
Abstract
Essential hypertension (EH) is one of the most widespread chronic diseases globally, with a multifactorial etiology. MicroRNAs (miRNAs) are key regulators in the development and progression of EH and hold great promise as biomarkers. However, reliably identifying EH-related miRNA biomarkers in small-sample datasets remains challenging. To address these limitations, we propose a novel computational framework, the Modular Probability-driven Global Analytical Method (MPGAM), tailored for biomarker discovery in small-sample settings. MPGAM integrates three key innovations: (1) the Dual-Index Nearest Neighbor Similarity Measure (DINNSM), which captures local similarity structures more accurately than conventional correlation-based methods; (2) a multi-dimensional module evaluation strategy that incorporates gene significance, module membership, and known hypertension-associated miRNAs; and (3) a Probability-based Global Sorting Method (PGSM), which ranks miRNAs across modules based on probabilistic enrichment. Using the GSE75670 dataset from the GEO database, MPGAM identified ten candidate miRNA biomarkers. In this study, identification refers to the data-driven selection of miRNAs that exhibit potential associations with EH. These may include both previously reported EH-related miRNAs and novel candidates that have not been documented in existing literature. Among these, eight have been previously reported to be associated with blood pressure, including four (hsa-miR-107, hsa-miR-210, hsa-miR-665, and hsa-miR-449a) cited in more than five independent studies. Target gene interaction analysis further suggests that these miRNAs may exert coordinated regulatory effects on EH-related pathways. Compared to existing methods, MPGAM demonstrated greater effectiveness in miRNA biomarker identification and offers an interpretable approach.
Similar content being viewed by others

Introduction
Essential hypertension (EH) is a complex multifactorial condition and a major independent risk factor for cardiovascular, cerebrovascular, and renal diseases1. According to the Global Trends in Hypertension Prevalence and Control: A Comprehensive Analysis published by the World Health Organization (WHO) in 2020, the number of adults aged 30 to 79 years living with hypertension has nearly doubled over the past three decades, increasing from 650 million to 1.28 billion2. This dramatic rise in prevalence poses a serious global public health challenge. Therefore, it is imperative to conduct in-depth research into the mechanisms underlying the EH to develop more effective prevention and treatment strategies.
Extensive research over the past few decades has significantly advanced our understanding of the pathogenesis of EH, revealing that miRNAs play a critical regulatory role in both the development of EH and the progression of target organ damage3,4,5. miRNAs regulate the expression of specific proteins involved in cardiovascular function by binding to the 3’ untranslated region (3’ UTR) of target mRNAs, thereby influencing post-transcriptional regulation. Studies have shown that certain miRNAs can modulate genes implicated in vascular tone, inflammation, and cardiac function, thereby impacting the progression of EH6,7,8,9. Moreover, miRNA-based therapeutic strategies have demonstrated considerable potential in regulating the expression of EH-related genes, providing novel avenues for improving blood pressure control and cardiovascular function10,11,12. Although there is substantial evidence supporting the involvement of miRNAs in various aspects of EH pathogenesis, such as hyperactivation of the renin-angiotensin-aldosterone system (RAAS), endothelial dysfunction, abnormal vascular smooth muscle cell function, and disruptions in ion channel activity on cell membraness13,14,15,16,17. Further studies are needed to elucidate the precise mechanisms underlying miRNA function in EH and uncover additional insights into its pathophysiology.
In studying miRNA biomarkers for the disease, bioinformatics approaches have become crucial tools for exploring and identifying potential biomarkers18. Compared with traditional biological experimental methods, bioinformatics techniques enable comprehensive screening of candidate miRNAs at low cost and high efficiency through the analysis of high-throughput data and the prediction of computational models19. These methods are particularly valuable in the preliminary exploration of unknown miRNA functions, revealing their potential biological roles and target genes20. Therefore, efficient miRNA prediction and functional inference based on bioinformatics analysis methods and algorithms have become the vital driving forces to promote miRNA research3.
In recent years, researchers have made significant progress in developing computational models to predict miRNA-disease associations, which play a vital role in identifying disease biomarkers21,22,23. Ha et al. proposed a probabilistic matrix factorization model that incorporates miRNA expression as implicit feedback to predict potential miRNA-disease associations24. This approach was later extended by introducing disease similarity constraints into the matrix factorization framework, which substantially improved the model’s predictive performance in identifying relevant associations25. Building upon this work, the authors developed an advanced model that integrates Node2vec-based representation learning with a deep neural collaborative filtering architecture, enabling more effective inference of miRNA-disease relationships26. Beyond matrix factorization-based methods, Chen et al. introduced a deep belief network model for association prediction, utilizing a restricted Boltzmann machine to learn latent features between miRNAs and diseases, thereby enhancing predictive accuracy27. Additionally, Ha et al. developed a metric learning framework that captures complex nonlinear associations through learned distance metrics, achieving precise identification of disease-associated miRNAs28. The same author also proposed a similarity-driven matrix factorization approach that incorporates miRNA and disease similarity information further to improve the integration of biological knowledge during model construction29.
In hypertension biomarker research, approaches for analyzing small-sample miRNA expression data often rely on differential expression analysis (DEA). For instance, Qi et al. performed a comprehensive RNA-Seq analysis on samples from six salt-sensitive and six salt-resistant hypertension patients, identifying 1,250 known and 570 novel miRNAs through DEA. Subsequent qRT-PCR validation suggested that hsa-miR-361-5p may serve as a potential biomarker for salt-sensitive hypertension30. Similarly, Ye et al. and Riffo et al. employed DEA to identify differentially expressed miRNAs or ncRNAs, followed by validation using experimental or computational downstream analyse31,32. While DEA remains a valuable tool in miRNA biomarker discovery, it has inherent limitations when used in isolation. Specifically, DEA depends on arbitrarily set thresholds, which may affect reproducibility and lead to the omission of biologically meaningful miRNAs with subtle expression changes, particularly in multifactorial diseases like EH. Moreover, differential expression does not necessarily imply functional or regulatory relevance33. Consequently, relying solely on DEA may fail to comprehensively capture the complexity of miRNA-mediated regulation34. Therefore, it is necessary to combine multiple analytical methods to improve the accuracy and reliability of miRNA marker identification.
