
Abstract
The post pandemic period has witnessed a notable increase in invasive Streptococcus pyogenes (GAS) infections across several countries. This surge has been partly attributed to the emergence of more virulent strains, and factors like the non-pharmaceutical interventions or the immunity debt. Despite its dual role as both a commensal and pathogen, our understanding of GAS carriage and the dynamics influencing the transition to invasive disease remains limited. In particular, the role of the pharyngeal microbiota in modulating GAS colonization and infection warrants further investigation. We performed a comprehensive analysis of a pediatric cohort in the post pandemic period collecting clinical data alongside diagnostic evaluations by using pharyngeal swabs and Rapid Antigen Detection Tests (RADTs) to differentiate GAS-positive from GAS-negative cases. Comparative genomic analyses were performed on clinical isolates to identify the prevalence of specific clones or virulence determinants. Additionally, the pharyngeal microbiota was characterized using 16 S rRNA targeted sequencing to assess differences in microbial community composition between the two groups. The overall composition of the pharyngeal microbiota appeared comparable between GAS-positive and GAS-negative children. Alpha diversity metrics (Observed Species, Shannon diversity and Pielou’s Evenness) were not significantly different (p = 0.820, p = 0.280 and p = 0.240, respectively), while Bray-Curtis defined two distinct groups (PERMANOVA, p = 0.01, R² = 0.523). The relative abundance of S. pyogenes within the Streptococcus genus emerged as a key differentiator. Genomic analysis revealed a mosaic representation of GAS clones, with the emm12.0 that was the most represented type, suggesting genomic characteristics did not directly correlate with the upsurge of respiratory GAS infections in our cohort. Our study underscores the complexity of GAS pathogenesis, highlighting that the transition from colonization to infection (acute pharyngitis) remains largely elusive and pharyngeal microbiota may contribute to a multifaceted interplay between host immunity and S. pyogenes.
Similar content being viewed by others
Introduction
Streptococcus pyogenes (β-hemolytic, Lancefield group A Streptococcus, commonly known as GAS) is a human-adapted pathogen responsible for a wide spectrum of diseases. These range from self-limiting throat and skin infections to invasive infections that typically involve normally sterile sites. Notably, repeated or inadequately treated infections can lead to post-infectious suppurative complications such as rheumatic heart disease and acute post-streptococcal sequelae1,2,3,4.
Following the COVID-19 pandemic, several countries have reported a significant increase in invasive GAS (iGAS) infections. In particular, reports from the UK in 2022 documented a marked rise in both scarlet fever and iGAS, with severe outcomes in children5. This trend appears to be linked to the spread of the M1UK clone, which emerged in 2008 and rapidly expanded nationwide6,7,8,9,10,11,12. While the genomic features of M1UK may provide it with a fitness advantage, increased pathogenicity, and a notable capacity to persist in the population, COVID-19-related restrictions may also have impacted population immunity and GAS carriage, further contributing to this trend13,14.
Acute pharyngitis is a common reason for pediatric visits, although GAS accounts for only 20–30% of these cases15. Differentiating between GAS and viral pharyngitis based solely on clinical examination is challenging, and there is no unanimous international consensus on the utility of microbiological confirmation16. Some countries, including the United States, France, Italy, Spain, and Sweden, support microbiological testing guided by clinical assessments and scoring systems, whereas the UK and the Netherlands rely exclusively on clinical evaluation without microbiological verification17,18. Although the pharyngeal swab remains the gold standard for GAS detection, along with Rapid Antigen Detection Tests (RADTs)19GAS colonization is observed in 5–20% of asymptomatic healthy children, prompting debate over the need for prophylaxis15. This underscores the importance of health policies that critically analyse epidemiological data.
The decrease in Emergency Department visits across Europe during 2020–2022 led to fewer pharyngeal swabs being collected. However, an increased focus on respiratory infections in 2023 resulted in more samples being obtained, which in turn led to a higher incidence of GAS compared to the pre-pandemic period5. In particular, an enhanced trend in respiratory GAS infections has been observed in preschoolers20resembling patterns seen with viral infections14. Intriguingly, there was no corresponding rise in iGAS cases, suggesting a more complex epidemiological scenario in Europe and highlighting the need to standardize surveillance programs21.
In addition to the risk factors mentioned above, the pharyngeal microbial community may influence mucosal colonization, GAS overgrowth, and subsequent disease development22. The upper respiratory tract, as the primary portal for infectious droplets, is supported by a diverse microbiota that plays an active role in preventing respiratory tract infections23. The pharyngeal microbiome, composed of numerous bacterial species, interacts with host epithelial and immune cells to form a protective micro-ecological system24. For instance, in vitro studies have shown that Lactobacillus and Streptococcus salivarius K12 can antagonize GAS25. Disruption of the pharyngeal microbiota and the resulting impairment of the local epithelium may increase the risk of infections by altering mucosal immune interactions26.
Current evidence on the optimal antibiotic for eradicating pharyngeal S. pyogenes carriage is limited27and its effects on the pharyngeal microbiota remain largely unknown. Further studies are needed to better characterize the pharyngeal microbiota and its interaction with the pathogen, particularly in pediatric populations that are primarily investigated for viral infections28,29,30.
In this study, we investigated a pediatric post-COVID-19 cohort to assess GAS infection by evaluating clinical scores, microbiological evidence, and GAS genomic features, while also comparing the pharyngeal microbiota of GAS-positive individuals with those testing negative.
Results
Cohort description
Eighty-eight patients were enrolled in the study, of whom 45 (51.1%) had evidence of GAS infection documented by a positive throat swab culture, according to the inclusion and exclusion criteria described in the Materials and Methods section. GAS-positive subjects appeared slightly older than GAS-negative individuals (5.54 vs. 4.47 years). A closer look at the clinical features revealed that pharyngodynia was twice as common in GAS-positive patients (46.7% vs. 23.3%, p = 0.027). Abdominal pain (0% vs. 14%, p = 0.011) and rhinitis (13.3% vs. 44.2%, p = 0.002) were significantly more frequent in GAS-negative patients. Lymphadenopathy showed a non-significant trend towards being more common in GAS-positive individuals (35.6% vs. 16.3%, p = 0.053). Other signs, such as fever, pharyngeal hyperaemia, tonsillar exudate, and palatal petechiae, did not differ significantly between the two groups.
