
Abstract
The current study evaluates the diagnostic utility of copy number variation sequencing (CNV-Seq) in elucidating the genetic etiology of abnormal brain development (ABD) in pediatric patients. We analyzed 130 children diagnosed with ABD at Gansu Maternity and Child-care Hospital between December 2018 and October 2023. Patients were categorized into non-syndromic (NS-ABD) and syndromic (S-ABD) groups, and CNV-Seq was performed to identify chromosomal aneuploidies and copy number variations (CNVs). Of the 130 cases, 42 (32.3%) exhibited genetic abnormalities, comprising 3 aneuploidies (2.3%; 2 trisomy 21, 1 trisomy 18) and 39 CNVs (30%). In the NS-ABD group (n = 15), abnormalities included 1 aneuploidy (6.7%), 4 pathogenic CNVs (pCNVs; 26.7%), and 10 variants of uncertain significance (VUS; 66.7%). The S-ABD group (n = 27) showed 2 aneuploidies (7.4%), 19 pCNVs (70.4%), and 6 VUS (22.2%). Chi-square analysis confirmed a significantly higher pCNV detection rate in the S-ABD group compared to the NS-ABD group (χ² = 40.03, P < 0.001). CNV-Seq demonstrates a high diagnostic yield for ABD, particularly in S-ABD patients, where the detection rate of pCNVs is significantly elevated. NS-ABD patients, with subtler clinical phenotypes and lower positive detection rates, are at risk of underdiagnosis in clinical practice. Increased attention to NS-ABD cases, alongside timely prenatal diagnosis and genetic counseling, is essential to improve clinical outcomes.
Similar content being viewed by others


Introduction
Abnormal brain development (ABD) is characterized by deficits in cognitive function and delays in social awareness or adaptive behavior that arise during the developmental period prior to adulthood. The overall incidence of ABD is approximately 3%, with moderate to severe cases accounting for 0.3–0.4%1,2. The female-to-male prevalence ratio ranges from 0.71 to 0.63:1. Clinically, ABD can be classified into two subtypes: non-syndromic ABD (NS-ABD) and syndromic ABD (S-ABD), the latter often accompanied by distinctive phenotypic features and congenital malformations3. The etiology of ABD is highly heterogeneous, encompassing genetic, nutritional, and endocrine factors, all of which may contribute to disease onset4,5.
Copy number variations (CNVs) are structural variations in the genome that involve gains or losses of DNA segments, typically larger than 50 base pairs. CNVs can include duplications, deletions (homozygous or hemizygous), triplications, and complex rearrangements6. When these genomic alterations disrupt gene function or dosage, they may be classified as pathogenic CNVs (pCNVs). Depending on their structure and size, pCNVs can be further categorized into microdeletions, microduplications, or complex CNVs, complex rearrangements, and other more complicated variations. Most CNV are considered benign (without clinical effect), and only a few CNVs were related to genetic diseases. However, micro-deletion/replication syndrome (MMS) caused by pCNVs still played an important role in human genetic diseases. Copy number variations (CNVs) are known to cause a broad range of syndromic disorders, including well-characterized conditions such as Williams-Beuren syndrome (WBS, ~ 1.6 Mb heterozygous deletion at 7q11.23), Angelman/Prader-Willi syndrome (AS/PWS, deletions at 15q11–q13), and DiGeorge syndrome (DGS, 22q11.2 deletions)7,8,9. These pathogenic CNVs vary in both genomic location and fragment size, underscoring the genomic heterogeneity underlying CNV-associated syndromes. CNV-Seq, based on next-generation sequencing, enables genome-wide detection of such variations with high resolution and throughput. The technology of CNV-Seq adopted next generation sequencing to perform low-depth whole-genome sequencing of the DNA of samples, which could detect abnormal fragments with a size of larger than 0.1 MB, and it had become one of the most suitable methods for prenatal diagnosis because of its large detection throughput, high resolution, easy operation, and short detection time10.
