
Abstract
Chinese red steppe cattle, a highly developed breed for their meat and milk, are not well known about the formation mechanisms for high-quality beef. Furthermore, many fundamental investigations of cattle are hampered by deficient full-length cDNAs. Here, we utilized ONT full-length transcript sequencing from 9 bovine preadipocytes during 3 periods to find 2502 novel genes and 3253 novel transcriptomes, further improving the quality of reference genome annotation. At the same time, based on diverse expression patterns in 3 periods (early, middle and late), we identified 754 significantly differentially expressed genes (DEGs) and 932 significantly differentially expressed transcripts (DETs) in the first contrast group (0d_vs_4d), 197 significantly DEGs and 297 significantly DETs in the second contrast group (0d_vs_8d), 465 and 555 significantly DEGs and DETs in the third contrast group (4d_vs_8d). These DEGs mainly enriched the pathways related to cell differentiation such as Wnt signaling pathway and positive regulation of fat cell differentiation. A total of 35,616 alternative splicing (AS) events were detected, in which 111, 109 and 101 significantly different AS events were found in the group 0d_vs_4d, 0d_vs_8d and 4d_vs_8d respectively. In particular, two essential DEGs, N4BP2L1 and PRKAA1 related to cell differentiation were shown to take part in distinct AS events and were therefore considered as key candidate genes for improving fat deposition and meat quality in Chinese red steppe cattle. These findings provide novel insights into the adipocyte differentiation in cattle and metabolic processes that regulate fat storage, which is essential for optimizing growth and production.
Similar content being viewed by others
Introduction
With the development of the national economy, the improvement of living standards and the increase of people’s awareness of health, high-quality and more nutritious beef has gained more and more recognition1. Marbled beef, which is composed of fat deposited in the middle of the muscle fibers and distributed with white fat, has become one of the most popular meats among customers due to its higher nutritional value2. Intramuscular fat (IMF) in the skeletal muscle is the main factor affecting beef quality, including marbling, flavor, juiciness and tenderness3,4. The IMF content is rich in polyunsaturated fatty acids and its deposition is influenced complexly by several factors such as genetics, nutrition, and environment3,5.
The growth and development of adipose is a complicated biological process that includes the transition from fertilized egg to fibroblast and the determination from preadipocyte into mature adipocyte6. During the series of development periods, the proliferation and differentiation of pre-adipocytes are closely related to the formation of IMF, which is regulated by several transcription factors and genes related to metabolism1. So far, numerous studies have demonstrated that the bovine adipocyte differentiation was regulated by multiple molecules and signaling pathways including AMPK, PI3K-Akt and PPAR involved in lots of critical genes such as CDKs7, KLF68, PLIN19, FATP110, CD4411, Snail112, PPARG13,14 and DGAT215. Extensive miRNAs and lncRNAs were also reported to participate in the regulation of adipocyte differentiation16,17,18.
In recent years, Oxford Nanopore Technologies (ONT) sequencing technology has gained popularity because of its high throughput, ultra-long read length and accuracy in comparison to next-generation sequencing19,20. The technology is dedicated to translating specific electrical signals into nucleotide sequences based on the current flowing through the nanopore21,22. By detecting and validating alternative splicing (AS) events, numerous studies on the RNA transcriptome using ONT sequencing have revealed molecular genetic mechanisms. Additionally, some of these studies have significantly improved annotation of the domestic livestock genome, providing new insights into filling in gaps of the reference genome and uncovering additional genomic variation information23,24,25.
Chinese red steppe cattle, as one of the excellent cultivated breeds of “yellow” class of Chinese cattle have good meat quality, strong stress resistance and other outstanding advantages26,27. Although one study indicated MED4 differentiation may affect the preadipocyte differentiation and adipogenesis in Chinese red steppe cattle by regulating the PPARγ gene28, the regulatory mechanisms affecting fat deposition are complicated and difficult to claim. Therefore, our study is aimed to investigate the differences in intramuscular adipocyte differentiation at different stages in the adipocytes of Chinese red steppe cattle to understand the mechanism of intramuscular fat formation, the expression pattern of lipid metabolism genes, and the role of lipid metabolism genes in intramuscular adipocyte deposition.
Methods
Preadipocytes isolation and culture
Bovine preadipocytes, including days 0, 4, and 8, were acquired from Chinese red steppe cattle of the Jilin Academy of Agricultural Sciences. They were separated into two parts. One is for phenotype detection involved in Oil Red O staining and triglyceride content assaying; the other part is for ONT sequencing.
All instruments including centrifuge tubes, instrument trays, ophthalmic scissors, forceps, and sterile cotton materials were subjected to high-temperature sterilization via autoclaving and subsequently exposed to UV irradiation for 30 min in a biosafety cabinet. A sterile PBS solution containing 6% Penicillin-Streptomycin (Gibco), 0.2% type I collagenase (Sigma), 75% ethanol and sterile culture dishes. The sampling area and surgical instruments (ophthalmic scissors, forceps) were disinfected with 75% ethanol-saturated prior to subcutaneous adipose tissue harvest. The excised specimens underwent 5–6 sequential rinses with phosphate-buffered saline (PBS) supplemented with 6% Penicillin-Streptomycin to eliminate residual erythrocyte contamination. Tissue fragments were aseptically transferred to new P/S PBS solution in a Class II biosafety cabinet. Microdissection was performed using sterilized ophthalmic instruments to meticulously separate adipocyte clusters from associated vascular networks and fascial attachments. Following three PBS washes to remove connective tissue remnants, the purified adipose matrix was mechanically fragmented into 1–2 mm³ explants through cross-sectional mincing.