For eight and six individuals, respectively for GAS and CTR group, antibiotics were prescribed before the last 30 days from enrolment not for upper respiratory tract infection. However, following diagnosis, amoxicillin was predominantly prescribed in GAS-positive children (60.0% vs. 9.3%), while amoxicillin-clavulanate was lesser but noteworthy used in both groups. A 10-day course was preferred for the GAS-positive group, and a 7-day course for the GAS-negative group. None of the comorbidities analysed in our dataset showed a significant association with either group.
Of note, the RADT was not performed in most cases, regardless of whether the culture was positive or negative Adenovirus was slightly more detected in GAS-negative patients. C-reactive protein (CRP) values were available only for a minority of patients (9 GAS-positive patients and 11 GAS-negative controls). Furthermore, CRP values were positive in 8 out of 9 cases (88.9%) in the GAS-positive patients and 6 out of 11 cases (54.5%) in the GAS-negative controls and did not differ significantly between groups (p = 0.157). Given the small and selectively tested subgroup, CRP data were considered inconclusive and were not further analysed. A schematic flowchart of the study is presented in Supplementary Fig. 1, and all demographic and clinical data are summarized in Table 1.
Characterization of Streptococcus pyogenes isolates
In recent years, GAS has been linked to several community outbreaks31,32,33and national surveillance programs have particularly documented a post pandemic upsurge in iGAS7,34,35. Although in many European countries an increased trend of invasive GAS was observed in children, our findings indicated a predominant involvement of the upper respiratory tract20. Despite potential variations due to differences in national surveillance guidelines, an increased spread of more virulent strains, particularly of M1UK isolates, was also observed36,37.
To investigate the possible clonal transmission of GAS isolates, we performed whole genome sequencing on GAS strains from 36 out of 44 positive individuals enrolled in the study (see Supplementary Data 1 for details) and applied the high-resolution core-genome multilocus sequence typing (cgMLST) scheme proposed by Toorop and colleagues, which defines a cluster threshold of fewer than 5 allelic differences out of 1,095 target loci38. The maximum spanning tree revealed outbreak clusters grouping isolates with an allelic difference of ≤ 1. This was observed for ST28 (3 isolates) and ST242 (5 isolates), while clusters associated with ST36 appeared more complex, accounting for three mini-outbreaks (Fig. 1a). While cgMLST-based analysis provided a robust classification of strains into sequence types and outbreak, it failed to resolve finer genetic relationships within STs. For instance, ST101 and ST28 isolates appeared as compact clusters in the cgMLST tree, with limited internal structure. In contrast, the SNP-based matrix and phylogeny (Supplementary Fig. 2) showed a deep resolution. Notably, strains TF054, TF059, TF068, TF042, TF118, and TF021, grouped distantly in the cgMLST network, generated a tight monophyletic clade in the SNP-based tree, separated from the rest by thousands of SNPs. This highlighted recent common ancestry or selective sweeps not captured by allele-based methods. We further performed pangenome analysis on 36 isolates identifying a total of 8287 genes, categorized based on their distribution across genomes into 157 core genes, 334 soft core genes, 2289 shell genes and 5507 cloud genes. This composition suggests a potentially high degree of genomic plasticity, although tempered by the presence of a consistent core genome, as reflected in the Supplementary Figs. 2 and 3a.
Together, these analyses showed that SNP-based phylogenies can detect microevolutionary patterns and recent divergence that cgMLST misses, while pangenome analysis added a functional layer, helping to interpret divergence in terms of gene content variation. As a consequence, cgMLST-based epidemiological investigations may be not sufficient to describe transmission prompting to consider genetic variability contained in the pangenome39. In other words, we have the spread of strains that maintained low core genome variability so that accessory genome information, such as virulence factors, antimicrobial resistance genes (AMRs) and prophages regions may result informative due to host-mediated selective pressure. In this scenario we carried out a SNP based analysis to elucidate the relationship between isolates and to generate a high-resolution phylogenetic tree (Supplementary Fig. 3b).
Intriguingly, our cohort was not associated with a specific GAS sequence type (ST) and relative emm-typing (Fig. 1). Four emm-clusters were identified: AC-3: comprising emm1.0 (33.3%), emm1.25 (50%), and emm1.52 (16.7%); AC-4: including emm12.0 (66.7%), emm12.101 (28.6%), and emm12.19 (4.8%); E1: representing emm4.0; E4: encompassing emm22.21 (37.5%), emm28.0 (12.5%), emm77.0 (12.5%), and emm89.0 (37.5%) (Fig. 1b). Notably, only two strains (TF054 and TF068) belonging to emm1.0 were recognized as M1UK clones, each presenting 27 SNPs according to Li and colleagues36 (Supplementary Data 2). Furthermore, virulence and AMR genes appeared to be partially associated with the emm-type (Fig. 1b). For instance, the genes cpa, emm, fctB, grab, lepA, mf3, speA, and srtC1 were selectively found in the AC-3 cluster, independent of the emm1 group, while other genes were more randomly distributed among the strains. Of note, the superantigen-encoding gene ssa was detected in only 9 out of 36 clinical isolates, a point that is particularly noteworthy when compared to UK data. Additionally, the virulence factor speB was identified in 30 (83.3%) out of 36 strains. Particularly, 4 out of 14 emm12.0 strains, 1 out of 6 emm12.101 strains, 1 out of 3 emm22.21 strains, and 1 emm28.0 isolate did not carry the speB gene.
In total, 79 prophage regions were detected, accounting for 72 different prophages. The most frequently represented prophages were T12, P9, φ3396, and several belonging to the Streptococcus phages 315 family, each detected in more than 20 strains. Summarized data are reported in Supplementary data 1. Notably, prophage T12 was detected in 25 out of the 36 isolates, and the T12-SpeA association was found exclusively in ST28 (emm1.0 and emm1.25). Conversely, two integrative conjugative elements (ICEs) and two integrative mobilizable elements (IMEs) were detected in the 36 analysed genomes. ICEs with a type IV secretion system (T4SS) were observed in 10 isolates, while ICEs without an identified flanking direct repeat (DR) were observed in 4 isolates (Supplementary Data 1). IMEs were detected in 11 isolates, with 5 strains showing an IME without DR (Supplementary Data 1).
