heterozygosity among Zebu breeds in this study was moderate to high with an overall mean of 0.329, consistent with previous reports32. Genetic diversity in most local breeds across Asia has predominantly been assessed using microsatellite markers, with only a few studies utilizing genome-wide SNP data are available. The discrepancies in reported heterozygosity levels may therefore be due to differences in marker types and methodologies employed. Zebu cattle breeds from Cambodia and Myanmar showed moderate but relatively lower diversity as compared to Pakistani and Indian Zebu cattle. The moderate diversity in these breeds is consistent with previous reports9 and is likely shaped by a combination of natural adaptation to local environmental conditions, historical population bottlenecks, and limited crossbreeding with exotic breeds. Further, domestication of Zebu cattle has been reported to have occurred in the Indus Valley regions of Pakistan about 7000–9000 years ago1. Studies on various livestock species have demonstrated a progressive reduction in genetic diversity among populations while geographically moving away from the domestication site. This trend is reflected in the comparatively lower genetic diversity observed in Myanmar and Cambodian cattle in the present study. Such findings underscore the influence of domestication, historical demographic events, and geographic isolation in shaping the genetic diversity of regional Zebu populations.
The relationships among Zebu breeds were inferred using dimension reduction techniques such as principal component analysis (PCA). The PCA incorporating commercial taurine breeds revealed three primary clusters: one encompassing all Zebu breeds and two separate clusters for commercial and Mongolian taurine breeds. Notably, some Zebu individuals displayed overlap with taurine clusters, likely due to historical crossbreeding between Zebu and taurine populations. The second PCA analysis without the taurine outgroups positioned Zebu breeds according to their geographic origin Cambodia-Myanmar, Indian-Sri Lankan breeds, and Pakistani breeds1,8,47,48,49. Similar geographic clustering patterns have been observed in studies on cattle and other livestock species.
Pairwise FST analysis further corroborated the geographic clustering, showing weak differentiation among breeds from the same regions. For instance, Myanmar breeds exhibited near-zero differentiation, suggesting historical gene flow among these populations. This lack of differentiation may also be attributed to the traditional reliance on physical traits, such as coat color, for breed classification, which often lacks sufficient genetic resolution. The FST values observed in this study ranged from 0.01 to 0.03 among closely related breeds, with the highest differentiation recorded at 0.11. These estimates are consistent and comparable with low FST values reported in Ethiopian Zebu populations50. However, higher FST values have been observed in comparisons such as Gir versus East African Zebu51, as well as other Asian populations52. The reported close genetic relationship among Zebu breeds in comparison to taurine populations in this study has been consistent with past reports47,53. Admixture analysis supported the findings from PCA and FST, revealing consistent genetic relationships among Zebu breeds. The divergence between Bos indicus and Bos taurus breeds was clearly present at all K values, this finding was consistent with previous reports on bovine domestication events54,55. The observed taurine introgression into non-descript Zebu populations (e.g., BGL and PND) highlights the impact of ongoing crossbreeding in these populations. In contrast, well-defined breeds such as Vechur (IVE), Batu Harak (LBH), and Achai (PAC) exhibited moderate taurine admixture, possibly reflecting limited but targeted historical gene flow aimed at improving specific traits such as milk production and adaptability. Quantification of taurine admixture levels in Zebu breeds showed negligible introgression in purebred populations from Myanmar (e.g., YPZ, YSG, YSN) and Cambodia (e.g., KDR, KDW), as well as in indigenous breeds from South India, Sri Lanka, and Pakistan. This purity indicates limited external influence on these breeds, likely due to geographic isolation and the preservation of traditional breeding practices.