Transfer the small tissue fragments to a centrifuge tube and add 0.2% type I collagenase. Invert the tube to mix thoroughly and digest in a 37 °C water bath for 60 min, inverting gently every 5–10 min to ensure mixing. Following sufficient digestion, add an equal volume of complete culture medium (DMEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin) to terminate the reaction. Filter the digested mixture sequentially through a 200-mesh cell strainer to remove large undigested tissue fragments, followed by a 400-mesh cell strainer to remove larger cell aggregates and debris. Collect the filtrate in a centrifuge tube and centrifuge at 1500 rpm for 15 min. Discard the supernatant. Resuspend the pellet in an appropriate volume of red blood cell lysis buffer by gentle pipetting, incubate for 1–2 min at room temperature, and centrifuge again at 1500 rpm for 5 min. Discard the supernatant. Wash the resulting cell pellet by resuspending it in DMEM complete culture medium, centrifuging at 1500 rpm for 10 min, and discarding the supernatant; repeat this wash step three times. Finally, resuspend the cell pellet in fresh DMEM complete culture medium by gentle pipetting. Seed the cell suspension into T25 culture flasks and incubate at 37 °C in a humidified atmosphere containing 5% CO₂. After 1–2 days of culture, replace the medium to remove non-adherent cells. Subsequently, change the medium every 48 h.
Oil red O staining and triglyceride content assay
Discard the medium in the fat cells that need to be stained, wash them twice with PBS, then add the Oil Red O fixative solution. After 20 to 30 min in the incubator, discard the fixative solution. After washing twice with distilled water, add the configured 60% isopropanol, soak it for 5 min, and discard the isopropanol. The filtered Oil Red O staining solution was added and placed in the incubator for 10 ~ 20 min. We discard the staining solution and wash it with distilled water for 5 times until there is no residual staining solution. After counterstaining the nucleus for 1 ~ 2 min with Mayer staining solution, discard the staining solution and wash it 5 times with distilled water. Finally, add Oil Red O buffer to soak for 1 min, discard, cover the cells with distilled water, and proceed to the microscope for photographing. Following photo capture, the distilled water in the culture was aspirated, 1 mL of isopropanol solution was added, and the lipid droplets were extracted at room temperature for 10 min. A wavelength of 490 mm was chosen for the OD measurement after 100 µL of extract was applied to each well of a 96-well plate and put in a microplate reader.
The medium in the adipocytes was discarded and washed twice with PBS. The cells were collected to the bottom of the dish with a cell scraper. After standing at room temperature for 10 min, a metal bath at 70 °C for 10 min, and centrifugation at room temperature of 2000 r/min for 5 min, 200 µL of cell lysate was added, and the proper amount of supernatant was taken in a 1.5 mL centrifuge tube. The supernatant was taken for triglyceride content determination. After adding the supernatant to the 96-well plate, the standards, samples, and working solution were added in accordance with kits instructions, mixed, and incubated for 30 min. They were then placed in the microplate reader, and the 550 mm wavelength was selected for OD determination and subsequent calculation.
ONT sequencing and quality control
Total RNA was extracted from bovine preadipocytes in the lab of the Jilin Academy of Agricultural Sciences for ONT sequencing, including 1 control group, 1 TRIAL1 group, and 1 TRIAL2 group, respectively, during days 0, 4, and 8 of cell differentiation with 3 repeats for each group. The standard differentiation protocol spans an 8-day cycle. During the initial 2 days, cells are cultured in induction medium to initiate differentiation, followed by maintenance medium for days 3–4 to support early differentiation events. From days 5–8, cells are switched to normal medium to facilitate maturation. Lipid droplets become visibly detectable by day 4, with the subsequent phase (days 5–8) primarily involving lipid accumulation and terminal differentiation. Thus we chose days 0, 4, 8 as early, middle and late cell differentiation. The purity of RNA was assessed by spectrophotometry, with OD260/280 ratios ranging between 1.9 and 2.1. Agarose gel electrophoresis further confirmed RNA integrity, revealing three distinct bands corresponding to the 28 S, 18 S, and 5 S ribosomal RNA subunits.
A 500 ng qualified total RNA sample was taken and the volume was adjusted to 9 ul using nuclease free water. Link the reverse transcription primer to carry out reverse transcript. Then mix 5 µL reverse-transcribed RNA sample, 1.5 µL PCR barcode, 18.5 µL nuclease-free water and 25 µL 2x LongAmp Taq Master Mix to start to amplify. AMPure beads were used for purification. 500 ng purified cDNA and Rapid Adapter were used to connect sequencing splices. Finally load the library onto the R9.4 sequencing chip for computer sequencing for 48 ~ 72 h after configuring the library with 24 µL cDNA connected products, 75 µL squencing buffer and 51 µL loading beads.
The raw fast5 data, which contains all the raw sequencing signals after Nanopore sequencing, was converted into the fastq format after using GUPPY (version 5.0.16)19,29 for base calling. Raw reads were filtered with read quality score < 7 (~ 20% error probability) and read length < 500 bp using NanoFilt (version 2.8.0, parameters: -q 7 -l 50)30. Seqkit (version: 0.12.0, parameters: default) was used for data statistics31.
Full-length sequences and consensus sequences identified
The full-length sequences were identified using Pychopper (version: 2.4.0, parameters: -Q 7 -z 50)30. Pinfish (version: 0.1.0, parameters: default) was utilized to quickly construct non-redundant transcript sets for full-length sequences by calling the program minimap2 (version 2.17-r941, parameters: -ax splice -uf -k14) to map the full-length sequences to the reference genome and the program spliced_bam2gff to convert bam alignments to gff format, then calling assembly cluster_gff, collapse_partials and polish_clusters for clustering, eliminating redundancy and checking32. The reference genome and the gff annotation file were LU_Bosgru_v3.0 (GCA_005887515.1) from the annotation database Ensembl Genes 114. We used StringTie (version 2.1.4, parameters: -conservative -L -R) to remove redundancy of transcripts and merge alignment with differences in exons only at the 5’ end33.