Taken together, our data suggest that there is no association between a specific clonal GAS genotype and the increasing trend of GAS respiratory infections.
Pharynx microbiotas in CTR and GAS group
The increased incidence of GAS infections observed in many European countries since late 2022 remains poorly understood. While the rising trend of iGAS cases may be explained by the spread of more virulent strains5,9 or various other factors21respiratory infections may be more closely linked to the microbial populations colonizing the oropharyngeal tract. In fact, genus-level associations have been described, both specific to particular respiratory conditions and common across different conditions40.
To investigate the role of microbial communities, we analysed pharyngeal specimens collected during the first months of 2023 from a clinical cohort of paediatric patients admitted to the emergency department. This cohort included individuals microbiologically diagnosed with GAS infection, as well as GAS-negative controls from the same setting and time period (Supplementary Fig. 1). The same clinical cohort was used for both the cgMLST genomic analysis and the pharyngeal microbiota study.
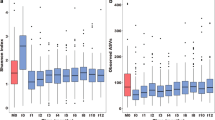
Following rarefaction, alpha diversity was quantified using Richness (Observed Species), the Shannon index, and Pielou’s index, revealing no significant differences between the control (CTR) and GAS microbiotas (Supplementary Figs. 4–5 and Supplementary Data 3). Conversely, beta diversity, measured by Bray-Curtis distance and visualized using Principal Coordinate Analysis (PCoA), demonstrated a separation between the two groups (Fig. 2a). Although PERMANOVA analysis confirmed a statistically significant difference between CTR and GAS groups (p = 0.01, R² = 0.523), the distance between centroids (representing the average characteristics of samples) suggested only a slight difference in microbial composition between the two groups, while the dispersion from centroids (indicating variability among samples) showed comparable homogeneity within each group (Supplementary Fig. 6).
Consistent with these observations, relative abundance at the phylum (Supplementary Fig. 7 and Supplementary Data 6) and genus levels (Supplementary Fig. 8 and Supplementary Data 6) showed only minor differences between the two groups, except for Enterobacteriaceae and the Streptococcus genus (p = 0.006 and p = 0.009, respectively). Differential abundance analysis revealed that a specific amplicon sequence variant (ASV), representing S. pyogenes (ASV: 89bdc53178d73e9d6314839f496ae4cc), along with Porphyromonas, Haemophilus, Corynebacterium, Abiotrophia, and Capnocytophaga, was enriched in GAS-positive samples, whereas Alloprevotella, Prevotella, and a different Streptococcus genus were more abundant in the CTR group (Fig. 2b). Each ASV enriched in GAS and CTR group was blasted to identify the corresponding species (Supplementary data 5).
Although taxonomic profiles were largely limited to the genus level due to sequencing constraints, we selected all Streptococcus taxa and analysed their prevalence and relative abundance between the CTR and GAS groups. Each ASV was then blasted to identify the corresponding Streptococcus species (Fig. 2c and Supplementary Data 6). While most ASVs were equally distributed between the two groups, S. pyogenes ASV showed increased relative abundance in the GAS group, whereas ASVs corresponding to non-S. pyogenes species were more abundant in the CTR group (Supplementary Data 6). To corroborate the reliability of the relative and differential abundances in both GAS and CTR groups, we quantified S. pyogenes and non-S. pyogenes burdens using digital PCR. Indeed, this quantitative insight might have clinical relevance, for example in distinguishing high- versus low-burden carriers or understanding microbial load in relation to symptoms or microbiota composition. Additionally, culture-based methods of pharyngeal swabs do not provide a quantitative measure. We observed that GAS-positive samples exhibited significant S. pyogenes overgrowth compared to non-S. pyogenes species and total bacteria, while stochastic distribution was detected within GAS group (Fig. 2d).
Additionally, ASV1 had a major impact on sample distribution. Biplot analysis demonstrated that ASV1, along with ASVs corresponding to Peptostreptococcus and Haemophilus genera, primarily influenced the distribution of GAS samples, although the angles between the three arrows suggested that these genera were independent of each other. Conversely, Lachnospiraceae and Prevotellaceae appeared to be correlated and influenced CTR distribution (Supplementary Fig. 9 and Supplementary Data 9).
We subsequently explored the common core microbiota shared between CTR and GAS specimens (Supplementary Fig. 10 and Supplementary Data 8). The number of ASVs included in the common core decreased as parameter values increased (relative abundance and prevalence across samples). Considering the range of parameter values (relative abundance of 0.001–0.2 and prevalence of 5–100%), the common core consisted of 14 ASVs in both CTR and GAS groups. At a detection threshold of 0.1 and prevalence of 0.5, approximately 25 ASVs were identified, suggesting that the core microbiota across all CTR and GAS specimens was numerically similar. The common core between CTR and GAS included 11 ASVs, while 3 ASVs represented core taxa specific to either CTR or GAS. The CTR-specific core included ASVs from the Streptococcus genus (excluding S. pyogenes) and Veillonella, which occurred at lower abundances but were highly prevalent across samples. Conversely, the GAS-specific core included ASVs from the Streptococcus genus matching S. pyogenes, which was found at high relative abundance and prevalence across all samples, as well as Porphyromonas and Haemophilus, which occurred at lower abundances and with lower prevalence across samples.
Network analysis revealed a different structure of microbial correlations in the GAS group compared to the CTR group (Supplementary Fig. 11). In the CTR group, the network exhibited a more balanced structure, with interactions distributed among various genera, including Streptococcus, Granulicatella, and Prevotella_7, which emerged as central nodes in both groups. However, the GAS network appeared more complex than the CTR network. Veillonella and Fusobacterium showed increased connections in the GAS group, suggesting a possible role in GAS infection. Additionally, Lautropia and Abiotrophia played a less prominent role in the GAS group compared to the CTR group. Interestingly, Porphyromonas established new positive connections with Fusobacterium, while the Streptococcus genus exhibited positive links with certain Proteobacteria (Acinetobacter, Pseudomonas). The elements describing the network analysis are summarized in Supplementary Data 9.