Admixture analysis further revealed distinct genetic ancestries of Southeast Asian (Myanmar and Cambodia) and South Asian (Pakistan, India, Sri Lanka, and Bangladesh) cattle. This regional differentiation aligns with the results of PCA and reflects the influence of geographic and cultural barriers in shaping genetic diversity. The results also revealed more refined population structures that emerged within the South Asian group. For example, Kangayam (IKA), Cholistani (PCH), and Sahiwal (PSH) cattle were predominantly assigned to single, distinct clusters, demonstrating their genetic homogeneity. Conversely, other South Asian breeds exhibited membership in multiple clusters, indicative of historical gene flow and shared ancestry. The differentiation between Pakistani breeds, located near the Zebu domestication center, and South Indian or Sri Lankan breeds further highlights the genetic and historical divergence within the region. Significant Brahman admixture was detected in Cambodian Kdarm White (KDW) cattle, highlighting the influence of targeted crossbreeding initiatives aimed at enhancing beef production. These programs have likely incorporated genetic material from Brahman cattle, a widely distributed Zebu breed known for its superior meat yield and adaptability. Similar strategies have been employed to improve the performance of many local cattle populations in South Asia by integrating traits from established commercial breeds.
Understanding the effective population size (Ne) is essential for assessing genetic diversity, inbreeding, and the evolutionary dynamics of livestock populations. Our results on Ne were similar but slightly lower to Ne reported for Bangladesh breeds47, and other zebuine studies across the globe. Historical trends in Ne exhibited a non-linear decline starting from generations 5–15, consistent with patterns of demographic contraction, recently reported in Indian breeds like Kangayam and Ongole and Korean and Chinese local breeds33,49,53,56. Contemporary Ne estimates calculated using CurrentNe highlighted significant variability and aligned with known demographic patterns of the investigated breeds. The critically low Ne values in South Indian breeds such as Punganur (IPN) and Vechur (IVE) are reflective of their endangered status, underscoring the urgency for conservation measures to prevent further genetic erosion. The relatively stable Ne in Tharparkar (PTP) cattle indicate successful management strategies that could serve as a model for other populations at risk. Meanwhile, the fluctuating Ne values in breeds such as Kdarm Red (KDR) and Shwe Ni Gyi (YSG) suggest a need for more targeted breeding strategies to ensure long-term genetic viability. In contrast, the highest contemporary Ne values were observed in Myanmar breeds such as Pyar Zein (YPZ), Shwe Ni (YSN), and Shwe Ni Gyi (YSG), suggesting relatively larger and stable breeding populations in these groups. This could be attributed to the extensive use of these breeds for agricultural operations, maintaining larger effective populations over time. These variations in Ne across breeds and regions highlight the influence of historical demographic events, geographic isolation, and breeding practices on genetic diversity. Overall, these findings emphasize the critical importance of monitoring both historical and contemporary Ne in Asian Zebu cattle populations. Effective management and conservation strategies tailored to each breed’s demographic history and current status are essential to maintain genetic diversity and mitigate the risks of inbreeding and extinction. With the above interpretations of Ne estimates obtained in the present study, it also needs to be noted that the differences in Ne estimates can also be due to population structure, admixture, gene flow and sampling methods. Hence the estimates of Ne in most population genetic studies including this study needs to be considered carefully as they are sensitive to the above factors particularly if there are overlapping generations and varied mating strategies57. With the availability of different algorithms to estimate LD and Ne, caution is advised when interpreting the results to make informed decisions related to conservation and management.
The analysis of population history relationships and gene flow provided further valuable insights into the genetic interconnections and migratory patterns among Zebu cattle breeds. By focusing on Zebu individuals with high membership coefficients (≥ 0.9375), the study effectively minimized confounding influences from recent admixture and ensured robust investigation of historical migration events. Gene flow from Ongole (ION) to Brahman (KBR) corroborates the historical development of the Brahman breed as a composite population derived from Ongole, Guzerat, and Gir breeds. This result is consistent with established records of the Brahman breed’s creation as a heat-tolerant Zebu breed adapted for commercial meat production globally. Among Cambodian cattle, the observed gene flow from Kdarm Red (KDR) to Kdarm White (KDW) suggests these populations, despite their distinct coat colors, represent a single genetic pool with minimal differentiation. This admixture aligns with the relatively low FST values between these two populations. The additional gene flow from Brahman (KBR) to KDW is consistent with the results of admixture analysis indicating the ongoing crossbreeding programs aimed at enhancing local cattle productivity in Cambodia. The gene flow between Pakistani Tharparkar (PTP) and Indian Deoni (IDN) cattle is particularly notable given the geographic proximity and similar phenotypic characteristics of these breeds. Both are dairy Zebu breeds characterized by a predominantly white coat, which may reflect shared ancestry and breeding practices. Another intriguing observation was the gene flow between India’s Kangayam (IKA) cattle and Sri Lankan White cattle (LWC). The Kangayam breed, native to the Mysore type cattle group, is reared predominantly in Tamil Nadu, South India, a region with cultural and linguistic ties to the Tamil-speaking population of eastern Sri Lanka, where Sri Lankan White cattle are maintained. The observed gene flow between IKA and LWC likely reflects the historical interactions facilitated by geographic proximity, cultural connections, and shared breeding practices. The morphological and functional similarities between these populations further support this gene flow.