As research has advanced, it has become increasingly clear that molecules within biological systems do not act in isolation but instead operate through interconnected networks, forming complex modules35. The modules are defined as clusters of genes, proteins, or other molecules that exhibit similar functional or expression patterns. These modules are often co-regulated and play critical roles in specific biological processes. Co-expression module analysis enables researchers to uncover the functional relationships among molecules and to identify key regulatory elements involved in specific biological pathways36. Consequently, module-based biomarker discovery has emerged as a growing focus in biomedical research. Clustering techniques and dense subgraph detection within molecular networks are widely employed to identify these modules37. Increasingly, network-based approaches are applied to extract disease-associated biomarkers. For example, Li et al. utilized Weighted Gene Co-expression Network Analysis (WGCNA) to construct miRNA and mRNA co-expression networks in EH, identifying two key gene modules and one miRNA module. They prioritized EH-associated genes and miRNA biomarkers by evaluating factors such as gene significance, module membership, and network centrality, offering insights into miRNA-gene interactions in the context of EH38. Similarly, Zhang et al. integrated community detection with K-means clustering to identify gene modules, and applied the PageRank algorithm to rank genes within key modules, ultimately pinpointing ten hub genes as candidate EH biomarkers39. Chun et al. combined WGCNA with enrichment tools such as Metascape to identify molecular biomarkers across different hypertension phenotypes, including EH, left ventricular remodeling, and uncontrolled hypertension, offering a more comprehensive view of hypertension-associated molecular alterations40.
Despite the evolution from static clustering to dynamic network-based strategies36, existing methods still exhibit notable limitations. First, the similarity measures used prior to module detection often fail to capture the full complexity of biological relationships41. Second, many approaches evaluate module relevance or intra-module biomarker importance based on a single metric or limited criteria, which may reduce reliability and robustness. Finally, many publicly available miRNA datasets for EH contain only a small number of samples, posing serious challenges for traditional machine learning-based biomarker discovery. In such small-sample contexts, issues such as overfitting, instability, and biased estimation can hinder the generalizability and interpretability of results42,43.
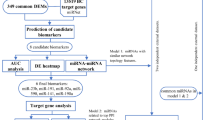
We propose a novel computational framework, The Modular Probability-driven Global Analytical Method (MPGAM), specifically designed for biomarker discovery in small-sample expression datasets. The overall workflow is illustrated in Fig. 1 First, we introduce the Dual-Index Nearest Neighbor Similarity Measure (DINNSM), which improves upon traditional correlation-based methods by capturing more nuanced local similarity structures between miRNAs. Second, a multi-dimensional module evaluation strategy is employed, integrating gene significance (GS), module membership (MM), and prior biological knowledge to comprehensively assess the importance of each miRNA within its respective module. Third, the Probability-based Global Sorting Method (PGSM) is applied to prioritize miRNAs across all modules using a probabilistic enrichment framework, rather than relying on isolated ranking criteria. For reproducibility, all model parameters used in the MPGAM pipeline are provided in Supplementary Table S1.

Overall framework of the MPGAM algorithm.
Materials and methods
Data source and preprocessing
The miRNA expression dataset GSE75670 was obtained from the Gene Expression Omnibus (GEO) database. This dataset was generated using the Exiqon miRCURY LNA microRNA Array, 7th generation platform, based on miRBase version 18. Accordingly, all miRNA annotations in this study were standardized using miRBase v18 to ensure consistency with the array platform.
The dataset includes six samples from patients with hypertension and six from healthy controls. Prior to analysis, the expression data were log2-transformed and normalized. Probes lacking annotation were excluded. For miRNAs represented by multiple probes, the probe with the highest expression value was retained. An expression matrix was then constructed, with miRNAs as row identifiers and sample IDs as column identifiers.
Clustering algorithm based on DINNSM
The division of co-expression modules based on clustering algorithms relies on the co-expression relationships of miRNAs, where such relationships reflect the similarity of their expression patterns. As the method used to quantify similarity directly affects the effectiveness of module detection, accurately characterizing the similarity between miRNAs is a critical step in clustering analysis.
DINNSM similarity measurement method
miRNAs exhibit complex interrelationships, which are often inadequately captured by conventional similarity metrics. These traditional measures may overlook the intrinsic characteristics of expression pattern similarity. To address this, more informative similarity estimation methods are required before performing co-expression module analysis. Association-based metrics, which capture co-varying expression trends between miRNA pairs, often offer advantages over distance-based approaches.
PC effectively identifies linear relationships between miRNAs44while the SPC is suitable for non-normally distributed data and more robust to outliers45. We propose a novel similarity measure that integrates both PC and SPC, combined with a nearest-neighbor strategy, to enhance biological relevance. This method generates a pairwise similarity matrix to quantify the relationships among n miRNAs based on m samples.
Let the expression dataset contain m samples and n miRNAs, and denote two miRNAs as X and Y. The initial similarity between them is computed using Eq. (1), resulting in an n × n similarity matrix, referred to as Disim.
where di is the difference in rank value of the i-th miRNA pair, and n is the total number of miRNAs.
Compared to similarity matrices computed solely based on PC or SPC, the Disim matrix demonstrates improved performance. However, due to the lack of transitivity in high-dimensional data spaces, such similarity metrics may fail to accurately capture the true relationships among miRNAs. To address this limitation, we incorporate a neighborhood-based strategy, which introduces two key factors into the similarity computation: (1) the number of shared neighbors between two miRNAs, and (2) the relative positions of these shared miRNAs within their respective nearest-neighbor tables.