Taken together, these results demonstrate a slight but significant distinction between the microbiotas of CTR and GAS individuals. However, this variation appears to be primarily driven by S. pyogenes overgrowth and the subsequent restructuring of microbial interactions, which may contribute to maintaining inflammation and promoting disease progression.
The associations between clinical variables, alpha diversity metrics, and Streptococcus ASVs were assessed using Spearman’s correlation and represented in a correlation plot (positive and negative correlations shown in blue and red, respectively, with stronger correlations depicted as larger and more colourful circles). In GAS-negative individuals, alpha diversity metrics exhibited a negative correlation with ASV11, as well as with maculopapular rash, ASV3, and ASV7 in relation to pharyngeal hyperaemia. Interestingly, a strong negative correlation was found between ASV10 and ASV1. Notably, the most significant positive correlations were observed between GAS detection (SBEGA, using RADT) and antibiotic therapy (not related to GAS infection) in the past month, as well as between ASV9 and ASV2 (Fig. 3a).
The analysis was repeated for GAS-positive patients, revealing a significantly different pattern (Fig. 3b). A strong negative correlation was detected between ASV6 and other Streptococcus genera (ASV1, ASV3, ASV8, ASV10, ASV11, and ASV14), as well as with alpha diversity metrics. This finding suggests a substantial impact of ASV6 on these parameters and on the modification of the pharyngeal microbiota. Interestingly, no strong correlations were observed with clinical variables a role for the microbiota in GAS colonization and infection rather than in the development of symptoms or clinical manifestation.

Genomic characterization of group A Streptococcus pyogenes clinical strains isolated from oropharynx swabs. Streptococcus pyogenes isolates obtained following admission in first aid care unit for respiratory symptoms from oropharynx specimen were genotypically characterized by whole genome sequencing. The experimental design, including the isolate cultured and sequenced number, is schematically represented in Supplementary Figure 1. Each genomic DNA was achieved from pure culture, processed to obtain an Illumina tagmented-based library and finally sequenced using an Illumina MiSeq instrument. Short reads raw data were quality checked to exclude contamination, de novo aligned and the final obtained contigs used as template to search for virulence genes and antimicrobial determinants. Sequencing details are reported in Supplementary data 1, whereas the bioinformatic tools used for the analysis are summarized in the Supplementary data 8. a. Minimum spanning tree shows phylogenetic distances among GAS isolates. Colours are associated to sequence type identification. Each number indicates the distances between two or more isolates, while a node with more isolates or the marked connection between more isolated highlights clonal strains (less than six single nucleotide variants). b. A maximum likelihood phylogenetic tree of the GAS isolates was generated with the cgMLST by using RidomSeqSphere. The heatmap summarizing sequence type (ST, tip points), emm-type, emm-cluster, virulence (blue: gene presence) and antimicrobial resistance (red: gene presence) genes and prophages and ICEs/IMEs was provided by using ggtree package. Virulence genes associated with prophage regions were evidenced with the symbol asterisk.

Oropharynx microbiotas in children admitted to first aid care unit for respiratory symptoms. The oropharynx microbiotas in children positive (GAS) or not (CTR) for S. pyogenes, microbiologically diagnosed, following admission in first aid care unit for respiratory symptoms were used in the diversity analyses. The experimental design, including case number, is schematically represented in Supplementary Figure 1. a. Comparative analysis of the microbial communities between the CTR and GAS groups was obtained by measuring Bray-Curtis distance and visualized by the Principal Coordinate Analysis (PCoA) plot. Samples dispersion within each group is represented by ellipses, while the size and the sharpness of the points reflects Shannon and Pielou’s alpha diversity indices, respectively. Alpha diversity metrics (Richness, Shannon diversity index and Pielou’s evenness) are reported additionally reported in Supplementary Figure 3. Pairwise analysis of the difference between two groups was performed using the Adonis test with default parameters. b. The volcano plot displays differentially abundant taxa between the two groups, with significantly enriched genera in CTR highlighted in blue and those in GAS in red. Amplicon sequence variants (ASV) representing the Streptococcus genus strongly enriched in the GAS group identifies (100% identity) Streptococcus pyogenes following blast analysis. Differential abundance was tested by the DESeq2 package with default parameters and alpha 0.01. Adjusted p values were generated by the Benjamini-Hochberg correction of the ANOVA test. Log2FoldChange and -log10(p value) were reported on the x and y axis, respectively. The vertical dotted red lines correspond to 2 Log2FoldChange up and down and the horizontal dotted red line shows represents an adjusted p value of 0.05. The relative abundances at Phylum and Genus level in the CTR and GAS groups are shown by a bar plot in Supplementary Figure 2–3 and Supplementary data 3. c. The heatmap shows the ASVs representing Streptococcus genera identified in the dataset. Circles size represents ASV frequency while colour intensity indicates their relative abundance. Supplementary data 6 summarized the plotted data with the blast the identification of each ASV following blast analysis. Adjusted p values were generated by the Bonferroni correction of the Mann–Whitney U test. d. Dot plot panels report the quantification of microbial DNA through quantitative PCR, comparing total bacterial load (16S rDNA), non-S. pyogenes bacteria, and S. pyogenes copies among CTR and GAS groups. Adjusted p values were generated by the Bonferroni correction of the Mann–Whitney U test. Bioinformatic tools used for the analysis are summarized in the Supplementary data 10.

Correlation among clinical variables, alpha diversity and Streptococcus genus associated ASVs. a. Correlation matrix for GAS negative group. b. Correlation matrix for GAS positive group. Each cell in the matrix represents Pearson’s correlation coefficient between two variables. The color scale indicates both the direction and strength of the correlation: blue hues denote positive correlations, red hues denote negative correlations, and white or lighter tones indicate values closer to zero (i.e. weaker correlations). Alpha diversity values and Streptococcus genus ASVs association are reported in Supplementary data 3 and 4.