In this study we employed two complementary genomic approaches, cross population extended haplotype homozygosity (EHH) and population differentiation (FST) to identify candidate regions under selection in Zebu cattle. These regions were associated with economically important traits such as growth, immune response, thermotolerance, reproduction, milk production and physiological adaptation. The identified pathways and genes provided insights into the evolutionary and functional mechanisms that shaped the adaptation of Zebu cattle to diverse production environments. Through comparative analysis of draught and dairy Zebu cattle, several significantly enriched gene ontology (GO) biological processes and KEGG pathways were identified. The JAK-STAT pathway was significantly enriched (P < 0.05), a key signaling mechanism for various cytokines and growth factors that target development and homeostasis7. Studies in Bos taurus have revealed the association of this pathway in carcass traits58. Additionally, Toll-like receptor (TLRs), natural killer cell mediated cytotoxicity, necroptosis, and cytokine-mediated signaling pathways were enriched. These pathways highlight the ability of Zebu cattle to adapt to tropical environments by enhancing pathogen recognition and immune defense. The interferon type 1 signaling pathway was represented by two genes: interferon alpha and beta receptor subunit 1 and 2 (IFNAR1, IFNAR2) encode proteins related to maintenance of bone mass indicating their critical role in mechanical functions of draught breeds. These genes are associated with the activity of osteoblasts and osteoclasts cells that coordinate the processes of bone formation and the resorption of old damaged bone tissue. These genes have also been reported to be responsible for polledness in taurine cattle, average daily gain and muscle gain in Hanwoo cattle59,60,61. Amongst other genes in the JAK-STAT signaling pathway were IL23A related to inflammatory resistance to Johne’s disease in cattle caused by Mycobacterium avium subspecies paratuberculosis (MAP)62. Studies have reported the close interaction between the JAK-STAT pathway with prolactin to regulate milk production traits63,64. The necroptosis pathway involved genes such as RIPK1 which releases cytokines or chemokines resulting in inflammation, and wound healing processes65,66.
Most draught breeds have a large or normal stature size, and they were used to compare large and miniature Zebu. The significantly enriched GO biological processes (p < 0.05) were related to regulation of heart rate, heart growth and muscle development. The genes MYH6 and MYH7 were enriched in the biological processes related to regulation of ATPase activity, muscle filament sliding, visceral muscle development, skeletal muscle structure and function. These genes are probably related to phenotypic features such as size of cattle and regulation of heart rate which is an important biological process related to large sized animals. DUSP10 and RIPK3 genes were enriched under regulation of adaptive immune response. Reproduction related genes specific to fertilizations (REC8, CLIC4, TSSK4) were observed to be significant in our study as reported earlier in taurine cattle and yak67,68. The REC8 gene is related to male recombination and the TSSK4 encodes kinases regulating sperm motility and fertility69.