The nearest-neighbor table for each miRNA is defined as a list of its \(\:{k}_{m}\) most similar miRNAs, derived from the corresponding row in the Disim matrix. For each row in Disim, the top-\(\:{k}_{m}\) miRNAs are selected to form individual nearest-neighbor tables. The final similarity score between two miRNAs is then computed by considering both the number of overlapping neighbors and their positional proximity within each other’s tables. This approach is formalized in Eq. (2). The resulting similarity matrix is subsequently used for clustering analysis. Further details of this methodology are provided in our previous work46.
where T(X) represents the nearest-neighbor table of X, Gn denotes the set of shared miRNAs between X and Y, and \(\:{P}_{g,T\left(X\right)\:}\) represents the position of the shared miRNAs g in T(X).
This similarity measure integrates the strengths of PC, SPC, and nearest-neighbor topology, effectively capturing local and nonlinear association patterns. By mitigating the non-transitivity issue in high-dimensional data, it provides a more biologically informed and reliable metric for miRNA similarity, especially valuable in small-sample datasets.
To address the computational demands of DINNSM, particularly for pairwise similarity computation and nearest-neighbor construction, we implemented a parallelized framework in R using the foreach package. Two custom functions, Constrsim() and Constrodsnn(), were developed to distribute computations across multiple CPU cores. This optimization substantially reduces runtime and was validated in our previous study46.
Clustering algorithm for module division
-
1.
Clustering Algorithm
In this study, the DINNSM-based similarity matrix was integrated with the K-means clustering algorithm to identify co-expression modules from miRNA expression data. K-means is one of the most widely used iterative clustering algorithms, known for its simplicity, computational efficiency, and relatively high clustering accuracy, making it popular in data analysis47. To evaluate the effectiveness of the proposed method, we conducted comparative experiments against WGCNA48 and DC-WGCNA41. In addition, clustering algorithms-including K-means, Fuzzy C-means (FCM), and Hierarchical Clustering (HC)-were tested in combination with five similarity measurement methods: Pearson Correlation (PC), Spearman’s Rank Correlation Coefficient (SPC), Euclidean Distance (ED), Maximum Information Coefficient (MIC), and Distance Correlation (DC).
-
2.
Clustering Performance Evaluation Method.
The performance of the DINNSM-based K-means clustering approach was assessed using three internal evaluation metrics: the Silhouette Index (SI), Calinski–Harabasz Index (CHI), and Davies-Bouldin Index (DBI). These metrics quantitatively evaluate clustering quality based on intra-cluster compactness, inter-cluster separation, and overall structural integrity. All evaluations were implemented using the clusterCrit package in R.
Methods for assessing module importance
This study introduces a multidimensional strategy for evaluating the importance of co-expression modules. Specifically, module relevance is quantified from four complementary perspectives: (1) the correlation between the module and the clinical phenotype; (2) the statistical significance of this module-phenotype association; (3) the number of differentially expressed miRNAs within the module; and (4) the number of miRNAs with known biological relevance contained in the module.
The correlation between each module and the phenotype is calculated using Eq. (3), where Module Eigengenes (MEs) represent the overall expression profiles of miRNAs within each module. To derive MEs, principal component analysis (PCA) is performed on all miRNAs within the module, and the first principal component is extracted as the module eigengene.
Here, \(\:{M}_{j}\) denotes the j-th module, \(\:\text{c}\text{o}\text{r}(\cdot\:)\)is the Pearson correlation coefficient, \(\:{MEs}_{j}\) is the eigengene of module Mj, and \(\:ph\) represents the phenotype.
The significance of the association between each module and the phenotype is assessed based on the Gene Significance (GS) values of the miRNAs within the module. Module Significance (MS) is defined as the average GS of all miRNAs in the module. Specifically, the GS of a given miRNA is computed from the P-value of its Pearson correlation with the phenotype across different sample groups. For the i-th miRNA in the j-th module, the GS is calculated as:\(\:{GS}_{j,i}=-\text{l}\text{o}\text{g}\left({p}_{j,i}\right)\). The module significance of the j-th module is then computed as the average GS of all miRNAs in that module, as shown in Eq. (4).
where \(\:{n}_{j}\) is the total number of miRNAs in module j, \(\:i\) represents the number of miRNA in the module, and k is the total number of the modules.
The number of differentially expressed miRNAs within each module was determined through DEA. The miRNA expression data were analyzed using the limma package in R. The analysis involved constructing the experimental design and contrast matrix using model.matrix() and makeContrasts(), respectively. A linear model was then fitted to the expression data to detect potential associations. After model fitting, F-statistics were computed, and a significance test was performed based on t-statistics to identify miRNAs with statistically significant differences. For each miRNA in module j, its differential expression status was defined as:
where p denotes the P-value obtained from the statistical test.
The proportion of differentially expressed miRNAs in module j was then computed as:
Where \(\:{n}_{j}\) is the total number of miRNAs in the module\(\:{\:M}_{j}\), and k denotes the total number of modules. This approach enables a quantitative assessment of the significance of each module.
The prior miRNAs within a module refer to the number of known hypertension-associated miRNAs present in that module. In this study, 262 experimentally validated hypertension-related miRNAs were compiled by querying the PubMed and HMDD v4.0 databases. Among these, 75 miRNAs were found in the dataset used in our analysis (see Supplementary Table 2). These 75 miRNAs were considered prior miRNAs and served as reference markers for subsequent evaluations.
The significance of each module was further quantified by calculating the proportion of hypertension-related prior miRNAs contained within the module, as defined in Eq.
where num(dj) denotes the number of known hypertension-related miRNAs in module Mj, and nj is the total number of miRNAs in that module.