Discussion
S. pyogenes (GAS) can cause a wide spectrum of infections, ranging from mild conditions to severe diseases, and contributes significantly to morbidity and mortality41. In recent years, growing interest in this bacterium has focused on its dual role as both a commensal organism and a pathogen42. GAS colonizes various mucosal surfaces, primarily the throat and skin, with estimates indicating that many individuals carry it asymptomatically42,43,44. Despite its prevalence, our understanding of GAS carriage remains limited, and conflicting data persist regarding the efficacy of antibiotic treatment for eradicating asymptomatic pharyngeal carriage27. Intriguingly, many countries have reported an increased trend in iGAS infections during the post-pandemic period45,46which has been partially attributed to the spread of more virulent strains such as the M1UK clone10,47,48. However, little attention has been paid to the primary anatomical site of GAS colonization and its role as the first line of defence against the bacterium.
In this context, our study provides a comprehensive analysis of GAS infections in a paediatric cohort following the COVID-19 pandemic. We evaluated the clinical features and management of patients along with microbiological findings, including comparative genomics of clinical isolates and characterization of the oropharyngeal microbiota. Notably, we found no major differences in the overall composition of the oropharyngeal microbiota between GAS-positive and GAS-negative children. All subjects were enrolled during the same period and were systematically investigated for GAS infection, an approach that is especially relevant given the heightened focus on respiratory infections and increased detection of GAS after COVID-19. Multiple factors may have contributed to this shift, including the “immunity debt” hypothesis13changes in non-pharmaceutical interventions49and altered seasonality of respiratory viruses21.
The pathogenic potential of GAS is largely attributed to its array of virulence factors, notably the M protein, which facilitates immune evasion, as well as various toxins and regulatory genes that exacerbate disease severity41. These factors likely aid GAS in transitioning from a benign colonizer to a dangerous pathogen, particularly under conditions of immunocompromise or environmental stress41,50. Understanding the epidemiology of GAS is essential for effective disease management. To this end, we collected clinical isolates and performed comparative genomic analyses to explore whether the increased incidence of respiratory GAS infections exhibits specific seasonal patterns, peaking during the colder months20and to assess potential geographic and demographic differences relative to data from other countries. While several studies have focused on invasive GAS (iGAS), there is still limited information on respiratory strains. Our data clearly indicate that subtle changes in the incidence of GAS infections have occurred in Italy in recent years, although differences in surveillance programs may influence these observations21.
Only two strains belonging to the M1UK cluster were identified, each carrying the 27-SNP signature characteristic of this clone, which suggests a moderate spread of M1UK in Italy, a finding supported by previous studies that call for deeper investigation47,48,51. Additionally, the phage-encoded superantigen gene ssa, commonly associated with scarlet fever, was detected in a limited number of isolates (approximately 25% of strains, primarily within ST36 -emm12.0) and at lower rates compared to reports from other countries52. This difference may be linked to the absence of mutations in the covR/covS two-component regulatory system, which are frequently observed in iGAS strains and ssa-positive isolates53,54. These genomic characteristics might help to explain the relatively low number of iGAS infections observed in Italy compared with other regions21,55,56. Moreover, the similar frequency of streptococcal pyrogenic exotoxins in our isolates aligns with other studies57. Of note, previous studies have documented rare instances of speB negative isolates, suggesting possible horizontal gene loss or recombination events, whereas more studies focused on expression loss58. Indeed, inactivation of speB occurred due to mutations or deletions, particularly in emm1 and emm3 lineages59. Similarly, Sumby and colleagues observed speB inactivation in emm3 strains due to mutations in the covR/covS regulatory system60.
The observation of several mini-outbreaks without a clear transmission link further indicates that the current cgMLST cluster threshold of < 5 allelic differences may be insufficient for accurately distinguishing related cases in the absence of classical epidemiological data and the use of SNP-based approach to characterize the pangenome38. Of note, a recent study evidenced only 355 up to 1390 core genes with defined essential role for S. pyogenes survival, but few is known about the involvement of the accessory genes in the bacterial fitness61. Furthermore, the extensive heterogeneity observed in emm-types suggests that there is no robust association between a specific strain type and the site of infection, a notion supported by previous investigations62,63. For example, while T4SS-associated ICEs have been linked to emm28 strains in iGAS cases64our data indicate that these elements are present across diverse emm-types. Similarly, the T12 phage appears to be clinically relevant only in ST28 isolates, particularly those carrying SpeA, which is strongly associated with scarlet fever and streptococcal toxic shock-like syndrome65. Although phage φ315.2-6 has been associated with several virulence genes and the emergence of hypervirulent M3 serotypes66in our study the presence of these virulence determinants was not consistently predictive of pathogenicity. This observation suggests that some genomic elements may not fully capture the complexity of GAS virulence, highlighting the need for integrating genomic data with clinical evidence when developing global vaccines.
Emerging research has also highlighted the presence of GAS as part of the normal respiratory microbiota, raising questions about its overall impact on human health42,67. GAS can colonize the upper respiratory tract without causing disease, as evidenced by asymptomatic carriers68,69. The transition from a commensal to an invasive state may be triggered by failures in immune defences or concurrent viral infections70,71. As a member of the complex pharyngeal ecosystem, GAS coexists with other microorganisms, and this interplay may be essential for maintaining a balanced microbiota, preventing infections, and supporting overall health23,72. Unfortunately, characterizing the pharyngeal microbiota remains challenging due to its dynamic and variable composition23. Interestingly, in our cohort, both GAS-positive and GAS-negative microbiotas were relatively homogeneous and showed only minor differences, suggesting that external factors such as viral infections or antibiotic treatments did not significantly alter the indigenous microbial communities29.
Our analyses indicate that the major difference in the microbiota signature is driven by the Streptococcus genus. Despite the inherent limitations of 16 S rRNA targeted sequencing, we demonstrated that GAS-positive children exhibit a distinct prevalence and abundance of S. pyogenes compared to non-S. pyogenes ASVs. Streptococcus species are among the dominant colonizers from early life and appear relatively stable regardless of feeding practices, underscoring their key role in the oropharyngeal niche73,74. In contrast to viral infections, where the pharyngeal microbiota is often enriched in opportunistic pathogens26our findings suggest that the relative abundance of Streptococcus is the primary differentiator between GAS-positive and negative individuals. Complementary studies have even demonstrated direct antagonism of S. pyogenes by probiotics such as Lactobacillus and S. salivarius (e.g. RS1, ST3 and K12) inhibiting GAS adhesion, upregulating epithelial barrier functions and triggering NF-kB cascade to modulate inflammation25,75. The altered equilibrium between GAS and protective microbial species might influence biofilm formation and ultimately affect GAS colonization and evasion of the host immune response68,75,76. Unfortunately, due to limited sample size and lack of severity-related metadata, we could not explore the microbiota’s role in GAS infection severity.