In this study, CNV-Seq was performed on samples from 130 individuals diagnosed with ABD at Gansu Maternal and Child Health Hospital, including both fetuses (amniotic fluid samples) and postnatal children (peripheral blood samples). Among them, 55.32% (n = 26) were diagnosed prenatally, and 44.64% (n = 21) postnatally. Based on phenotypic presentation, participants were stratified into S-ABD and NS-ABD groups. CNV-Seq was then used to detect and characterize genomic abnormalities across the cohort. Genetic and statistical analyses were performed to assess the relationship between clinical features and CNV profiles. These findings were used to evaluate the diagnostic utility of CNV-Seq in ABD cases and to enhance clinical understanding of its genetic underpinnings. Ultimately, this work aims to provide a theoretical and empirical foundation for improving diagnosis and management of ABD—particularly among pediatric populations in Northwest China—and to inform future strategies for prevention and intervention.
Materials and methods
Study oversight
This study was conducted at the Medical Genetics Center authorized by the Gansu Provincial Health Commission, which is licensed for genetic testing. All participants were enrolled following the standard CNV-seq protocol and provided written informed consent. The consent form included a detailed explanation of the testing methodology, sample requirements, eligibility criteria, and potential risks. It also covered insurance information, legal disclaimers, national ethical compliance statements, laboratory procedures, and pre-test consultation sections. To ensure transparency and scientific rigor, participants were informed of the positive predictive value (PPV), negative predictive value (NPV), sensitivity, and specificity associated with CNV-seq results.
Sample collection and DNA extraction
Informed consent was obtained from all participants or their legal guardians after clear communication of the study’s purpose and procedures. Guardians of pediatric participants acknowledged and agreed to the publication of anonymized data for research purposes.
Peripheral blood samples (5 mL) were collected from pediatric participants and transferred into EDTA-containing anticoagulant tubes (KIRGEN Medical Equipment Co., Ltd., China). For amniotic fluid samples, approximately 15 mL of fluid was obtained under ultrasound guidance and collected into sterile centrifuge tubes. Chorionic villus samples were obtained via syringe aspiration and cultured briefly in vitro before DNA extraction.
Genomic DNA was extracted using the QIAamp DNA Micro Kit (Qiagen, Germany), following the manufacturer’s standard operating procedures (SOP). DNA concentration was measured using a Qubit 3.0 Fluorometer (Thermo Fisher Scientific, USA).
Sequencing and analysis
Low-depth whole-genome sequencing was performed on the CN-500 NGS platform (Illumina, USA). Raw sequencing reads were filtered and processed to remove low-quality reads using standard quality control (QC) procedures. The resulting clean data consisted of 36 bp reads at an average sequencing depth of 0.1× and were exported in BAM format for analysis. Chromosome copy number analysis was conducted using the CNV analysis system (version 2.0; Berry Genomics, Beijing).
Post-QC reads were aligned to the human reference genome, and CNVs were identified through standardized analytical pipelines. Variants were annotated based on public mutation and population frequency databases, including ClinVar, HGMD, gnomAD, DECIPHER, and the 1000 Genomes Project. These annotations were used to evaluate the clinical significance of each CNV. Notably, to ensure result reliability, only CNV fragments larger than 100 Kb were considered. The pathogenicity of CNVs was classified in accordance with the guidelines from the American College of Medical Genetics and Genomics (ACMG) and ClinGen11,12.
Statistical analyses were conducted using R software (version 4.2.1; https://www.r-project.org/). Associations between categorical variables were assessed using the chi-square test (χ²) via the “chisq.test” function. Results are reported as percentages (%) with corresponding p-values, and statistical significance was defined as p < 0.0513.
Results
Overview of CNV-Seq findings in ABD cohort
The study analyzed 130 samples from 42 individuals, representing 32.31% (42/130) of the cohort. A total of 50 abnormal copy number variants (CNVs) were identified, including both aneuploidies and subchromosomal CNVs. Among these, three cases were diagnosed with aneuploidy—two with trisomy 21 and one with trisomy 18. The number of detected CNVs exceeded the number of patients due to the presence of multiple pathogenic CNVs (pCNVs) in some individuals.