Novel genes and novel transcripts authenticating
We ultilized gffcompare (version 0.12.1, parameters: -R -C -K -M)34 to compare the identified transcripts with the known transcripts in the reference genome. Transcripts with class code types of “i, j, o, u, x” by gffcompare are considered new transcripts, and those with class code types of “u” are regarded as new genes. Newfound transcript coding sequences were predicted by TransDecode (version 5.5.0, parameters: -m 50, -single_best_only)35.
Analysis of different expressed transcripts and genes
To evaluate the repeatability and correlation of samples, Principal Component Analysis (PCA) and Pearson correlation coefficients were carried out using the expression quantity of transcripts and genes. PCA was performed to evaluate global transcriptional or gene expression across samples and assess the reproducibility of biological replicates using the princomp function in the software package R (v4.0.5) stats (v4.0.5) based on normalized expression values. The Pearson correlation coefficients were calculated by using the built-in function “cor” of R. For biological duplication, the Pearson correlation coefficient is at least greater than 0.9. To minimize technical noise and improve the statistical power of downstream differential expression analysis, low-abundance genes or transcripts were excluded: only those with a total read count ≥ 10 across all samples in both experimental and control groups were retained. The differentially expressed genes and transcripts between different groups were identified with padj < 0.05 or p value < 0.05 and |log2FoldChange| > 1 using DESeq2 (version 1.26.0)36, applying the Wald test to assess the statistical significance of expression changes between experimental conditions and applied the Benjamini-Hochberg procedure to control the false discovery rate (FDR) at a threshold of < 5% into DESeq236. These identified DEGs were analyzed for GO term37 and KEGG38,39 pathway analysis using ClusterProfiler (version 3.14.3) via the hypergeometric test with Benjamini-Hochberg correction to control the FDR40.
Transcripts structure analysis
Gffcompare (version 0.12.1, parameters: -R -C -K -M) was exploited to optimize the transcript structure of the original annotation by correcting transcript boundaries34. SUPPA2 (parameters: -f ioe -e SE SS MX RI FL) was used to classify the AS events41. For the AS events analysis, we first used Salmon (version: 1.4.0) to perform the expression quantity analysis for transcripts and genes to gain transcripts per million (TPM) values42, then calculated a percentage or proportion spliced-in (PSI) matrix which quantify relative abundances of the splicing events or transcript isoforms based on TPM matrix using SUPPA2, and finally obtained all AS events in each sample. Different AS events between each period were stated based on the module DiffSplice of SUPPA241. Long noncoding RNAs (LncRNA), defined with transcripts longer than 200nt and not encoding proteins, were identified with three strategies: CNCI (version 2.0, parameters: default)43 aimed at predicting incomplete transcripts and antisense transcript pairs, CPC2 (version: standalone_python3 v1.0.1)44 based on the biological sequences of each transcript coding frame to evaluate the encoding potential, and Pfam45.
Results
Differences in preadipocyte differentiation during different periods
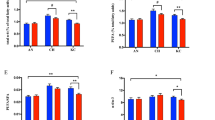
The preadipocytes on the 0th, 4th, and 8th days showed obviously distinct differentiation patterns and abilities following Oil Red O staining (Fig. 1a). The Oil Red O staining and triglyceride assay results demonstrated that both lipid droplet content and triglyceride levels on day 4 were higher than those on day 0, though not to a statistically significant extent (Fig. 1b). However, by day 8, the lipid droplet accumulation and triglyceride concentrations showed highly significant (P < 0.001), and significant (P < 0.01) increases compared to day 0 and day 4, respectively.

Oil Red O and triglyceride contents assaying. (A) The plots for preadipocyte differentiation at days 0, 4, and 8 after staining Red Oil O; (B) The lipid drop contents and triglyceride contents examined at days 0, 4, and 8. NS represented not significant (P > 0.05); *** significant (P < 0.01); **** highly significant (P < 0.001).
ONT sequencing and full-length reads identifying
Based on the visibly different appearances above during 3 periods, ONT sequencing was performed from 9 bovine preadipocytes on days 0, 4, and 8 to excavate the different expressed genes in different stages. In total, 89,507,373 raw reads were generated via high-throughput sequencing (Table S1). Following the removal of low-quality reads, 83,193,120 clean reads with an average read length of 667 bp were obtained and 71,146,002 full-length reads were identified (Table S1, S2). Finally, 78,602 circular consensus sequences were generated after mapping on the LU_Bosgru_v3.0 reference genome and removing the redundant (Table S3).
Novel genes and novel transcripts authenticating
The analysis of all the samples resulted in the identification of a total number of 47,237 transcripts and 28,561 genes (Fig. 2A). Among them, there were 1,548 genes that exhibited novel transcripts and were consequently classified as novel genes. Ultimately, it was determined that 8.76% of the identified genes could be verified as novel, improving the cattle genome annotation to some extent. On average, more than 3,000 genes were identified in each sample. A significant proportion of genes exhibit 1 to 4 transcripts, while only a minority of them display more than eight transcripts (Fig. 2B). About 67.43% of the identified genes produced only one single transcript, suggesting that a portion of the genes underwent variable splicing. We further found that all transcripts are evenly distributed across the chromosomes (Fig. 2C). Compared with known transcripts, 3,253 transcripts were newly found, in which the code “i,” which denotes a transcript that is fully contained within a reference intron, accounts for the largest proportion, reaching 49.65% (Fig. 2D). The second largest portion, or almost 30% of the transcripts, is accounted for by the code “u,” which denotes unknown transcripts. The quantities of other codes “j” (multiple exons in which at least one matched), “x” (exon overlapped on the anti-chain) and “o” (other parts in the same chain that overlap with the reference exon) are 423, 258 and 2 transcripts representatively (Fig. 2D).