However, these findings underscore the emerging role of the pharyngeal microbiota in GAS colonization and infection and point to additional factors that should be considered when identifying immune correlates essential for vaccine development77. A limitation of our study is the lack of host immunological data, which could have provided further insights into the immune response against GAS. Moreover, while the concept of “immunity debt” has been widely discussed, we could not compare our findings with historical data78 and we could not measure the impact of non-pharmaceutical interventions during the COVID-19 pandemic on the host immunity79.
Seasonal trends remain a critical factor in GAS epidemiology, as infections typically increase during colder months, likely due to increased indoor crowding and enhanced viral survivability in dry, cold conditions80. Our study showed a shift in seasonality compared to previous years, a change that may be attributed to pandemic containment measures20. Notably, while clinical manifestations were largely similar between GAS-positive and -negative patients, GAS-positive individuals were generally older, a finding that aligns with previous reports showing lower GAS incidence in children under three years of age and supports our earlier observations on respiratory GAS infections20. Many symptoms were observed in both groups, consistent with the known low specificity of clinical signs in diagnosing streptococcal pharyngitis, which complicates the differentiation between viral and bacterial infections based solely on clinical examination16. For instance, adenovirus infections can present with exudative pharyngitis, characterized by tonsillar exudates and cervical lymphadenopathy, and are often clinically indistinguishable from GAS infections, a challenge further complicated by other viruses such as Epstein–Barr virus (EBV)17. In children, adenoviral pharyngitis often features fever and a sore throat, sometimes accompanied by tonsillitis that appears exudative, making it difficult to differentiate from bacterial causes based purely on symptoms and signs. While the presence of conjunctivitis, rhinitis, or systemic viral symptoms may suggest a viral aetiology, adenovirus (especially serotypes 3 and 7) can cause a distinct clinical syndrome known as pharyngoconjunctival fever, which includes pharyngitis with exudate, fever, and conjunctivitis, further blurring clinical differentiation81. Moreover, EBV can present with even more prominent pharyngeal exudates and massive anterior and posterior cervical lymphadenopathy, as well as splenomegaly, leading to further diagnostic confusion. This overlap has substantial clinical implications. Routine clinical criteria alone are inadequate to distinguish viral from bacterial aetiologies with confidence. The diagnostic challenge is further amplified in young children, where symptoms may be nonspecific or atypical (such as irritability, refusal to eat, or abdominal complaints), and the consequences of misdiagnosis carry potential risks of both untreated GAS complications and the hazards of antibiotic overuse. In this context, the use of RADT assays and pharyngeal swabs remains essential for confirming GAS infection and improving our understanding of its epidemiology.
Whereas serious bacterial infections, including but not limited to GAS, and viral infections can cause leucocytosis. The finding that leucocytosis is more common in GAS-negative patients suggests that other, potentially more systemic or non-bacterial causes of inflammation (such as viral or non-GAS bacterial pathogens, or non-infectious conditions) may result in greater overall leukocyte increases in this population82. In GAS-positive patients, the increased percentage of neutrophils reflects the acute, often localized inflammatory response to this bacterium, in line with the classical concept of a “left shift” in bacterial infections, where neutrophilia predominates83. Unfortunately, because CRP measurement was applied inconsistently and only in a small subgroup of patients, no meaningful conclusions can be drawn from these data.
Our antibiotic usage data indicate that amoxicillin remains the most frequently prescribed treatment (60%), compared with amoxicillin-clavulanate. This trend, observed both in Italy and across Europe, highlights the need for targeted interventions to improve adherence to treatment guidelines and promote rational antibiotic use12. Although a standard 10-day course of 50 mg/kg oral amoxicillin is generally recommended for optimal GAS eradication, the ideal treatment duration remains debated84,85. Revised treatment protocols could also be necessary, involving the combination of β-lactams with clindamycin to counteract toxin-mediated severity, particularly when gene variants associated with rising penicillin MIC values are reported86,87,88. Moreover, given the carrier state and our microbiota findings, extending treatment may have implications for the broader Streptococcus community. Current evidence suggests that chronic carriers pose minimal risk for transmitting GAS and have a negligible likelihood of developing complications89,90. Differentiating between recurrent GAS pharyngitis and repeated viral infections in chronic carriers remains challenging, and without genomic data, it is difficult to determine whether subsequent episodes represent true recurrences or new infections21.
Overall, the expanding research on GAS emphasizes the need of continued surveillance and to integrate genomic findings and microbiota associations into public health policies. A deeper understanding of GAS resistance patterns and epidemiological trends can guide interventions to address immediate health threats, while insights into genomic diversity and microbial interactions will be crucial for developing novel treatments and effective vaccines.
Methods
Cohort
We enrolled patients aged < 18 years who showed tonsillopharyngitis, scarlet fever, and other signs of GAS infection at the Pediatric Emergency Department (PED) of the Fondazione Policlinico A. Gemelli in Rome over the course of the year 2023. All the patients with a clinical suspicion of GAS pharyngitis were considered eligible with symptoms of acute respiratory tract infection such as cold, pharyngitis, laryngitis, tracheitis, otitis, epiglottitis, laryngotracheitis, influenza, bronchitis and pneumonia. We excluded patients that followed antibiotic therapies in the last 30 days, treatments with prebiotics or probiotics in the last 60 days (in particular containing Streptococcus salivarius), hormone therapies in the last 60 days, local therapies in the last 60 days, treatments with oral cavity disinfectants in the last 30 days, presence of other pre-existing or ongoing respiratory pathologies and current severe sepsis91. Demographic and clinical data were collected for all participants, including age, gender, symptoms, laboratory tests, management details including Rapid Antigen Detection Test (RADT). Both antibiotic therapies administered before the last 30 days and those initiated after admission to PED were recorded. Patients were dichotomized into two different categories, ‘positive’ or ‘negative’, based on the microbiological result obtained by oropharynx swab culture. The RADT is a point-of-care diagnostic tool that provides rapid identification of GAS in pharyngeal swabs within few minutes. According to Di Mario and colleagues92management of paediatric pharyngitis should combine the McIsaac clinical score (with testing advised for scores of 3–4) and the RADT, reserving antibiotic therapy (typically amoxicillin) for patients with positive RADT results or very high clinical scores93. Despite these recommendations, in our hospital setting RADT was rarely available (see Table 1) in the emergency department during the study period. Consequently, antimicrobial treatment decisions were primarily based on clinical evaluation using the McIsaac score, in accordance with national guidelines when RADT was not feasible.