Finally, this study integrated the four module-level characteristics into a unified module importance score, calculated using Eq. (8). Modules were then ranked based on this score to identify those with the highest potential biological significance to hypertension. In this study, biological significance refers to the degree to which miRNAs within a module are functionally or experimentally linked to hypertension, as supported by literature evidence and functional enrichment analysis of their predicted target genes.
Method for assessing the importance of MiRNAs within a module
The proposed method for evaluating the importance of miRNAs within a module integrates three key information: (1) the GS of each miRNA within the module; (2) the correlation between miRNA expression profiles and the module eigengene, referred to as module membership (MM); and (3) the correlation between miRNAs and known hypertension-related miRNAs. Among these, The computation of \(\:{GS}_{j}\left({X}_{i}\right)\) is the same as described in Sect. 2.3.
The MM value is calculated using Eq. (9), where a higher value indicates a stronger association between the miRNA and the module eigengene.
where \(\:{X}_{i}\)denotes the expression value of the i-th miRNA in module j, and k represents the total number of modules.
The function \(\:\text{i}\text{n}\text{f}\text{o}\left({M}_{j}{,X}_{i}\right)\), defined in Eq. (10), quantitatively evaluates the relevance of miRNA Xi within module Mj based on its similarity to known hypertension-associated miRNAs.
Here, \(\:\text{n}\text{u}\text{m}\left({M}_{j}{d}_{j}\right)\) denotes the number of known hypertension-associated miRNAs in module \(\:{M}_{j}\), and \(\:{X}_{\phi\:}\) is the φ-th such miRNA in the module. The function\(\:\:\text{D}\text{I}\text{N}\text{N}\text{S}\text{M}({X}_{i},{X}_{\phi\:})\) computes the similarity between miRNAs Xi and Xφ using the DINNSM method.If no known hypertension-related miRNAs are present in \(\:{M}_{j}\), then \(\:\text{i}\text{n}\text{f}\text{o}\left({M}_{j}{,X}_{i}\right)=0\).
Finally, The overall significance of miRNA Xi within module Mj is defined as Eq. (11).
where \(\:j\text{ϵ}\{\text{1,2},...,k\}\) represents the total number of modules, and \(\:i\text{ϵ}\{\text{1,2},...,{n}_{j}\}\).
The global MiRNA ranking method based on PGSM
To account for the relationship between miRNAs and their corresponding modules, we proposed a global miRNA ranking method, termed PGSM, which integrates the importance of each module. The core idea is as follows: during the miRNA ranking process, the rank of each miRNA is influenced not only by its intrinsic properties, but also by the contribution of previously ranked miRNAs. Suppose the top counti miRNAs in the global ranking have already been identified. Let M(i,j) denote the number of miRNAs from module Mj among these counti miRNAs. E(i,j) denotes the expected number of miRNAs from module Mj among the top counti ranked miRNAs, and p(i,j) is a probability value indicating the likelihood that the currently ranked miRNA (i.e., the counti+1-th miRNA) originates from module Mj.
According to Sect. 2.4, imp(Mj) represents the probability that miRNAs originate from module Mj. This value is normalized using Eq. (12) to obtain the standardized module importance score \(\:{\text{i}\text{m}\text{p}\left({M}_{j}\right)}^{{\prime\:}}\).
In the PGSM, the relationships among \(\:{count}_{i}\), \(\:\text{M}(i,j)\), \(\:\text{E}(i,j)\), \(\:\text{p}(i,j)\) and \(\:{\text{i}\text{m}\text{p}\left({M}_{j}\right)}^{{\prime\:}}\) are described as shown in Eqs. (13) and (14).
The pseudocode of the proposed PGSM algorithm is provided in Algorithm 1.

PGSM algorithm.
Results
MiRNA module detection and validation
MiRNA module detection
The preprocessed miRNA expression matrix was analyzed using the procedure described in Sect. 2.2 to construct a pairwise similarity matrix. A dynamic tree cut algorithm was applied to identify co-expression modules, and the optimal number of clusters (k) was determined by evaluating the Silhouette Index. Based on this evaluation, the optimal value was found to be k = 10, resulting in the partitioning of all miRNAs into ten distinct co-expression modules. The detailed clustering results are presented in Table 1.
Validation of clustering performance
To assess the effectiveness of the proposed similarity measurement method, comparative clustering experiments were conducted using K-means, FCM, and HC in combination with five different similarity metrics: PC, SPC, ED, MIC, and distance correlation DC. For WGCNA and DC-WGCNA, clustering parameters were set as follows: minimum module size (minClusterSize = 30), module partition depth (deepSplit = 2), and merging of modules with similarity greater than 80%.
As shown in Table 2, the DINNSM-based K-means method achieved the highest SI score of 0.55, indicating superior intra-cluster compactness and inter-cluster separation relative to all other approaches. In addition, it achieved the lowest -DBI = score (0.878), further supporting its ability to generate well-separated clusters. Although the CHI score of DINNSM (1075.05) was marginally lower than that of ED-Kmeans (1080.30), the difference was negligible. Overall, these quantitative results demonstrate that DINNSM exhibits strong and competitive clustering performance.
To further evaluate the clustering quality, t-distributed stochastic neighbor embedding (t-SNE) was employed to project the high-dimensional data into a two-dimensional space for visualization. Figure 2 illustrates the clustering outcomes of PC-Kmeans, ED-Kmeans, and DINNSM. Among these, DINNSM produced the most compact and well-separated clusters with minimal overlap, visually corroborating its superior SI and DBI scores.

t-SNE visualization of clustering results. (a) Visualization of PC-Kmeans (b) Visualization of ED-Kmeans (c) Visualization of DINNSM-based.
These findings suggest that the DINNSM-based approach more effectively captures the underlying biological structure of miRNA expression data, resulting in clusters that are both statistically robust and biologically meaningful.