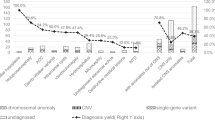
Of the 50 abnormal CNVs, 23 (17.69%) were classified as pathogenic (P) or likely pathogenic (LP) according to current clinical guidelines (see Supplementary Table S1 for details). These included eight CNV fragments larger than 10 Mb (encompassing both deletions and duplications), three fragments between 5 and 10 Mb, and twelve fragments smaller than 5 Mb (see Supplementary Table S2 for details). Notably, several patients harbored multiple pCNVs, leading to a total CNV count greater than the number of affected individuals (Fig. 1). The chromosomal distribution of pCNVs is illustrated in Fig. 2, with higher detection frequencies observed on chromosomes X, 15, 2, and 17.

CNV-seq detection distribution. B/LB represents Benign or Likely Benign; T21 represents Trisomy 21; T18 represents Trisomy 18; VUS represents Variant of Unknown Significance; Au CNVs represents Autosomal CNVs; S chr CNVs represents Sex chromosome CNVs; S chr CNVs mos. represents Sex chromosome CNVs mosaicism.

Distribution of CNVs in each chromosome.
Comparison of CNV detection rates between NS-ABD and S-ABD groups
Among the 130 ABD cases analyzed, genomic abnormalities were identified in 42 individuals. In the non-syndromic ABD (NS-ABD) group (n = 15), 15 CNVs were detected, comprising 1 aneuploidy (6.67%), 4 pCNVs (26.67%), and 10 CNVs of uncertain clinical significance (66.67%). In contrast, the syndromic ABD (S-ABD) group (n = 27) showed abnormalities in all 27 individuals, including 2 aneuploidies (7.41%), 19 pCNVs (70.37%), and 6 CNVs of uncertain significance (22.22%).
Chi-square analysis revealed a statistically significant difference in pCNV detection rates between the NS-ABD and S-ABD groups (χ² = 40.03, p < 0.05). Furthermore, the overall positive detection rate was markedly higher in the S-ABD group (77.78%) compared to the NS-ABD group (33.33%) (χ² = 40.97, p < 0.05; see Table 1 for details).
Identification of candidate genes for ABD within pCNV regions
To identify candidate genes potentially associated with ABD, we focused on genes located within the detected pCNV regions. First, candidate genes were retrieved from the OMIM database based on their genomic coordinates and known disease associations. Next, genes were prioritized according to their expression profiles and functional relevance to neurodevelopment. Ultimately, eight candidate genes were identified as potentially implicated in ABD: UBE3A, AUTS4, SATB2, GLSS, SMCR, ARSL, and CDPX1 (Table 2).
Discussion
The advancement of next-generation sequencing (NGS) has increasingly driven its adoption in research and clinical diagnostics to investigate genomic variations14,15. While next-generation sequencing (NGS) offers advantages such as high resolution and the potential for simultaneous detection of multiple variant types, microarray-based CNV detection remains the gold standard in certain clinical contexts, particularly for identifying copy-neutral abnormalities and low-level mosaicism. Low-depth genome sequencing, as applied in this study, offers a cost-effective and rapid method for genome-wide CNV detection with relatively uniform coverage. While its resolution is generally comparable to that of some array platforms, it may not detect small exonic events that are identifiable by high-resolution arrays. Therefore, NGS and microarray are best viewed as complementary rather than mutually exclusive approaches. CNV-Seq is increasingly utilized for detecting submicroscopic chromosomal abnormalities. This method employs statistical models to estimate CNV proportions within confidence intervals, enabling robust comparison and identification of genomic variations16.Unlike microarray-based genotyping, which targets specific fragments, CNV-Seq uses reference sequences (e.g., READS) as templates for alignment17. It performs two-dimensional sequence comparisons by pairing shotgun sequences and employs a sliding window approach for data analysis. However, this method is less effective for long sequences, and the accuracy of large fragment analysis requires further improvement18.