We focused on the oropharynx microbiotas, assessed by 16s rRNA targeted sequencing, and variables influencing this biological niche. The schematic experimental design is reported in Supplementary Fig. 1 and all participants and samples metadata available for this study are reported in Supplementary Data 14. Differences between GAS and CTR groups were assessed by using Welch T-test and Chi-Square or Fischer exact T-test according to variable type using IBM SPSS Statistics v.26. The statistical methods are introduced in the other parts of the Methods as shown below. All procedures performed were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Ethical approval for this study was obtained from the Ethics Committee of the Fondazione Policlinico Universitario Agostino Gemelli IRCCS in Rome (ID6603, 0021428/24-approval date 03/07/2024). The parents of the patients were informed about the purpose of this study and signed an informed consent form for consenting access to children’s medical records and for processing personal data.
GAS specimens’ manipulation, genomic DNA extraction and whole genome sequencing
GAS clinical strains were isolated and stored in 20% glycerol and stored at -80 °C at the time of the diagnosis. To perform whole genome sequencing stored strains were streaked onto Columbia blood agar (Oxoid) prior to broth culture. We recovered thirty-six strains out of fourty-five. Genomic DNA was extracted from cultures of GAS grown overnight in Todd-Hewitt broth (Oxoid) using a CTAB protocol and DANAGENE Microbial DNA kit (Danagen-Bioted). Briefly, 10 ml of overnight cultures was centrifuged and suspended in CTAB lysis buffer according to manufacturer’s instruction. All procedures were performed in a biosafety level 2 laboratory. DNA concentration and purity were assessed with a NanoDrop One spectrophotometer before proceeding with sequencing (ThermoFisher).
Whole genome sequencing libraries were generated using the DNA Prep kit (Illumina)94,95,96. Paired end short reads were generated by an Illumina MiSeq DX platform following Illumina’s recommendation. All raw sequence data have been deposited in the NCBI Sequence Read Archive (BioProject accession number: PRJNA1241328, https://submit.ncbi.nlm.nih.gov/subs/bioproject/SUB15197955/overview).
Whole genome sequencing bioinformatic analysis
Raw data were processed using CLC Genomic Workbench (Qiagen). Each sample quality checked and then was both quality (quality limit: 0.05, maximum number of ambiguities: 2) and adapter trimming process. All pre-processed reads were processed for a k-mer analysis to identify possible contamination, and best Streptococcus pyogenes matched reads were collected for the next steps.
The high-quality paired end reads resulted from pre-processing were then used for de novo genome assembly (bubble size:50, word size: 20, minimum contig length 200). To assess the assembly stats and quality reads were mapped to contigs. The summary of all analyses is reported in the Supplementary Data 10. De Novo assemblies were then used to detected virulence genes and antimicrobial resistance genes by using both AMRFinderPlus (https://github.com/ncbi/amr) v.3.11.18 and ABRicate v.1.0.1 (https://github.com/tseemann/abricate). The latter was used with Comprehensive Antibiotic Resistance Database (CARD, 2631 sequences updated to 2024-Jul-17)97 and Virulence Factor Database (VFDB, 2597 sequences updated to 2024-Jul-18)98respectively. Short-read assemblies were finally analysed to determine the ST using a 1095-loci multilocus sequence typing (MLST) scheme38as implemented in Ridom SeqSphere+ (ridoma bionformatics), as above described99. A minimum spanning tree, based on the core-genome MLST profiles, was generated to visualize phylogenetic distance between clinical strains. Pangenome analysis was performed by annotating de novo aligned contigs using Prokka v.1.14.6 100 before analysing the output results using Roary v. 3.13.0 101 (both retrieved in https://usegalaxy.org/). The results were displayed in Phandango102. SNP based analysis was further performed by mapping reads with S. pyogenes MGAS5005 (Accession number CP000017.2) reference genome in CLC Genomic Workbench (Qiagen). Consensus sequences were finally used to obtain a comparison matrix.
Emm-typer was used to detect emm-type and emm-cluster for each strain. Mobilome analysis was carried out as follows: the presence of integrative and conjugative elements (ICEs) and of integrative and mobilizable elements (IMEs) in the assemblies was investigated with ICEfinder (https://bioinfo-mml.sjtu.edu.cn/ICEfinder/ICEfinder.html)103while the presence of prophages was investigated with PHASTET (https://phastest.ca)104. M1UK Clone was finally detected mapping each strain sequence with S. pyogenes MGAS5005 (Accession number CP000017.2) reference genome according to Li et al.36. A neighbour-joining tree was generated and used to show generated to summarize the phylogenetic relationships, emm-types, AMR, virulence genes, and prophages and ICEs/IMEs elements by using ggtree v.3.8.2. Default parameters were used for all software unless otherwise specified.
Sample manipulation, DNA extraction and 16s targeted sequencing
The oropharynx specimens, used for the microbiological diagnosis of GAS, were collected and immediately processed in a strictly controlled level 2 biological safety workplace. Each specimen was vigorously vortexed and 600 µl were used to extract bacterial DNA using the DANAGENE MICROBIOME Salive DNA kit (Danagen-Bioted) according to manufacturer’s indication105,106. The DNA was eluted in 100 µL of pre-heated nuclease-free water and stored at − 20 °C until it was processed. DNA quantification and purity was measured by using a NanoDrop One spectrophotometer (ThermoFisher).