Evaluation of module and Intra-Module MiRNA importance
In this study, the proposed DINNSM method was used to partition miRNAs into ten distinct co-expression modules. The overall importance of each module was subsequently calculated using Eq. (8), with the results summarized in Table 3.
Following module-level evaluation, miRNAs within each module were ranked based on their individual importance scores, as computed using Eq. (11). Table 4 presents the top 10 most important miRNAs from Module 1, along with their corresponding importance values.
To further generate a global ranking of miRNAs across all modules, the PGSM algorithm was applied. The top 10 miRNAs with the highest global importance scores are listed in Table 5.
Algorithm validation and results analysis
Comparative analysis of algorithms
Due to the absence of laboratory infrastructure and funding constraints, wet-lab validation could not be performed. Instead, a literature-supported validation strategy-commonly adopted in biomarker discovery studies-was employed as a practical alternative. To evaluate the effectiveness of the proposed MPGAM algorithm in identifying hypertension-related miRNA biomarkers, we compared its results with those of several benchmark methods published in recent years. The comparison was conducted using two evaluation criteria: (1) the number of miRNAs previously reported to be associated with blood pressure, and(2) the number of miRNAs cited in more than five independent studies.
The comparative results are summarized in Table 6, where miRNAs supported by more than five publications are highlighted in bold to denote higher confidence. Among the top 10 miRNAs identified by MPGAM, eight had prior evidence in hypertension-related literature, yielding a validation rate of 80%, including four that were cited in over five independent studies49,50,51,52. In contrast, the performance of the other three methods in these two indicators is all lower than that of the MPGAM algorithm. These results confirm the advantage of MPGAM in identifying relevant miRNA biomarkers.
Interaction analysis among MiRNA biomarkers
To explore the interactions between the ten miRNA biomarkers and their target genes, we utilized the miRWalk database (https://mirwalk.umm.uni-heidelberg.de/) to retrieve high-confidence miRNA-target gene pairs. Interactions with an association score greater than 0.95 were selected. This score, provided by miRWalk, integrates predictions and experimentally validated interactions from multiple computational tools and ranges from 0 to 1, with higher values indicating stronger predicted binding affinity and greater consensus among algorithms. To ensure high reliability, we adopted a conservative threshold of 0.95 and retained only those miRNA-gene pairs with experimental validation. Based on these criteria, we constructed a miRNA-target gene interaction network comprising 654 nodes, including 10 miRNAs and 644 unique target genes. The network was visualized using Cytoscape.
As shown in Fig. 3, the target genes of the ten miRNAs exhibit dense interconnectivity, suggesting that these miRNAs are functionally associated through shared or overlapping regulatory targets. This implies potential cooperative regulation among the miRNAs, which may play a synergistic role in the pathogenesis of EH.

Interaction network of miRNA biomarkers and their target genes. Genes are shown in blue, and miRNAs in red.
Functional enrichment analysis revealed that the target genes of the identified miRNAs are involved in multiple biological processes and signaling pathways closely associated with blood pressure regulation. GO analysis showed that these genes were primarily enriched in biological processes such as cellular response to DNA damage stimulus, regulation of vascular permeability, labyrinthine layer blood vessel development, and cellular response to lipopolysaccharide. These biological processes may contribute to EH pathogenesis by promoting vascular damage, affecting vascular remodeling, and regulating vascular tone. At the molecular function level, the target genes were enriched in functions including protein binding, metal ion binding, protein kinase binding, and myosin phosphatase activity. These molecular functions may be fundamental to intracellular signaling and physiological regulation, and are tightly linked to key mechanisms of EH, such as vascular smooth muscle contraction/relaxation and signal transduction dysregulation.
KEGG pathway analysis further indicated that these genes are involved in several critical signaling pathways, including the p53 signaling pathway, chemokine signaling pathway, adrenergic signaling in cardiomyocytes, and the JAK-STAT signaling pathway. These pathways play essential roles in inflammation, cell proliferation, stress responses, and cardiovascular regulation. Collectively, these findings highlight the biological significance of the identified miRNAs and their target genes in the molecular mechanisms underlying EH.
It is worth noting that although the GSE75670 dataset contains only miRNA expression data, the miRNA-target gene interactions analyzed in this study were derived from experimentally validated sources (miRWalk). While matched mRNA expression data are not available to directly confirm co-expression under identical conditions, the biological functions of the predicted target genes, as revealed by enrichment analysis, are highly consistent with known hypertension-related pathways. This concordance indirectly supports the regulatory relevance of the selected miRNA-mRNA interactions in the context of EH.
Discussion
This study addresses the challenges of biomarker identification from small-sample miRNA expression data by proposing a novel algorithm, MPGAM. Using this approach, ten hypertension-related miRNA biomarkers were successfully identified, among which eight have been previously reported to be associated with blood pressure, resulting in a literature-based validation rate of 80%. Compared with three benchmark methods, MPGAM demonstrated superior reliability and greater potential for practical application. We further focused on four miRNA biomarkers (hsa-miR-107, hsa-miR-210, hsa-miR-665, and hsa-miR-449a) that have been repeatedly cited in the literature and exhibit strong pathophysiological relevance. These miRNAs are implicated in key processes including vascular remodeling, hypoxia adaptation, and inflammation regulation. The remaining miRNAs, although highly ranked by MPGAM, were not discussed individually due to space constraints and the relatively limited literature available to support their associations.
hsa-miR-107 is a widely studied miRNA that plays a key role in multiple pathological processes related to hypertension, including hypertension-induced left ventricular hypertrophy, hypertensive nephropathy, and hypertension associated with obstructive sleep apnea syndrome. Previous studies have shown that hsa-miR-107 is significantly upregulated in patients with hypertensive nephropathy and has been identified as an important biomarker for this condition49. Additionally, Yang et al. validated the diagnostic and prognostic potential of miR-107 in patients with obstructive sleep apnea-associated hypertension using DEA and qRT-PCR experiments53. Further research has revealed that miR-107 significantly influences the pathophysiological mechanisms of hypertension by regulating sympathetic nervous system activity54,55. Moreover, miR-107 is closely associated with left ventricular hypertrophy56.