The FST results from this study were consistent with the EHH results on association with growth, immune response, thermotolerance, reproduction, milk production and physiology related characteristics. The results identified additional pathways related to environmental adaptations. For instance, the JAK-STAT cascade involved in growth hormone signaling pathway harbored genes such as STAT2, STAT3, STAT5A, STAT5B and STAT6 associated with growth and neuroendocrine system that has an important role in physiological adaptation to stress70,71,72,73. Candidate genes involved in significantly enriched pathways such as GnRH signaling, progesterone-mediated oocyte maturation, PI3K-Akt signaling were associated with male and female fertility. MAPK genes (MAPK10, MAPK13, MAPK14) reported in our study are important to produce gonadotrophins by stimulating GnRH neurons in the hypothalamus74. The growth hormone synthesis and secretion pathway is a crucial pathway that provides insights on both production and stature related traits. The growth hormone has been reported to influence the physical growth of cattle61 and production of alpha casein protein related to milk production75,76,77.
The growth hormone synthesis and secretion pathway were significantly enriched, implicating genes such as IGF1, GH1, and GHRH, which are well-established contributors to physical growth and milk production in bovines. These findings align with studies in taurine cattle, such as Holstein, and highlight the evolutionary importance of these genes in cattle productivity78,79,80. Interestingly, the PI3K-Akt signaling pathway, crucial for cell cycle regulation, emerged as a key adaptive mechanism for thermotolerance. The heat shock protein HSP90AA1, widely reported in bovine81,82,83,84, goats, and chickens85, underscores the ability of these breeds to thrive in extreme temperatures. Additionally, genes such as HOXB1-HOXB9, linked to embryonic skeletal system morphogenesis, may explain stature differences between large draught and miniature Zebu breeds. However, the allelic variants of PLAG1, NCAPG-LCORL reported to be associated with stature in multiple taurine breeds were not found to be significantly differentiated in Zebu cattle populations86,87,88.
The enrichment of growth and skeletal system pathways gave insights into the regions positively selected in the Zebu populations in relation to their uses and stature. The interaction between pathways related to immune response, fertility and reproduction also explain the superiority of Zebu cattle to thrive in extreme hot temperatures yet reproduce, thrive on poor quality feeds, and build resilience to diseases that prevail in these environments. While the identified selection signatures provide valuable insights, several methodological limitations need to be highlighted as well. The use of a taurine-biased SNP chip may introduce ascertainment bias, potentially leading to the exclusion of low-frequency alleles specific to Zebu cattle. This could result in the loss of rare but critical alleles or an overrepresentation of false positives. Additionally, sample sizes might have constrained the detection of subtle selection signals. Future studies leveraging whole-genome sequencing of Zebu genomes could mitigate these biases and provide a more comprehensive understanding of selection processes.
Conclusion
Zebu cattle play a critical role in the livelihoods of smallholder farmers and indigenous communities of South Asia. Investigation of Zebu cattle genomic diversity is essential to make informed decisions regarding their breeding and conservation programs. Our results showed significant differentiation of Southeast Asian Zebu cattle (Myanmar and Cambodian) from that of South Asian Zebu cattle breeds from Pakistan and India. Gene flow and migration analyses provided evidence of historical events, such as the genetic contribution of Ongole cattle to the Brahman breed and gene exchange between Pakistani and Indian breeds prior to the establishment of modern national boundaries. The analysis also highlighted the extent of taurine introgression in Zebu breeds, reflecting widespread crossbreeding programs aimed at improving productivity. However, the observed decline in effective population sizes among certain Zebu breeds raises concerns about potential genetic erosion. The identification of selection footprints related to growth, physiology, immune response, and production traits underscored the genetic basis for the adaptability and productivity of Zebu cattle, irrespective of their stature or use. Notably, genes associated with immune response, fertility, and reproduction pathways explain the resilience of Zebu cattle to thrive under extreme heat, withstand disease pressures, and sustain productivity on low-quality nutrition. The results emphasize the importance of conserving Zebu cattle breeds and maintaining their genetic purity, particularly in the context of uncontrolled crossbreeding programs to improve milk production and changing climatic conditions. Lack of quality bulls and absence of semen from native zebu bulls for artificial insemination (AI) under field conditions results in admixture and consequent genetic erosion of these important germplasms. Breeding programs should aim to balance the need for improving productivity while preserving key adaptive traits. Availability of frozen semen from elite true to the type bulls needs to be ensured so that farmers have access to AI and genetic improvement of purebred zebu cattle. Future research should focus on comprehensive whole-genome sequencing of Asian Zebu cattle to overcome these limitations. Such efforts could expand the existing genomic database, support the development of a Zebu-specific SNP chip, and enhance genetic studies on Zebu cattle across a broader range of breeds and populations.