Gene and transcript distribution results across all samples. (A) The number of genes and transcripts mapped to the reference (the red represents gene; the cyan represents transcript); (B) The bar plot for gene numbers with varying transcript counts; (C) The transcript density distribution plot (from the inside to outside of the circle are the novel transcripts, the known transcripts and all transcripts); (D) The number distribution of novel transcripts including five types (the code “i” meaning the transcripts completely contained in the introns of the reference transcripts, “j” meaning the transcripts with multiple exons in which at least one matched, “o” meaning other parts in the same chain that overlap with the reference exon, “u” meaning unknown novel transcripts and “x” meaning exons overlapped on the anti-chain).
Analysis of differentially expressed genes (DEGs) and transcripts (DETs)
To investigate the correlations among all the samples, PCA analysis was performed using the gene and transcript expression amounts. Samples with three biological repeats are clustered together, making it easy to identify distinct groupings (Figure S1). Additionally, the Pearson correlation coefficient heat map revealed that the coefficient was above 0.9 in different individuals at the same stage of differentiation, showing better relativity (Figure S2).
Finally, we totally identified 754 significantly DEGs and 932 significantly DETs in the first contrast group (0d_vs_4d), including 401 up regulated genes, 501 up regulated transcripts, 353 down regulated genes, and 431 down regulated transcripts. In the second contrast group (0d_vs_8d), we found 197 significantly DEGs and 297 significantly DETs, including 120 up regulated genes, and 178 up regulated transcripts and further identified 77 down regulated genes and 119 down regulated transcripts. In the third contrast group (4d_vs_8d), 465 and 555 significantly DEGs and DETs, respectively, were identified, which include 246 up regulated genes, 293 up regulated transcripts and 219 down regulated genes, 262 down regulated transcripts (Fig. 3A, C). Genes were ranked by |log2FoldChange| > 5 and padj < 0.05. The top 10 DEGs in the 0/4-day and 4/8-day comparisons included NTS, SOST, OXT, SLPI, VEPH1, ASB9, C16H1orf158, CD24, PDK4, ACTC1, and SOST, MT1E, CCL19, CD24, MEOX1, MMP9, WFDC10A, HAS2, ASB9, C16H1orf158, whereas only one extremely significant DEG MMP9 was identified in the 0/8-day comparison (Fig. 3A). There are 29 shared DEGs and 37 shared DETs among three different contrast groups (Fig. 3B, D). Due to some DEGs being not annotated, we just displayed the number of annotated genes here. And the results on the identified DEGs between each period were shown in Table S4-S6. Particularly, some certain transcription factors and candidate genes involved in adipocyte differentiation, adipogenesis and lipid metabolism were identified in our results, such as PEG10, ADIRF, PLSCR4, BAMBI, FST, FOS, INHBA, RUNX2, SERPINE1, LTBP1, HIF1A, CCL2, EGR1, and MAF (Table S4-S6). The cluster analysis heatmap of DEGs indicated different gene expression patterns during the early, middle and late cell differentiation, as is illustrated in Figure S3. Especially, genes exhibiting high expression levels such as ENSBTAG00000019203 (S100A4), ENSBTAG00000033319 (CD200), ENSBTAG00000000088 (ALCAM) and ENSBTAG00000016766 (TMEM176B) during the early phase of cell differentiation demonstrated significantly reduced expression at the mid-differentiation stage, whereas those with low initial expression showed reciprocal upregulation during the same transitional period (Figure S3). The clustering dendrogram with these highly expressed genes revealed coordinated biological functions, particularly for S100A4, CD200 and TMEM176B genes enriched in extracellular region (GO:0005576), negative regulation of cell population proliferation (GO:0008285) and cell differentiation (GO:0030154), which may function in stage-specific regulatory switches during early stage of differentiation.

The identified DEGs and DETs in different groups. (A) The volcano map for DEGs; (B) The Venn diagram for DEGs; (C) The volcano map for DETs; (D) The Venn diagram for DETs; (E) The Sankey diagram for parts of pathways of DEGs.
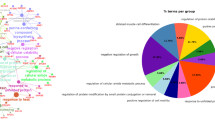
Additionally, the results of the enrichment analysis of significantly DEGs (adjust p value < = 0.05) of the three groups by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) revealed similar pathways related to the cellular response, cell migration, and many other biological processes in cell differentiation. The results of the GO and KEGG enrichment analyses of these DEGs are displayed in the Supplementary material (Figure S4). There are 123 pathways notably enriched in three groups with p.adjust value ≤ 0.05, mainly including Wnt signaling pathway (hsa04310), Cellular senescence (hsa04218), positive regulation of fat cell differentiation (GO:0045600) and regulation of lipid metabolic process (GO:0019216), which showed predominant association with cell proliferation, migration, differentiation and lipid metabolic processes. (Fig. 3E). Particularly, we found some overlapped DEGs were enriched into some common pathways, such as PLK2, TCIM, POSTN, CXCL8 and IL1RN. We focused on the regulation of the lipid metabolic process (GO:0019216) and found that it completes the biological process by regulating the primary metabolic and cellular process (Figure S5).
Structure analysis
We totally identified 7 AS forms in the preadipocyte of cattle and obtained 35,616 AS events in all samples, including 6,992 Alternative 3’ splice site (A3), 6,064 Alternative 5’ splice site (A5), 6,081 Alternative First exon (AF), 2,325 Alternative Last exon (AL), 520 Mutually exclusive exon (MX), 4,014 Retained intron (RI) and 9,620 Skipping exon (SE) respectively, using SUPPA2 (Fig. 4A, Table S7). Among all the AS events, SE is the most dominant splicing form in the differentiation of preadipocytes, accounting for about 27.01%. MX has the lowest probability of occurrence during all periods, approximately 1.46%.