Another significant miRNA biomarker is hsa-miR-210, a hypoxia-inducible miRNA implicated in various hypoxia-related hypertensive pathological processes50. It has been shown that hsa-miR-210 plays a critical regulatory role in vascular smooth muscle cell proliferation, anti-apoptotic effects, and angiogenesis, all of which are closely associated with the pathophysiological mechanisms of EHs50. Yang et al. identified significant upregulation of hsa-miR-210 in hypertensive patients through miRNA expression microarray studies and confirmed these findings in two independent cohorts57. Furthermore, hsa-miR-210 has been shown to suppress apoptosis in pulmonary vascular smooth muscle cells under hypoxic conditions by targeting and downregulating the E2F3 gene58. This miRNA is also considered a potential biomarker for hypertensive disorders during pregnancy59,60.
hsa-miR-665 is another important miRNA, significantly upregulated in patients with chronic thromboembolic pulmonary hypertension. It contributes to vascular remodeling by regulating the proliferation and migration of vascular smooth muscle cells51. The role of hsa-miR-665 in early vascular aging and aging-related cardiovascular diseases has been validated using clinical samples and animal models61. Additionally, studies have shown that hsa-miR-665 affects ischemic myocardial injury by activating the Pak1/Akt signaling pathway, suggesting its potential as an important biomarker for the early diagnosis and prognosis of cardiovascular diseases62.
hsa-miR-449a is downregulated in arterial smooth muscle cells of pulmonary arterial hypertension patients and is thought to play a crucial role in vascular remodeling and the regulation of mitochondrial metabolism63. It has been identified as a potential therapeutic target for pulmonary arterial hypertension64. Studies have shown that hsa-miR-449a is involved in vascular inflammation and remodeling, particularly in the context of atherosclerosis and ischemia65,66. Additionally, hsa-miR-449a has been implicated in the pathogenesis of metabolic stress and hypertension associated with low birth weight52.
Target gene network analysis further revealed that the target genes of the identified miRNAs are involved in critical processes such as inflammation, cell proliferation, stress responses, and cardiovascular regulation. These target genes also exhibit significant interactions with each other, suggesting that the miRNAs may collectively influence the pathophysiology of EH through coordinated regulatory mechanisms.
Conclusion
This study proposes a novel algorithm, MPGAM, for the identification of miRNA biomarkers associated with EH. By incorporating an enhanced similarity measurement strategy, a multidimensional evaluation framework, and global miRNA ranks based on probabilistic enrichment, MPGAM overcomes the limitations of traditional methods and improves biomarker identification accuracy. A total of ten EH-related miRNAs were identified, all of which demonstrated strong literature-based support and exhibited significant biological relevance in both functional enrichment and network analyses.
Despite the supportive evidence from existing studies, the precise regulatory mechanisms of these miRNAs remain unclear and require further experimental validation. Future research should involve comprehensive in vivo and in vitro investigations to elucidate their molecular functions and biological roles in the context of EH. Although MPGAM is specifically designed to perform well under small-sample conditions, future work will aim to validate its performance in larger cohorts to evaluate its generalizability and robustness across varying sample sizes and biological scenarios. To support large-scale applications, future implementations will also consider GPU acceleration and distributed computing strategies to improve computational efficiency. Moreover, the modular and interpretable design of MPGAM makes it readily adaptable to other cardiovascular or multifactorial complex diseases, providing a promising and extensible framework for the discovery of disease-associated biomarkers.
Data availability
The GSE75670 dataset was obtained from the GEO database (http://www.ncbi.nlm.nih.gov/geo/).
References
Kokubo, Y. & Iwashima, Y. Higher blood pressure as a risk factor for diseases other than stroke and ischemic heart disease. Hypertension 66, 254–259 (2015).
Zhou, B. et al. Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: a pooled analysis of 1201 population-representative studies with 104 million participants. Lancet 398, 957–980 (2021).
Bátkai, S. & Thum, T. MicroRNAs in hypertension: mechanisms and therapeutic targets. Curr. Hypertens. Rep. 14, 79–87 (2012).
Weber, G. J., Purkayastha, B., Ren, L., Pushpakumar, S. & Sen, U. Hypertension exaggerates renovascular resistance via miR-122-associated stress response in aging. J. Hypertens. 36, 2226–2236 (2018).
Wang, Y. S., Jin, P. P., Liu, J. J. & Xie, X. Exosomal microRNA-122 mediates obesity-related cardiomyopathy through suppressing mitochondria ADP-ribosylation factor-like 2. Clin. Sci. 133, 1871–1881 (2019).
Hou, Z. X. et al. Longterm exercise-derived exosomal miR-342-5p A novel exerkine for cardioprotection. Circ. Res. 124, 1386–1400 (2019).
Baker, M. A. et al. MiR-192-5p in the kidney protects against the development of hypertension. Hypertension 73, 399–406 (2019).
Jia, Y. J., Wang, Q., Liang, M. L. & Huang, K. KPNA2 promotes angiogenesis by regulating STAT3 phosphorylation. J. Transl Med. 20, 1 (2022).
Yaacoub, S. et al. The pharmaco-epigenetics of hypertension: a focus on microRNA. Mol. Cell. Biochem. 479, 3255–3271 (2024).
Romaine, S. P. R., Charchar, F. J., Samani, N. J. & Tomaszewski, M. Circulating microRNAs and hypertension - from new insights into blood pressure regulation to biomarkers of cardiovascular risk. Curr. Opin. Pharmacol. 27, 1–7 (2016).
Jusic, A. & Devaux, Y. CA17129, E.-C. C. A. Noncoding RNAs in hypertension. Hypertension 74, 477–492 (2019).