Data availability
All relevant data has been stated in the manuscript. SNP genotype data generated in the study is available in Supplementary Information S2 file.
Change history
25 November 2025
The original online version of this Article was revised: In the original version of this Article the affiliation 1 “1Animal Production and Health Laboratory, Joint FAO/IAEA Centre of Nuclear Techniques in Food and Agriculture, Department of Nuclear Sciences and Applications, International Atomic Energy Agency, Vienna, Austria,” was incorrectly given as “Animal Production and Health Laboratory, Joint FAO/IAEA Division of Nuclear Techniques in Food and Agriculture, International Atomic Energy Agency, Vienna, Austria.” The original article has been corrected.
References
S Chen 2010 Zebu cattle are an exclusive legacy of the South Asia neolithic Mol. Biol. Evol. 27 1 6
YT Utsunomiya 2019 Genomic clues of the evolutionary history of Bos indicus cattle Anim. Genet. 50 557 568
V Manomohan 2021 Legacy of draught cattle breeds of South India: Insights into population structure, genetic admixture and maternal origin PLoS ONE 16 e0246497
Breed Wise Report on Livestock and Poultry Based on 20th Livestock Census. https://dahd.gov.in/sites/default/files/2023-07/BreedReportEnglish04-08-2022.pdf (2022).
Ganbold S. Cattle stock in the Asia-Pacific region in 2022, by country or territory. https://www.statista.com (2022).
Sajjad K.M, Rehman, Z.-U., K, M. A. & Ahmad, S. Genetic resources and diversity in Pakistani cattle. Vet. J. 28 (2008).
N Iqbal 2019 Genomic variants identified from wholegenome resequencing of indicine cattle breeds from Pakistan PLoS ONE 14 e0215065
LGS Lokugalappatti 2023 Indigenous cattle of Sri Lanka: Genetic and phylogeographic relationship with Zebu of Indus Valley and South Indian origin PLoS ONE 18 e0282761
M Lwin 2018 Genetic diversities and population structures of four popular Myanmar local cattle breeds Anim. Sci. J. 89 1648 1655
G Giovambattista 2020 Characterization of bovine MHC DRB3 diversity in global cattle breeds, with a focus on cattle in Myanmar BMC Genet. 21 95
S Mam 2022 A survey of genome-wide genetic characterizations of crossbred dairy cattle in local farms in Cambodia Animals 12 2072
M Barbato 2020 Adaptive introgression from indicine cattle into white cattle breeds from Central Italy Sci. Rep. 10 1279
S Purcell 2007 PLINK: A tool set for whole-genome association and population-based linkage analyses Am. J. Hum. Genet. 81 559 575
CC Chang 2015 Second-generation PLINK: Rising to the challenge of larger and richer datasets Gigascience 4 s13742-015
J Goudet 2005 Hierfstat, a package for R to compute and test hierarchical F-statistics Mol. Ecol. Notes 5 184 186
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing. https://www.R-project.org/ (2021).
Alexander, D. H., Shringarpure, S. S., Novembre, J. & Lange, K. Admixture 1.3 Software Manual. (2020).
Pickrell, J. K. & Pritchard, J. K. User Manual for TreeMix v1.0. www.boost.org/; (2012).