ABD is characterized by impairments in cognitive function and adaptive behavior that emerge during early human development. The prevalence of ABD is estimated to be approximately 6–8%, making it a significant contributor to both physical and mental health burdens. The condition arises from a complex interplay of genetic and environmental factors, with chromosomal abnormalities, gene mutations, and pathogenic copy number variations (pCNVs) all implicated in its etiology. Chromosomal abnormalities alone account for an estimated 30–40% of ABD cases. In the general population, the prevalence of ABD is approximately 1–3%19. Given the large population size in China, the number of individuals affected by ABD is correspondingly substantial. However, effective and targeted strategies for the prevention and treatment of ABD remain lacking for most patients. Understanding the underlying causes of ABD is therefore of critical importance. Yet, the etiology remains elusive in many cases. According to data from the World Health Organization, over 50% of ABD cases worldwide have no clearly identified cause. This situation is even more pronounced in China, where approximately 67% of cases are of unknown origin20. Genetic factors are believed to play a pivotal role in the pathogenesis of ABD. Clinically, ABD can be divided into syndromic (S-ABD) and non-syndromic (NS-ABD) forms. Patients with NS-ABD typically present with isolated neurodevelopmental features such as language impairment, motor delay, and cognitive decline. In contrast, S-ABD encompasses these neurodevelopmental abnormalities in combination with additional systemic anomalies or comorbidities, including congenital heart defects, craniofacial dysmorphisms, and cleft lip and palate21.
In the present study, 130 patients diagnosed ABD were enrolled for copy number variation sequencing (CNV-seq) analysis. Based on clinical phenotypes and available prenatal ultrasound findings, patients were stratified into non-syndromic ABD (NS-ABD) and syndromic ABD (S-ABD) groups. CNV-seq identified abnormal genomic findings in 42 cases (42/130). Notably, several individuals carried multiple abnormal CNV regions, indicating the presence of two or more pathogenic CNVs (pCNVs) within a single patient. An increasing body of evidence supports the application of CNV-seq in detecting chromosomal aberrations associated with neurodevelopmental disorders such as intellectual disability and disorders of sex development, highlighting its diagnostic utility22. The present study reinforces this view, with a diagnostic yield of 32.3%, further demonstrating CNV-seq’s effectiveness as a clinical tool. Among the detected cases, genomic abnormalities included both aneuploidies and submicroscopic pCNVs. To further examine genotype–phenotype correlations, we conducted a subgroup comparison between the NS-ABD and S-ABD cohorts. The analysis revealed a statistically significant difference in the detection rate of pathogenic CNVs between the two groups (χ² = 40.03, P < 0.05). Moreover, when assessing the overall positivity rate—including both aneuploidies and pCNVs—the NS-ABD group showed a positivity rate of 33.33%, whereas the S-ABD group exhibited a significantly higher rate of 77.78% (χ² = 40.97, P < 0.05). These findings suggest that chromosomal abnormalities and pCNVs are more prevalent in syndromic forms of ABD. However, the current study is not without limitations. Due to incomplete clinical data and patient reluctance to disclose personal information, the sample size remains limited, which may affect the accuracy and generalizability of the observed detection rates. Future studies will aim to expand the cohort size and improve data completeness to further validate and refine these findings.