Paseban, M. et al. Modulation of microRNAs by aspirin in cardiovascular disease. Trends Cardiovas Med. 30, 249–254 (2020).
Chekka, L. M. S. et al. Circulating microRNA biomarkers of thiazide response in hypertension. J. Am. Heart Assoc. 13, 1 (2024).
Zhang, H. G. et al. The circulating level of miR-122 is a potential risk factor for endothelial dysfunction in young patients with essential hypertension. Hypertens. Res. 43, 511–517 (2020).
Wen, T. et al. miR-342-5p promotes vascular smooth muscle cell phenotypic transition through a negative-feedback regulation of Notch signaling via targeting FOXO3. Life Sci. 326, 1–14 (2023).
Wang, G. et al. MicroRNA regulation of phenotypic transformations in vascular smooth muscle: relevance to vascular remodeling. Cell. Mol. Life Sci. 80, 144 (2023).
Ren, X. S. et al. MiR155-5p in adventitial fibroblasts-derived extracellular vesicles inhibits vascular smooth muscle cell proliferation via suppressing angiotensin-converting enzyme expression. J. Extracell. Vesicles. 9, 1698795 (2020).
Chen, C. et al. Identification of candidate biomarkers for salt sensitivity of blood pressure by integrated bioinformatics analysis. J. Hypertens. 39, E45–E46 (2021).
Zhang, X. Y. et al. The diagnostic values of circulating MiRNAs for hypertension and bioinformatics analysis. Bioscience Rep. 38, BSR20180525 (2018).
Li, Y. Y. et al. Vascular smooth muscle cell-specific miRNA-214 knockout inhibits angiotensin II-induced hypertension through upregulation of Smad7. Faseb J. 35, e21947 (2021).
Z hao, Y., Wang, C. C. & Chen, X. Microbes and complex diseases: from experimental results to computational models. Brief. Bioinform. 22, bbaa158 (2021).
Ha, J., Park, C., Park, C. & Park, S. Improved prediction of miRNA-disease associations based on matrix completion with network regularization. Cells 9, 881 (2020).
Ha, J. LncRNA expression profile-based matrix factorization for predicting lncRNA-disease association. IEEE Access. 12, 70297–70304 (2024).
Ha, J., Park, C., Park, C. & Park, S. IMIPMF: inferring miRNA-disease interactions using probabilistic matrix factorization. J. Biomed. Inf. 102, 103358 (2020).
Ha, J. MDMF: predicting miRNA–disease association based on matrix factorization with disease similarity constraint. J. Pers. Med. 12, 885 (2022).
Ha, J. & Park, S. NCMD: node2vec-based neural collaborative filtering for predicting miRNA-disease association. IEEE ACM Trans. Comput. Biol. Bioinform. 20, 1257–1268 (2023).
Chen, X., Li, T. H., Zhao, Y. & Wang, C. C. Deep-belief network for predicting potential miRNA-disease associations. Brief. Bioinform. 22, bbaa186 (2021).
Ha, J. & Park, C. MLMD: metric learning for predicting miRNA-disease associations. IEEE Access. 9, 78847–78858 (2021).
Ha, J. SMAP: similarity-based matrix factorization framework for inferring miRNA-disease association. Knowl. -Based Syst. 263, 110295 (2023).
Qi, H. et al. micro-RNA screening and prediction model construction for diagnosis of salt-sensitive essential hypertension. Med. (Baltim). 96, e6417 (2017).
Ye, Y. Z. et al. Screening of differentially expressed MicroRNAs of essential hypertension in Uyghur population. Lipids Health Dis. 18, 1–13 (2019).
Riffo-Campos, A. L. et al. Exosomal and plasma non-coding RNA signature associated with urinary albumin excretion in hypertension. Int. J. Mol. Sci. 23, 823 (2022).
Cui, W. T. et al. Effect of high variation in transcript expression on identifying differentially expressed genes in RNA-seq analysis. Ann. Hum. Genet. 85, 235–244 (2021).
Tang, S. Z. et al. Differential gene expression analysis based on linear mixed model corrects false positive inflation for studying quantitative traits. Sci. Rep-Uk. 13, 16570 (2023).
De Smet, R. & Marchal, K. Advantages and limitations of current network inference methods. Nat. Rev. Microbiol. 8, 717–729 (2010).
Saelens, W., Cannoodt, R. & Saeys, Y. A comprehensive evaluation of module detection methods for gene expression data. Nat. Commun. 9, 1090 (2018).
Rappoport, N. & Shamir, R. Multi-omic and multi-view clustering algorithms: review and cancer benchmarkvol 46, pg 10546, Nucleic Acids Res 47, 1044–1044 (2019). (2018).
Li, Z. J. et al. Identification of hub genes associated with hypertension and their interaction with MiRNA based on weighted gene coexpression network analysis (WGCNA) analysis. Med. Sci. Monit. 26, e923514–e923511 (2020).
Zhang, Y. et al. A gene module identification algorithm and its applications to identify gene modules and key genes of hepatocellular carcinoma. Sci. Rep-Uk. 11, 5517 (2021).
Yu, C. Y., Gu, Y., Jiang, Y. C. & Zhang, X. W. Identification of intrinsic genes across general hypertension, hypertension with left ventricular remodeling, and uncontrolled hypertension. Front. Cardiovasc. Med. 9, 992284 (2022).
Hou, J. et al. Distance correlation application to gene co-expression network analysis. BMC Bioinform. 23, 81 (2022).
Navarro, C. L. A. et al. Risk of bias in studies on prediction models developed using supervised machine learning techniques: systematic review. Bmj-Brit Med. J. 375, n2281 (2021).
Rajput, D., Wang, W. J. & Chen, C. C. Evaluation of a decided sample size in machine learning applications. BMC Bioinform. 24, 48 (2023).