DH Huson 1998 SplitsTree: Analyzing and visualizing evolutionary data Bioinformatics 14 68 73
MG Sobell 2015 A practical guide to ubuntu linux Pearson Education
E Santiago 2020 Recent demographic history inferred by high-resolution analysis of linkage disequilibrium Mol. Biol. Evol. 37 3642 3653
E Santiago A Caballero C Köpke I Novo 2024 Estimation of the contemporary effective population size from data while accounting for mating structure Mol. Ecol. Resour. 24 e13890
L Zhang 2013 cgaTOH: Extended approach for identifying tracts of homozygosity PLoS ONE 8 e57772
M Ferenčaković 2013 Estimates of autozygosity derived from runs of homozygosity: Empirical evidence from selected cattle populations J. Anim. Breed. Genet. 130 286 293
PC Sabeti 2002 Detecting recent positive selection in the human genome from haplotype structure Nature 419 832 837
M Gautier R Vitalis 2012 Rehh An R package to detect footprints of selection in genome-wide SNP data from haplotype structure Bioinformatics 28 1176 1177
O Delaneau C Coulonges JF Zagury 2008 Shape-IT: New rapid and accurate algorithm for haplotype inference BMC Bioinform. 9 540
BT Sherman 2022 DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update) Nucleic Acids Res. 50 W216 W221
M Kanehisa M Furumichi Y Sato Y Matsuura M Ishiguro-Watanabe 2025 KEGG: Biological systems database as a model of the real world Nucleic Acids Res. 53 D672 D677
Ajmone-Marsan, P., C. L., G. C. K. J. & L. J. A., eds. 2023. Genomic characterization of animal genetic resources. (FAO, 2023). https://doi.org/10.4060/cc3079en
M Barbato 2020 A genetically unique Chinese cattle population shows evidence of common ancestry with wild species when analysed with a reduced ascertainment bias SNP panel PLoS ONE 15 e0231162
SP Dixit 2021 Genome analyses revealed genetic admixture and selection signatures in Bos indicus Sci. Rep. 11 21924
R Gargiulo 2024 Estimation of contemporary effective population size in plant populations: Limitations of genomic datasets Evol. Appl. 17 e13691
N Singh 2015 Single-copy gene based 50 K SNP chip for genetic studies and molecular breeding in rice Sci. Rep. 5 11600
P Kathiravan KN Raja GK Sachdeva 2006 Present status, genetic improvement and future of indigenous draught cattle—a review J. Remount. Vet. Corps 45 2 79 87
S Panneerselvam N Kandasamy 2008 The Kangayam Cattle- A retrospective and prospective study Department of Animal Genetics and Breeding
Singh PK, P. R. A. S. K. S. G. M. A. K. Phenotypic characterization and performance evaluation of Hallikar cattle in its native tract. Indian J. Anim. Sci. 78, 211–214 (2008).
Natarajan A, S. K. V. K. Productivity performance of Kangayam cattle. Indian J. Anim. Sci. 82, 1440–1441 (2012).
A Kumar U Singh R Singh R Vinoo 2016 Genetic studies on production and reproduction traits of Ongole cattle at organized farms Indian J. Anim. Sci. 86 826 830
P Ratwan AK Chakravarty M Kumar 2021 Lactation wise performance of Sahiwal cattle at an organized herd of northern India Indian J Anim Sci 90 1179 1185
MI Mustafa M Latif MK Bashir B Ahmad 2002 Productive performance of Red Sindhi cattle under hot and humid environment of Balochistan province of Pakistan Pak. Vet. J. 22 4 151 157
L George ID Gupta PB Nandhini A Verma JP Achankunju 2021 Enhancement of production performance of Tharparkar cattle using lactation persistency as a selection tool Indian J. Anim. Res. https://doi.org/10.18805/ijar.b-4439
D Pitt 2019 Demography and rapid local adaptation shape Creole cattle genome diversity in the tropics Evol. Appl. 12 105 122
M Nath 1993 Punganur—the miniature Bos indicus cattle Anim. Genet. Resour. Inf. 11 57 60
S Iype 1996 The Vechur cattle of Kerala Anim. Genet. Resour. Inf. 18 59 64
PK Singh A Sharma 2017 Assessment of degree of endangerment of livestock breeds in India Indian J. Anim. Sci. 87 316 323