In the present study, a total of 27 pathogenic copy number variations (pCNVs) were identified, including regions 15q11.2–q13.2, 2q33.1–q33.3, 17p11.2, and Xp22.33–p11.1. Notably, pCNVs such as the 15q11.2–q13.2 microdeletion syndrome23 and the 22q11.21 microdeletion syndrome24, both of which have been recurrently reported in the literature and are known to be associated with ABD were also detected in this cohort. What’s more, we also found rarely reported pCNV fragments associated with ABD such as 18p11.23. Many pathogenic CNVs associated with ABD were distributed across various chromosomal regions, including loci such as 7p11.23. While their genomic locations appear diverse, recurrent CNVs are known to arise through common mechanisms, such as non-allelic homologous recombination mediated by low-copy repeats or gene clusters (e.g., olfactory receptor regions), which are distributed throughout the genome. This genomic architecture may underlie both the recurrence and complexity of CNV formation in ABD. We review the information included in four public databases DECIPHER, OMIM, ClinGen and PubMed, and then investigated the highly associated genes located close to the region where the pCNVs fragments appeared, based on the above-mentioned pCNVs fragments. A total of 7 Several candidate genes associated with ABD were selected for further analysis. Within the 15q11.2–q13.2 region, loss of function of the maternally inherited UBE3A gene leads to Angelman syndrome (AS), a neurodevelopmental disorder characterized by severe intellectual disability, speech impairment, and ataxia. In contrast, deletions of the paternally inherited chromosome 15q11–q13 result in Prader-Willi syndrome (PWS), which is caused by the loss of expression of multiple genes, including the SNORD116 snoRNA cluster and the IPW non-coding RNA. Additionally, AUTS2, located on 7q11.22, has been implicated in autosomal dominant intellectual developmental disorder (OMIM: 615834), and was also included among the candidate genes evaluated in this study. These syndromes exhibit overlapping clinical features, most notably intellectual disability, ataxia, and motor delay25. Numerous studies have confirmed that UBE3A is the critical gene responsible for Angelman syndrome26. The GLSS and SATB2 genes, located within the 2q33.1–q33.3 region, are implicated in Glass syndrome, which is typically characterized by growth retardation and profound intellectual disability; these phenotypes frequently manifest during both the prenatal and postnatal periods27,28. The SMCR gene within the 17p11.2 region is associated with Smith-Magenis syndrome, whose cardinal clinical signs include mild to moderate intellectual impairment and delayed reflexes29. Notably, the ARSL and CDPX1 genes, located in the Xp22.33 region, are strongly linked to type I chondrodysplasia punctata. Although the syndrome is primarily defined by short phalangeal cartilage dysplasia and hypoplasia of the distal phalanges, multiple reports have also documented developmental delay and profound cognitive impairment in early infancy30,31. A limitation of our study is that clinical assessment of dysmorphic features and neurodevelopmental outcomes could not be performed in the prenatal diagnosis (PND) cases. However, including PND samples allowed us to evaluate the prenatal genomic landscape and identify potentially pathogenic CNVs, which may contribute to early genetic counseling and decision-making. Moreover, in several patients, large segmental duplications and deletions involving different chromosomes were detected, which may indicate unbalanced translocations. These CNV patterns are suggestive of derivative chromosomes arising from structural rearrangements such as t(17;18), t(9;20), or t(4;8). However, parental karyotype or FISH analyses were not performed, limiting further confirmation of inheritance patterns. In short, the candidate genes discovered in these crucial pCNVs regions were closely associated with the occurrence of ABD.
Conclusions
In summary, the current findings indicate that the occurrence of ABD is closely associated with chromosomal aneuploidies and pathogenic copy number variations (pCNVs). Both the pCNV detection rate and the overall chromosomal abnormality detection rate were significantly higher in patients with the S-ABD subtype compared to those with the NS-ABD subtype. These results suggest that individuals with NS-ABD are more likely to be overlooked in clinical evaluations. Accordingly, greater clinical attention should be directed toward patients with NS-ABD, and appropriate genetic testing should be strongly recommended. Moreover, the application of CNV-seq for the detection of submicroscopic chromosomal aberrations substantially enhances diagnostic efficiency. Integrating genotype–phenotype correlation analyses can further provide robust genetic evidence for prenatal diagnosis of ABD, while also offering a theoretical foundation and practical guidance for both eugenics and postnatal care.
Data availability
Sequence data that support the findings of this study have been deposited in the European Nucleotide Archive with the primary accession code PRJNA1297644 (https://dataview.ncbi.nlm.nih.gov/object/PRJNA1297644?reviewer=nadsbg0nthkq0mj53katef5kbq) and PRJNA1297151 https://dataview.ncbi.nlm.nih.gov/object/PRJNA1297151?reviewer=b5unlpt5hc9ukng190qhr1k0hr).