The distribution of AS events. (A) The pie chart of seven AS events in all samples; (B) The bar plot for AS events that are significantly different in each sample; C-I. The upset plot for A3, A5, AF, AL, MX, RI, and SE in each group (The horizontal bar chart at the bottom left shows the number of AS events for each group, and the dot plot at the bottom right, together with the bar chart at the top, represent the number of corresponding AS events shared between the corresponding groups).
Throughout the three stages, various types and diverse amounts of AS were detected. Nevertheless, the number of AS on days 0 and 4 was higher than on day 8 (Fig. 4B). Furthermore, for a group of packets, the AS events must be detected at the same time for different repeats to count as a valid event. Seven different and valid AS events also showed clear differences in three groups (0d, 4 d and 8 d) (Fig. 4C-I). Finally, we acquired 1,914 different AS events and 111 significantly different AS events in the first contrast group (0d_vs_4d), 1,781 distinct AS events and 109 significantly different AS events in the second contrast group (0d_vs_8d) and 1,744 different AS events and 101 significantly different AS events in the third contrast group (4d_vs_8d) (Fig. 5A, Table S8). The differential AS events between each period were illustrated in Table S9-S11.

The AS events and LncRNA across different periods of preadipocytes. (A) The bar plot for the number of significantly different AS events; (B) One example of MX (N4BP2L1); (C) One example of SE (PRKAA1); (D) Venn diagram illustrating the results of the LncRNA prediction.
Moreover, we identified significant DEGs that corresponded to different AS events. A total number of 768 significant DEGs were found in different stages of preadipocyte differentiation, among which 21 had significant alternative splicing events annotated as ARL4A, ACIN1, ATP2B1, DUT, ECHDC1, G6PD, INPP1, ME1, MPST, PKIG, TMEM119, TNIP1, CXCL12, N4BP2L1, GSTM3, ENAH, MCUB, PRKAA1, IFI6, RAP1GDS1 and TPM1 (Table S12). Among the 21 DEGs exhibiting significant alternative splicing, AF events were the most abundant (52.38%), followed by SE events (28.57%). During cell differentiation, TNIP1 undergone two distinct AS events: an alternative first exon (AF) event is observed between days 0–4, while a retained intron (RI) event occurs between days 4–8 (Table S12). These stage-specific splicing shifts suggested TNIP1 may generate functionally divergent isoforms to orchestrate transcriptional reprogramming critical for adipogenesis. Two DEGs, N4BP2L1 and PRKAA1, with remarkable different AS events were identified in two groups (Fig. 5B, C). The MX occurred across the first and second group with DPSI values greater than 0.1 and P values less than 0.05, annotated as the gene N4BP2L1 (Fig. 5B). The SE occurred on the gene PRKAA1, showing the diverse patterns across the second and third groups, but no significant changes during days 0 and 4 with a P value higher than 0.05, as also depicted in Fig. 5C (Fig. 5C). Additionally, the coding potential of newly identified transcripts was predicted, and we discovered 2,687 LncRNA shared across CNCI, CPC2 and pfam (Fig. 5D).
Discussion
Despite the fact that bovine reference genomes have been updated frequently in recent years from ARS-UCD1.1 to 2.0 assembly, compared to the genomes of other species, such as humans or mice, the annotation of the bovine genome is still incomplete46,47,48. Detailed functional information is still lacking in some regions, especially in some complex gene regulatory or non-coding regions. Due to a large amount of genetic variations and the complexity of the cattle genome, different expression patterns, AS and lncRNA are highly prevalent23. Understanding how regulatory gene expression, cellular differentiation and functional diversity are essential. However, short-read technologies (RNA-Seq) hinder the prediction of full-length transcriptomes and often struggle to resolve complex transcript isoforms. Here, ONT sequencing was performed in 9 preadipocytes of cattle across early, middle and late differentiation (3 duplicates in each group). Totally, 78,602 circular consensus sequences were generated. 2502 novel genes and 3253 novel transcriptomes were identified. These results all further fill in gaps in the reference genome annotation of cattle, providing new insights into searching for potential genetic variations. Furthermore, by using Oil Red O to stain the preadipocytes at these three distinct stages of differentiation, we were able to find distinct cell patterns, suggesting there may be different expression genes and regulatory mechanisms during the differentiation stages.