MSA Bhuiyan 2021 Unraveling the genetic diversity and population structure of Bangladeshi indigenous cattle populations using 50K SNP markers Animals 11 2381
W Barris 2012 Next generation sequencing of African and Indicine cattle to identify single nucleotide polymorphisms Anim. Prod. Sci. 52 133
FC Canavez 2012 Genome sequence and assembly of bos indicus J. Hered. 103 342 348
X Liao 2013 Whole genome sequencing of Gir cattle for identifying polymorphisms and loci under selection Genome 56 592 598
FA Corredor 2023 Genetic diversity and population structure of a Peruvian cattle herd using SNP data Front Genet. 14 1073843
H Mustafa 2017 Genome-wide SNPs analysis of indigenous zebu breeds in Pakistan Biotechnol. Anim. Husb. 33 13 25
EM Strucken 2021 Genetic diversity and effective population sizes of thirteen Indian cattle breeds Genet. Sel. Evol. 53 47
M Zerabruk 2012 Genetic diversity and admixture of indigenous cattle from North Ethiopia: Implications of historical introgressions in the gateway region to Africa Anim. Genet. 43 257 266
MGG Chagunda 2018 Use of High Density Single Nucleotide Polymorphism (SNP) Arrays to Assess Genetic Diversity and Population Structure of Dairy Cattle in Smallholder Dairy Systems: The Case of Girinka Programme in Rwanda Front Genet. 9 438
A Sharma 2016 A genome-wide assessment of genetic diversity and population structure of Korean native cattle breeds BMC Genet. 17 139
JE Decker 2014 Worldwide patterns of ancestry, divergence, and admixture in domesticated cattle PLoS Genet. 10 e1004254
N Chen 2018 Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia Nat. Commun. 9 2337
MN Mbole-Kariuki 2014 Genome-wide analysis reveals the ancient and recent admixture history of East African Shorthorn Zebu from Western Kenya Heredity (Edinb.) 113 297 305
S Kim 2018 Genetic diversity and divergence among Korean cattle breeds assessed using a BovineHD single-nucleotide polymorphism chip Asian-Australas. J. Anim. Sci. 31 1691 1699
AG Doran DP Berry CJ Creevey 2014 Whole genome association study identifies regions of the bovine genome and biological pathways involved in carcass trait performance in Holstein-Friesian cattle BMC Genom. 15 837
A Damerau T Gaber S Ohrndorf P Hoff 2020 JAK/STAT activation: A general mechanism for bone development, homeostasis, and regeneration Int. J. Mol. Sci. 21 9004
NA Sims 2020 The JAK1/STAT3/SOCS3 axis in bone development, physiology, and pathology Exp. Mol. Med. 52 1185 1197
S Sheet SS Jang JH Kim W Park D Kim 2024 A transcriptomic analysis of skeletal muscle tissues reveals promising candidate genes and pathways accountable for different daily weight gain in Hanwoo cattle Sci. Rep. 14 315
M Malvisi 2016 Responses of bovine innate immunity to Mycobacterium avium subsp. paratuberculosis infection revealed by changes in gene expression and levels of MicroRNA PLoS ONE 11 e0164461
SJ Arun 2015 Targeted analysis reveals an important role of JAK-STAT-SOCS genes for milk production traits in Australian dairy cattle Front Genet. 6 342
MZ Khan 2020 Role of the JAK-STAT pathway in bovine mastitis and milk production Animals 10 2107
YK Dhuriya D Sharma 2018 Necroptosis: A regulated inflammatory mode of cell death J. Neuroinflamm. 15 199
Z Yu N Jiang W Su Y Zhuo 2021 Necroptosis: A novel pathway in neuroinflammation Front Pharmacol. 12 701564
C Sandor 2012 Genetic variants in REC8, RNF212, and PRDM9 influence male recombination in cattle PLoS Genet. 8 e1002854
K Wang 2021 Whole-genome sequencing to identify candidate genes for litter size and to uncover the variant function in goats (Capra hircus) Genomics 113 142 150
Y Zhao 2022 Growth traits and sperm proteomics analyses of myostatin gene-edited Chinese yellow cattle Life 12 627
H Cheon 2013 IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage EMBO J. 32 2751 2763