References
Moeschler, J. B. & Shevell, M. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics 134, e903–e918 (2014).
Sebat, J. et al. Large-scale copy number polymorphism in the human genome. Science 305, 525–528 (2004).
Bokhoven, H. Genetic and epigenetic networks in intellectual disabilities. Annu. Rev. Genet. 45, 81–104 (2010).
van Ommen, G. J. Frequency of new copy number variation in humans. Nat. Genet. 37, 333–334 (2005).
Vissers, L. E. et al. Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am. J. Hum. Genet. 73, 1261–1270 (2003).
Hastings, P. J., Lupski, J. R., Rosenberg, S. M. & Ira, G. Mechanisms of change in gene copy number. Nat. Rev. Genet. 10, 551–564 (2009).
Neo, W. S., Tonnsen, B. L., Report, B. & Challenging behaviors in toddlers and preschoolers with angelman, Prader-Willi, and Williams syndromes. J. Autism Dev. Disord. 49, 1717–1726 (2019).
Nassisi, M. et al. Ocular features in Williams-Beuren syndrome: A review of the literature. Curr. Opin. Ophthalmol. 34, 514–521 (2023).
Lackey, A. E. & Muzio, M. R. DiGeorge Syndrome, StatPearls, StatPearls Publishing Copyright © 2024, StatPearls Publishing LLC., Treasure Island (FL) Ineligible Companies (Maria Rosaria Muzio declares no relevant financial relationships with ineligible companies, 2024).
Shi, P., Xia, Y., Li, Q. & Kong, X. Usefulness of copy number variant detection following Monogenic disease exclusion in prenatal diagnosis. J. Obstet. Gynaecol. Res. 47, 1002–1008 (2021).
Riggs, E. R. et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American college of medical genetics and genomics (ACMG) and the clinical genome resource (ClinGen). Genet. Medicine: Official J. Am. Coll. Med. Genet. 22, 245–257 (2020).
Kearney, H. M., Thorland, E. C., Brown, K. K., Quintero-Rivera, F. & South, S. T. American college of medical genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet. Medicine: Official J. Am. Coll. Med. Genet. 13, 680–685 (2011).
Aslam, M. Chi-square test under indeterminacy: an application using pulse count data. BMC Med. Res. Methodol. 21, 201 (2021).
Schmid, S. et al. How to read a next-generation sequencing report-what oncologists need to know. ESMO Open. 7, 100570 (2022).
Xuan, J., Yu, Y., Qing, T., Guo, L. & Shi, L. Next-generation sequencing in the clinic: promises and challenges. Cancer Lett. 340, 284–295 (2013).
Luo, H., Wang, Q., Fu, D., Gao, J. & Lu, D. Additional diagnostic value of CNV-seq over conventional karyotyping in prenatal diagnosis: A systematic review and meta-analysis. J. Obstet. Gynaecol. Res. 49, 1641–1650 (2023).
Li, N. et al. The performance of whole genome amplification methods and next-generation sequencing for pre-implantation genetic diagnosis of chromosomal abnormalities. J. Genet. genomics = Yi Chuan Xue Bao. 42, 151–159 (2015).
Muys, J., Blaumeiser, B., Janssens, K., Loobuyck, P. & Jacquemyn, Y. Chromosomal microarray analysis in prenatal diagnosis: ethical considerations of the Belgian approach. J. Med. Ethics 46, 104–109 (2020).
Iafrate, A. J. et al. Detection of large-scale variation in the human genome. Nat. Genet. 36, 949–951 (2004).
Kidd, J. M. et al. Mapping and sequencing of structural variation from eight human genomes. Nature 453, 56–64 (2008).
Stankiewicz, P. & Lupski, J. R. Genome architecture, rearrangements and genomic disorders. Trends Genet. 18, 74–82 (2002).