The process of adipocyte differentiation is complex and involves the interaction of extracellular signals, transcription factors, hormones, and components of the cellular microenvironment. Studies have shown peroxisome proliferator-activated receptor-γ (PPARγ) was the master adipogenic regulator49. Adipogenic stimuli trigger signaling cascades, such as WNT and TGFβ superfamily pathways, which regulate transcription factors to drive stem cell differentiation toward adipogenesis. In our study, we excavated a total of 768 significant DEGs and 1216 significant DETs related to preadipocyte differentiation across three mentioned periods using ONT sequencing. However, compared with more duplicates, using three biological replications will reduce call rate (the total number of reported positives/the total number of transcripts) and true positive rate (TPR) of DE calls, but have no major effect on false positive rate (FPR)50. That means the DEGs or DETs with low abundance in our study might not be detected. But in other words, what we use is cell duplication, not individuals, and the stability will be better. Three distinct contrast groups displayed 29 shared DEGs and 37 shared DETs, including PLK251, CNN152, TNFSF1553, POSTN54 and many other genes related to cell proliferation and differentiation. Polo-like kinases (PLKs), a family of serine-threonine kinases, were confirmed to participate in regulation on diverse cellular processes, in which PLK2 was predicted to regulate the proliferation and invasion of glioblastoma cells55 and promote microglial activation via regulation of the HSP90α/IKKβ pathway56. CNN1 was reported to inhibit the invasion, migration and EMT of lung squamous cell carcinoma cells by DKK1/Wnt/β-catenin/c-myc signaling pathway57, also involved in differentiation and thermogenesis of porcine beige adipocytes58. POSTN has been identified as the direct target gene of miR-489 and is involved in the formation and differentiation of adipocytes in mice59. Despite the well-documented roles of these genes in murine or other animals’ adipogenesis, their functional conservation in bovines has been persistently ambiguous due to insufficient comparative transcriptomic data. Our research proposed a hypothesis that the POSTN gene may affect the differentiation of bovine preadipocytes through the cellular response to fibroblast growth factor stimulus pathway, establishing a novel regulatory model for adipogenesis in ruminants. Also, the results of enrichment analysis of significant DEGs showed similar functions relevant to cellular process, immune response and metabolic process. We also identified the classical pathways that are important for cell proliferation, differentiation, migration and apoptosis, including the activation of MAPK (AVPI1|PEA15|FGF2|PRKAA1|ITGA1)60, the Wnt signaling pathway (RSPO3|WNT5B|WIF1|SERPINF1|SFRP4)61, and the positive regulation of fat cell differentiation (WNT5B|RARRES2|ID2|WIF1)62. The MAPK signaling is a highly conserved signal transduction pathway60. Previous studies have confirmed that MAPK can be activated by a variety of extracellular signals and trigger a cascade of protein phosphorylation events, resulting in the regulation of adipogenesis, migration, apoptosis and survival60,63,64. The Wnt signaling pathway is a fundamental regulator of cell development, differentiation, and tissue homeostasis. It has a role in regulating processes in the body such as embryogenesis, stem cell maintenance, cell proliferation, migration, and polarity65. The canonical pathway in Wnt can make the activation of the expression of Wnt target gene involved in promoting cell proliferation, survival, and differentiation.
The study of AS plays a crucial role in understanding gene expression regulation and its impact on cellular diversity. By choosing distinct exon combinations during transcription, AS enables a single gene to produce multiple mRNA variants. This process contributes to the generation of protein isoforms with potentially distinct functions, localization, and interactions, significantly expanding the functional capacity of the genome.
In the present study, 321 significantly different AS events were identified, and by annotating these AS events, 21 significant DEGs were obtained. Remarkably, during the early and middle stages of preadipocyte differentiation as well as the late stages, one DEG N4BP2L1 with divergent MX type was discovered. Previous studies indicated that N4BP2L1 regulated adipocyte differentiation, as its knockdown significantly reduced lipid accumulation and downregulated key adipogenic markers (e.g., C/ebpa, C/ebpb, and Pparg), highlighting its essential role in adipogenesis66. Additionally, it was revealed that tissues with lipid development showed a trend of high expression of this gene66. Moreover, USF1 can directly bind to the Ebox in the N4BP2L1 promoter involved in adipocyte differentiation67. These findings all suggested that our newly identified differentially expressed gene N4BP2L1 likely orchestrated adipocyte differentiation via alternative splicing-driven isoform switching (MX), generating transcripts with structurally and functionally distinct exon compositions, critical for lipid accumulation and cell maturation. We also found a significantly different expressed gene called PRKAA1 with the SE type across different periods. PRKAA1 plays a key role in cell cycle regulation, participating in cell cycle progression, cell proliferation, cell apoptosis and other processes. Studies have shown that it can promote proliferation and inhibit apoptosis of gastric cancer cells68. In this study, PRKAA1 expression exhibited temporal dynamics of splice variants across three differentiation stages. These isoforms may orchestrate differentiation through modulating apoptotic suppression, MAPK activation, and PI3K/AKT signaling, suggesting a multifaceted regulatory role in cellular commitment.
Conclusions
In total, our results greatly improved the annotation information of the cattle reference genome and laid a foundation for the application of the cattle genome in molecular breeding. Also, we identified abundant genes as key candidates for bovine adipocyte differentiation such as PLK2, CNN1, TNFSF15, POSTN, TNIP1, N4BP2L1 and PRKAA1, providing new insights for understanding the accumulation of fat and improving the meat quality of Chinese red steppe cattle.
Data availability
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive69 in National Genomics Data Center70, China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA026449) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa.
References
Luo, M. et al. IMF deposition CeRNA network analysis and functional study of HIF1a in Yak. Front. Vet. Sci. 10, 1272238 (2023).
Nguyen, D. V., Nguyen, O. C. & Malau-Aduli, A. E. O. Main regulatory factors of marbling level in beef cattle. Vet. Anim. Sci. 14, 100219 (2021).
Zhang, M. et al. The landscape of DNA methylation associated with the transcriptomic network of intramuscular adipocytes generates insight into intramuscular fat deposition in chicken. Front. Cell. Dev. Biol. 8, 206 (2020).
Park, S. J. et al. Genetic, management, and nutritional factors affecting intramuscular fat deposition in beef cattle - A review. Asian-Australas J. Anim. Sci. 31 (7), 1043–1061 (2018).
Ma, X. et al. RNA-seq analysis reveals the critical role of the novel LncRNA BIANCR in intramuscular adipogenesis through the ERK1/2 signaling pathway. J. Anim. Sci. Biotechnol. 14 (1), 21 (2023).
Ntambi, J. M. & Young-Cheul, K. Adipocyte differentiation and gene expression. J. Nutr. 130 (12), 3122S–3126S (2000).
Pan, C. et al. Genome-wide identification of cyclin-dependent kinase (CDK) genes affecting adipocyte differentiation in cattle. BMC Genom. 22 (1), 532 (2021).
Raza, S. H. A. et al. RNA-Seq reveals the potential molecular mechanisms of bovine KLF6 gene in the regulation of adipogenesis. Int. J. Biol. Macromol. 195, 198–206 (2022).