Shaheen, M. & Broxmeyer, H. E. Cytokine/receptor families and signal transduction. In Hematology 163–175 (Elsevier, 2018). https://doi.org/10.1016/B978-0-323-35762-3.00016-0
P Lemal K May S König M Schroyen N Gengler 2023 Invited review: From heat stress to disease—immune response and candidate genes involved in cattle thermotolerance J. Dairy Sci. 106 4471 4488
MS Tahir 2021 Meta-analysis of heifer traits identified reproductive pathways in Bos indicus Cattle Genes (Basel) 12 768
E Kasuya 2016 Secretory pattern and regulatory mechanism of growth hormone in cattle Anim. Sci. J. 87 178 182
Flint, D. J. Regulation of milk secretion and composition by growth hormone and prolactin. In Intercellular Signalling in the Mammary Gland 131–140 (Springer, 1995). https://doi.org/10.1007/978-1-4615-1973-7_29
K Sakamoto 2005 Growth hormone acts on the synthesis and secretion of α-casein in bovine mammary epithelial cells J. Dairy Res. 72 264 270
RM Akers 2006 Major advances associated with hormone and growth factor regulation of mammary growth and lactation in dairy cows J. Dairy Sci. 89 1222 1234
RA Curi HN Oliveira de AC Silveira CR Lopes 2005 Association between IGF-I, IGF-IR and GHRH gene polymorphisms and growth and carcass traits in beef cattle Livest Prod. Sci. 94 159 167
MP Mullen 2011 Single nucleotide polymorphisms in the insulin-like growth factor 1 (IGF-1) gene are associated with performance in Holstein-Friesian dairy cattle Front Genet. 2 3
B Zhang 2012 Polymorphism in GHRH gene and its association with growth traits in Chinese native cattle Res. Vet. Sci. 92 243 246
R Kumar ID Gupta A Verma N Verma MR Vineeth 2015 Genetic polymorphisms within exon 3 of heat shock protein 90AA1 gene and its association with heat tolerance traits in Sahiwal cows Vet. World 8 932 936
TM Badri 2018 Genetic polymorphism in Hsp90AA1 gene is associated with the thermotolerance in Chinese Holstein cows Cell Stress Chaperones 23 639 651
AC Bouwman 2018 Meta-analysis of genome-wide association studies for cattle stature identifies common genes that regulate body size in mammals Nat. Genet. 50 362 367
JL Gualdrón Duarte 2023 Sequenced-based GWAS for linear classification traits in Belgian Blue beef cattle reveals new coding variants in genes regulating body size in mammals Genet. Sel. Evol. 55 83
AS Abdelmanova 2022 Genome-wide screening for SNPs associated with stature in diverse cattle breeds Diversity (Basel) 14 692
Acknowledgements
We would like to express our gratitude to the local farmers and veterinarians in Bangladesh, Cambodia, Sri Lanka, India, Pakistan, Myanmar, and Mongolia for their assistance during survey and sample collection. The authors acknowledge the support of the International Atomic Energy Agency for their financial support to the project.
Funding
International Atomic Energy Agency coordinated research project CRP D31030, International Atomic Energy Agency Technical cooperation projects MON5025, KAM5009, SRL5046 and MYA5022.
Author information
Authors and Affiliations
Contributions
Conceptualization, K-P, J-S and G-M; Methodology M-TK and R-P, Sample collection and Investigation; S-R, V-M, R-K, K-A, T-H, M-EB, LGS-L, E-T, M-P, S-T, MMU-B and M-P; Formal analysis, M-TK; Data curation, TK-M, M-B and K-P; Writing-original draft preparation, TK-M; Review and editing K-P, J-S and G-M; Project administration, K-P; Funding acquisition, K-P. All authors have read, reviewed and agreed to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mavunga, T.K., Sölkner, J., Mészáros, G. et al. Genomic diversity and selection signatures in Asian Zebu Cattle: insights into adaptation and genetic erosion. Sci Rep 15, 33346 (2025). https://doi.org/10.1038/s41598-025-14744-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-14744-z