Sedgwick, P. Pearson’s correlation coefficient. Brit Med. J. 344, e4483 (2012).
Balasubramaniyan, R., Hüllermeier, E., Weskamp, N. & Kämper, J. Clustering of gene expression data using a local shape-based similarity measure. Bioinformatics 21, 1069–1077 (2005).
Li, Z. J. et al. Clustering algorithm based on DINNSM and its application in gene expression data analysis. Technol. Health Care. 32, S229–S239 (2024).
Jiang, D. X., Tang, C. & Zhang, A. D. Cluster analysis for gene expression data: A survey. Ieee T Knowl. Data En. 16, 1370–1386 (2004).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 1–13 (2008).
Wang, Z., Liu, Z. J., Yang, Y. X. & Kang, L. Identification of biomarkers and pathways in hypertensive nephropathy based on the CeRNA regulatory network. Bmc Nephrol. 21, 1–10 (2020).
Guan, Y. N., Song, X. J., Sun, W., Wang, Y. R. & Liu, B. Effect of hypoxia-induced microRNA-210 expression on cardiovascular disease and the underlying mechanism. Oxid. Med. Cell. Longev. 2019, 4727283 (2019).
Sulastomo, H., Dinarti, L. K., Hariawan, H. & Haryana, S. M. MicroRNA expression alteration in chronic thromboembolic pulmonary hypertension: A systematic review. Pulm Circ. 14, e12443 (2024).
Nemoto, T., Nakakura, T. & Kakinuma, Y. Elevated blood pressure in high-fat diet-exposed low birthweight rat offspring is most likely caused by elevated glucocorticoid levels due to abnormal pituitary negative feedback. Plos One. 15, e0238223 (2020).
Yang, X. P., Niu, X., Xiao, Y., Lin, K. & Chen, X. MiRNA expression profiles in healthy OSAHS and OSAHS with arterial hypertension: potential diagnostic and early warning markers. Resp. Res. 19, 1–12 (2018).
Improta-Caria, A. C. et al. MicroRNAs regulating renin-angiotensin-aldosterone system, sympathetic nervous system and left ventricular hypertrophy in systemic arterial hypertension. Biomolecules 11, 1771 (2021).
Zhang, K. X. et al. Molecular mechanism for hypertensive renal disease: differential regulation of chromogranin A expression at 3′-untranslated region polymorphism C + 87T by microRNA-107. J. Am. Soc. Nephrol. 26, 1816–1825 (2015).
Zeng, J. Y. et al. Molecular mechanisms in microRNA-mediated TRB3 gene and hypertension left ventricular hypertrophy. Exp. Ther. Med. 13, 1907–1911 (2017).
Yang, Q. B. et al. MicroRNA-505 identified from patients with essential hypertension impairs endothelial cell migration and tube formation. Int. J. Cardiol. 177, 925–934 (2014).
Gou, D. et al. miR-210 has an antiapoptotic effect in pulmonary artery smooth muscle cells during hypoxia (303, Pg L682, 2012). Am. J. Physiol-Lung C. 303, L1011–L1011 (2012).
Biró, O. et al. Various levels of circulating exosomal total-miRNA and miR-210 hypoxamir in different forms of pregnancy hypertension. Pregnancy Hypertens. 10, 207–212 (2017).
Rigó Jr, J., Alasztics, B., Molvarec, A., Nagy, B. & Biró, O. 11 expression analysis of circulating exosomalhsa-miR-210 in hypertensive disorders of pregnancy: biomarkers, prediction of preeclampsia. Pregnancy Hypertension: Int. J. Women’s Cardiovasc. Health. 6, 183 (2016).
Chen, T. B. et al. MiR-665 regulates vascular smooth muscle cell senescence by interacting with LncRNA GAS5/SDC1. Front. Cell. Dev. Biol. 9, 700006 (2021).
Li, X. Z., Zhou, J. H., Wang, M., Yang, C. M. & Sun, G. B. Cardiovascular disease and depression: a narrative review. Front. Cardiovasc. Med. 10, 1274595 (2023).
Roiz-Valle, D., Caravia, X. M. & Otín, C. L. Mechanisms of mitochondrial microRNA regulation in cardiovascular diseases. Mech. Ageing Dev. 212, 111822 (2023).
Badi, S. A. et al. Downregulation of ACE, AGTR1, and ACE2 genes mediating SARS-CoV-2 pathogenesis by gut microbiota members and their postbiotics on Caco-2 cells. Microb. Pathogenesis. 173, 105798 (2022).
Lin, S. G., Lin, R. R., Zhang, H. K., Xu, Q. B. & He, Y. Y. Peripheral vascular remodeling during ischemia. Front. Pharmacol. 13, 1078047 (2022).
Chen, L. B. et al. A single nucleotide polymorphism located in microRNA-499a causes loss of function resulting in increased expression of osbpl1a and reduced serum HDL level. Oncol. Rep. 38, 3515–3521 (2017).
Funding
This research was supported by the Hebei Provincial Medical Science Research Project (20260679), the Basic Scientific Research Project of Hebei Provincial Department of Education (JJC2024059), the Basic Research Fund Project for Provincial Universities in Hebei Province (JJC2024054), and Science Research Project of Hebei Education Department(QN2025011).
Author information
Authors and Affiliations
Contributions
Z.L., D.L., Y.J. and X.W. participated in study conception and design; Z.L. and H.Z. contributed significantly to the data extraction and analyses; C.S., Z.Y. and Q.C. helped in the results presentation and interpretation; Z.L. and X.W. contributed to write and revise manuscript; Z.L., Y.L. and A.S. provided advice for the study and edited the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Z., Liu, D., Jin, Y. et al. Identification of miRNA biomarkers for essential hypertension in small samples based on MPGAM. Sci Rep 15, 37378 (2025). https://doi.org/10.1038/s41598-025-16021-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-16021-5