Li, S., Raza, S. H. A., Zhao, C., Cheng, G. & Zan, L. Overexpression of PLIN1 promotes lipid metabolism in bovine adipocytes. Animals (Basel) 10(11), 1944 (2020).
Liu, X. et al. The effect of FATP1 on adipocyte differentiation in Qinchuan beef cattle. Animals (Basel) 11(10), 2789 (2021).
Li, G. et al. Lipid regulatory element interact with CD44 on mitochondrial bioenergetics in bovine adipocyte differentiation and lipometabolism. J. Agric. Food Chem. 72 (31), 17481–17498 (2024).
Tang, X., Sui, X., Weng, L. & Liu, Y. SNAIL1: linking tumor metastasis to immune evasion. Front. Immunol. 12, 724200 (2021).
Ramayo-Caldas, Y. et al. A marker-derived gene network reveals the regulatory role of PPARGC1A, HNF4G, and FOXP3 in intramuscular fat deposition of beef cattle. J. Anim. Sci. 92 (7), 2832–2845 (2014).
Costa, V. et al. PPARG: Gene Expression Regulation and Next-Generation Sequencing for Unsolved Issues. PPAR Res 2010. (2010).
Roe, N. D., Handzlik, M. K., Li, T. & Tian, R. The role of Diacylglycerol acyltransferase (DGAT) 1 and 2 in cardiac metabolism and function. Sci. Rep. 8 (1), 4983 (2018).
Guo, H. et al. RNA-Seq reveals function of Bta-miR-149-5p in the regulation of bovine adipocyte differentiation. Animals (Basel) 11(5), 1207 (2021).
Hu, C. et al. miR-10167-3p targets TCF7L1 to inhibit bovine adipocyte differentiation and promote bovine adipocyte proliferation. Genomics 116 (5), 110903 (2024).
Li, X. et al. LncRNA BlncAD1 modulates bovine adipogenesis by binding to MYH10, PI3K/Akt signaling pathway, and miR-27a-5p/CDK6 axis. Journal Agricultural Food Chemistry 72(19), 11094–11110 (2024).
Wang, Y., Zhao, Y., Bollas, A., Wang, Y. & Au, K. F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 39 (11), 1348–1365 (2021).
Goodwin, S., McPherson, J. D. & McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17 (6), 333–351 (2016).
Athanasopoulou, K., Boti, M. A., Adamopoulos, P. G., Skourou, P. C. & Scorilas, A. Third-Generation sequencing: the spearhead towards the radical transformation of modern genomics. Life (Basel) 12(1), 30 (2021).
Jain, M., Olsen, H. E., Paten, B. & Akeson, M. The Oxford nanopore minion: delivery of nanopore sequencing to the genomics community. Genome Biol. 17 (1), 239 (2016).
Gao, J., Xu, G. & Xu, P. Full-length transcriptomic analysis reveals osmoregulatory mechanisms in coilia nasus eyes reared under hypotonic and hyperosmotic stress. Sci. Total Environ. 799, 149333 (2021).
<13. Identifying and Validating Alternative Splicing Events.pdf>.
Beiki, H. et al. Improved annotation of the domestic pig genome through integration of Iso-Seq and RNA-seq data. BMC Genom. 20 (1), 344 (2019).
Hu, M. et al. Assessing Genomic Diversity and Signatures of Selection in Chinese Red Steppe Cattle Using High-Density SNP Array. Animals (Basel) 13(10). (2023).
Li, G. et al. RNA-Seq analysis identifies differentially expressed genes in the longissimus dorsi of Wagyu and Chinese red steppe cattle. Int J. Mol. Sci 24(1), 387 (2022).
Wei, M. et al. MED4 gene positively affects preadipocyte differentiation in Chinese red steppe cattle. Biocell 47 (9), 2115–2123 (2023).
Wick, R. R., Judd, L. M. & Holt, K. E. Performance of neural network basecalling tools for Oxford nanopore sequencing. Genome Biol. 20 (1), 129 (2019).
De Coster, W. et al. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34(15), 2666–2669 (2018).
Zou, Q., Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: A Cross-Platform and ultrafast toolkit for FASTA/Q file manipulation. PloS One 11(10), e0163962 (2016).
Li, H. et al. Genome project data processing S: the sequence alignment/map format and samtools. Bioinformatics 25 (16), 2078–2079 (2009).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33 (3), 290–295 (2015).
Pertea, G. & Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Res 9. (2020).
Tang, S., Lomsadze, A. & Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 43 (12), e78–e78 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated Estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550 (2014).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nat. Genet. 25 (1), 25–29 (2000).
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y. & Hattori, M. The KEGG resource for Deciphering the genome. Nucleic Acids Res. 32 (Database issue), D277–280 (2004).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53 (D1), D672–d677 (2025).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. ClusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16 (5), 284–287 (2012).
Trincado, J. L. et al. SUPPA2: fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 19 (1), 40 (2018).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 14 (4), 417–419 (2017).
Sun, L. et al. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 41 (17), e166 (2013).
Kong, L. et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res 35, W345–349 (2007).
Finn, R. D. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44 (D1), D279–D285 (2016).
Rosen, B. D. et al. De Novo assembly of the cattle reference genome with single-molecule sequencing. Gigascience 9(3), giaa021 (2020).
Harrow, J. et al. GENCODE: the reference human genome annotation for the ENCODE project. Genome Res. 22 (9), 1760–1774 (2012).
Frankish, A. et al. GENCODE: reference annotation for the human and mouse genomes in 2023. Nucleic Acids Res. 51 (D1), D942–D949 (2023).
Cristancho, A. G. & Lazar, M. A. Forming functional fat: a growing Understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol. 12 (11), 722–734 (2011).
Robles, J. A. et al. Efficient experimental design and analysis strategies for the detection of differential expression using RNA-Sequencing. BMC Genom. 13 (1), 484 (2012).