Wang, Q. et al. Application of copy number variation squencing for prenatal diagnosis. Zhonghua Yi Xue Yi Chuan Xue Za zhi = zhonghua Yixue Yichuanxue Zazhi = Chinese. Chin. J. Med. Genet. 39, 949–953 (2022).
Butler, M. G. Prader-Willi syndrome and chromosome 15q11.2 BP1-BP2 region: A review. Int. J. Mol. Sci. 24 (2023).
Leader, G. et al. Adaptive living skills, sleep problems, and mental health disorders in adults with 22q11.21 deletion syndrome. Res. Dev. Disabil. 136, 104491 (2023).
Jiang, X. L. et al. Prenatal diagnosis of 15q11.2 microdeletion fetuses in Eastern China: 21 case series and literature review. J. Maternal-fetal Neonat. Med.: Off. J. Eur. Assoc. Perinat. Med., Feder. Asia Ocean. Perinat. Soc., Int. Soc. Perinat. Obstet. 36 2262700 (2023).
Hicks, M. A., Lalonde, E., Zoladz, J., Gonik, B. & Ebrahim, S. A diagnosis of maternal 22q duplication and mosaic deletion following prenatal Cell-Free DNA screening. Case Rep. Genet. 2023, 9127430 (2023).
Yang, L. et al. Genotype-Phenotype correlations in Angelman syndrome. Genes (Basel) 12 (2021).
Duis, J. et al. A multidisciplinary approach and consensus statement to establish standards of care for Angelman syndrome. Mol. Genet. Genomic Med. 10, e1843 (2022).
Gregoric Kumperscak, H., Krgovic, D. & Vokac, N. K. Specific behavioural phenotype and secondary cognitive decline as a result of an 8.6 mb deletion of 2q32.2q33.1. J. Int. Med. Res. 44, 395–402 (2016).
Lewis, H. et al. Epilepsy and electroencephalographic abnormalities in SATB2-associated syndrome. Pediatr. Neurol. 112, 94–100 (2020).
Smith, A. C. M. et al. Smith-Magenis Syndrome. in (eds Adam, M. P., Feldman, J., Mirzaa, G. M., Pagon, R. A., Wallace, S. E., Bean, L. J. H., Gripp, K. W. & Amemiya, A.) GeneReviews(®). Copyright 1993–2023, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved., Seattle (WA), (University of Washington, Seattle, 1993).
Funding
This research: including experimental design, sample collection, data analysis, and manuscript writing, was funded by the Scientific Research Project of Gansu Provincial Health Commission (GSWSKY2024-24), the Lanzhou Youth Science and Technology Talent Innovation Project (2024-QN-31), the Gansu Natural Science Foundation (25JRRA333), the Major project of Gansu Maternal and Child Health Hospital (GSFY-2021), and Clinical application of non-invasive prenatal genetic testing technology in chromosomal microdeletion and microduplication syndrome (2017-04-50).
Author information
Authors and Affiliations
Contributions
We acknowledge Mr X.F. and F.L. for reading through the manuscript and giving valuable comments Conceptualization, S.Z.; Data curation, S.Z. and C.J.; Formal analysis, S.Z. and S.C.; Funding acquisition, X.F., F.L. and S.Z.; Investigation, J.W. and P.L.; Methodology, X.F.; Project administration, X.F. and F.L.; Resources, X.L. and C.J.; Software, S.Z. and P.Z.; Validation, S.C.; Visualization, P.L.; Writing—original draft, S.Z. and S.C.; Writing—review & editing, F.L. and X.F.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
All sample collection procedures in the study strictly complied with the national ethical guidelines and were approved by the Ethics Committee of Gansu Maternal and Child Health Hospital (Batch No.: 2021GSFY Ethical Review). The study also complied with the World Medical Association Declaration of Helsinki.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, S., Cheng, S., Jia, C. et al. Clinical application of CNV-seq for diagnosing abnormal brain development in children. Sci Rep 15, 33547 (2025). https://doi.org/10.1038/s41598-025-18350-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-18350-x