Zhang, C., Ni, C. & Lu, H. Polo-Like kinase 2: from principle to practice. Front. Oncol. 12, 956225 (2022).
Tang, J. et al. Arterial Sca1 + Vascular stem cells generate de Novo smooth muscle for artery repair and regeneration. Cell. Stem Cell. 26 (1), 81–96e84 (2020).
Xu, W. D., Li, R. & Huang, A. F. Role of TL1A in inflammatory autoimmune diseases: A comprehensive review. Front. Immunol. 13, 891328 (2022).
Wang, Z. et al. Periostin: an emerging activator of multiple signaling pathways. J. Cell. Commun. Signal. 16 (4), 515–530 (2022).
Tan, S., Zhao, J. & Wang, P. DYRK1A-mediated PLK2 phosphorylation regulates the proliferation and invasion of glioblastoma cells. International J. Oncology 63(2), 94 (2023).
Cheng, J. et al. Polo-like kinase 2 promotes microglial activation via regulation of the HSP90α/IKKβ pathway. Cell. Rep. 43 (10), 114827 (2024).
Liu, W., Fu, X. & Li, R. CNN1 regulates the DKK1/Wnt/β-catenin/c-myc signaling pathway by activating TIMP2 to inhibit the invasion, migration and EMT of lung squamous cell carcinoma cells. Experimental Therapeutic Med. 22 (2), 855 (2021).
Zhang, L. et al. Functional and genetic characterization of Porcine beige adipocytes. Cells 11(4), 751 (2022).
Qi, R. et al. MicroRNA-489-3p promotes adipogenesis by targeting the Postn gene in 3T3-L1 preadipocytes. Life Sci. 278, 119620 (2021).
Li, B. et al. MAPK family genes’ influences on myogenesis in cattle: Genome-wide analysis and identification. Res. Vet. Sci. 159, 198–212 (2023).
Hayat, R., Manzoor, M. & Hussain, A. Wnt signaling pathway: A comprehensive review. Cell. Biol. Int. 46 (6), 863–877 (2022).
Sul, H. S., et al. Regulation of Fat Synthesis and Adipose Differentiation. In: Progress in Nucleic Acid Research and Molecular Biology. Edited by Moldave K, vol. 60: Academic Press; : 317–345. (1998).
Wang, L. et al. MiR-145 reduces the activity of PI3K/Akt and MAPK signaling pathways and inhibits adipogenesis in bovine preadipocytes. Genomics 112 (4), 2688–2694 (2020).
Widmann, C., Gibson, S., Jarpe, M. B. & Johnson, G. L. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol. Rev. 79 (1), 143–180 (1999).
Yu, M. et al. The evolving roles of Wnt signaling in stem cell proliferation and differentiation, the development of human diseases, and therapeutic opportunities. Genes Dis. 11 (3), 101026 (2024).
Watanabe, K., Yokota, K., Yoshida, K., Matsumoto, A. & Iwamoto, S. A novel upstream transcription factor 1 target gene N4bp2l1 that regulates adipogenesis. Biochem. Biophys. Rep. 20, 100676 (2019).
Watanabe, K., Matsumoto, A., Tsuda, H. & Iwamoto, S. N4BP2L1 interacts with dynactin and contributes to GLUT4 trafficking and glucose uptake in adipocytes. J. Diabetes Investig. 12 (11), 1958–1966 (2021).
Zhang, Y. et al. PRKAA1 promotes proliferation and inhibits apoptosis of gastric cancer cells through activating JNK1 and Akt pathways. Oncol. Res. 28 (3), 213–223 (2020).
Chen, T. et al. The genome sequence archive family: toward explosive data growth and diverse data types. Genom. Proteom. Bioinform. 19 (4), 578–583 (2021).
Database Resources of the National Genomics Data Center. China National center for bioinformation in 2022. Nucleic Acids Res. 50 (D1), D27–d38 (2022).
Acknowledgements
The authors acknowledge the funding support received from Agricultural Science and Technology Innovation Project of Jilin Province (CXGC2023TD103) and National Beef Cattle and Yak Industrial Technology System (CARS-37).
Author information
Authors and Affiliations
Contributions
Wenwen Fang: Writing – original draft, Visualization, Software, Methodology, Formal analysis, Data curation, Conceptualization. Shaoying Yang: Writing – editing, Visualization, Validation. Xuanyu Li: Data curation, Resources, Investigation. Asadollahpour Nanaei: Writing – review & editing, Supervision. Yu Liu and Yang Cao: Validation, Resources, Investigation. Cheng Xiao, Xin Li and Haiguo Jin: Project administration, Methodology, Data curation. Yumin Zhao: Writing – review & editing, Supervision, Funding acquisition. Yang Cao: Writing – review & editing, Supervision, Resources, Investigation, Project administration, Methodology, Investigation, Conceptualization.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ethical approval and declarations
The preadipocytes of Chinese red steppe cattle were obtained from Molecular Breeding Research Laboratory, Institute of Animal Husbandry and Veterinary Medicine, JAAS, China. This study has been approved by the Institutional Animal Ethics Committee of Jilin Academy of Agricultural Sciences (JAAS). The Number of Permit is JNK20230307-02. This study strictly adhered to the “Guidelines for animal science and technology ethics” published by Jilin Academy of Agricultural Sciences (Northeast Agricultural Research Center of China).
Approval for animal experiments
The approval ID is JNK20230307-02.
Arrive guidelines statement
The study is reported in accordance with ARRIVE guidelines.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fang, W., Yang, S., Li, X. et al. Long-read sequencing uncovers key regulatory genes involved in the differentiation of preadipocytes of Chinese red steppe cattle. Sci Rep 15, 29459 (2025). https://doi.org/10.1038/s41598-025-15106-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-15106-5