V5–V6 hypervariable regions from the 16S rRNA gene were amplified by using the following primers: V5_Next_For, 5’- TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG[ATTAGATACCCYGGTAGTCC] -3’ and V6_Next_Rev, 5’- GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG[ACGAGCTGACGACA RCCATG] -3’, which were designed to contain (from 5’ to 3’), the sequences for the Nextera transposon and for BV5 (Next For) and AV6 (Next Rev) priming107,108. Extracted DNA (5 ng) was used as the template in a 50-µL PCR volume, which contained 1U Phusion High-Fidelity DNA polymerase (Thermo Fisher), 1X High-Fidelity buffer (ThermoFisher), 200 µM dNTPs, and 0.3-µM each primer. Thermal cycling conditions were set as follows: (i) 98 °C for 30 s; (ii) 20 cycles, each consisting of 98 °C for 10 s, 55 °C for 30 s, and 72 °C for 15 s; (iii) 15 cycles, each consisting of 98 °C for 10 s, 62 °C for 30 s, and 72 °C for 15 s; and (iv) 72 °C for 7 min. Amplicons were purified using Agencourt AMPure XP beads (Beckman Coulter) and were eluted in 35-µL nuclease-free water. Amplicons were then checked for quality on 1% agarose gel electrophoresis (ThermoFisher), and DNA concentration was determined using the above-mentioned method. To incorporate unique Nextera XT i5 and i7 indexes to both amplicon ends, we used 40 ng of purified amplicons as the template in a 50-µL PCR volume, which contained 1U Phusion High-Fidelity DNA polymerase, 1X High-Fidelity buffer, 100-µM dNTPs, and 5-µL each of i5 and i7 indexes. Thermal cycling conditions were set as follows: (i) 98 °C for 30 s; (ii) 5 cycles, each cycle consisting of 98 °C for 10 s, 63 °C for 30 s, and 72 °C for 3 min. Indexed amplicons were purified using Agencourt AMPure XP beads and eluted in 25 µL nuclease-free water, and amplicon quality and concentration was assessed. Each sample’s indexed amplicons were equimolarly diluted, and the final pool was subjected to 2 × 250 paired-end sequencing (Illumina) onto an Illumina MiSeq instrument. To increase the base-diversity degree, an internal control (PhiX v3; Illumina) was added to the DNA library109. All raw sequence data have been deposited in the NCBI Sequence Read Archive (BioProject accession number: PRJNA1241326, https://submit.ncbi.nlm.nih.gov/subs/bioproject/SUB15197939/overview).
Bioinformatic and statistical analysis
Raw sequencing data were processed with Qiime2 (version 2023.5) in a homemade pipeline above described94,109,110. Demultiplexing and quality assessment of the paired-end reads were performed using the “demux” plugin, while trimming of Illumina adapter sequence (5’-CTGTCTCTTATACACATCT-3’) was performed using the “cutadapt trim-paired” plugin. Denoising of paired-end reads was performed using the “dada2 denoised-paired” plugin, which allowed to adjust the number of 5’- and 3’-end trimmed bases to remove primer sequences or low-quality sequences (trim-left-f: 20, trim-left-r: 20, trunc-len-f: 220, trunc-len-r: 200, max-ee-f: 3, max-ee-r: 5, trun-q: 2)111,112. This led to approximately 70% good-merged reads output. Amplicon sequence variants (ASVs) were assembled using the “feature-table summarize” plugin, while we applied the “feature-classifier” plugin to classify ASVs at the taxonomic level by the VSEARCH global consensus alignment and the SILVA 138 full length database at a 99% sequence similarity threshold113. ASVs and taxonomy are reported as Supplementary Data 12 and Supplementary Data 13.
Data analysis was performed using R v.4.3.1 (https://www.rstudio.com/) and the phyloseq software package v.1.46.0 114.
ASVs for which a bacterial taxonomic assignment could not be achieved (i.e., unassigned ASVs) were removed and one sample was discharged because showed less than 1000 reads. Finally, bacterial taxa with relative abundances less than 0.01% if not in more than 5% of the samples were removed. Rarefaction curves indicated that the number of observed taxa in the studied microbiotas remained stable once the sequencing depth reaches 10400 (Supplementary Fig. 2). Taxonomic feature tables normalized by rarefaction to the depth of the lowest number of reads in the samples was used for the diversity analysis. Alpha diversity, assessed as Richness (Observed species), Shannon index, and Pielou’s Evenness were evaluated using the MicrobiotaProcess package v1.12.4 115. The two-sided Mann–Whitney U test was used to test the difference between alpha diversities of two microbiotas. Beta diversity was measured and visualized by Principal coordinate analysis PCoA of Bray-Curtis distances using the MicrobiotaProcess package v.1.12.4 115. The difference in beta diversity between two microbiotas quantified by the Bray-Curtis distance was measured by a PERMANOVA analysis using the adonis2 function in the vegan package v.2.6-8 116. The difference in sample dispersion was quantified by the PERMDISP test using the betadisper and adonis2 functions in the vegan package. Biplot analysis was performed at Genus level centered log ratio transformation and Principal Component Analysis ordination) by using microViz v.11.0 and microbiome v.1.22.0 117 packages. Divergence was measured by using microbiome v.1.22.0 packages117.
Relative abundances were calculated at Phylum and Genus taxonomic level and Mann–Whitney U test was used for statistical significance assessment. Features identifying Streptococcus genera were analyzed by blasting to NCBI database retrieving the best matched Species (see specific Supplementary data for each dataset), while statical significance was assessed as previously described. Differential abundance was evaluated by using DESeq2 package v.1.40.2 and represented as Vulcano plot graph118. Network analysis was performed at Genus level by using NetCoMi package v.1.1.0 with default parameters119. Microbiota core composition was assessed by using microbiome package v.1.22.0 and plotted as lineplot and heatmap120. Correlation analysis among alpha diversity measures, relative abundances of the Streptococcus ASVs, and clinical variables was performed by using Hmisc v.5.2-2 and corrplot v.0.95 packages. A summary of the packages used for the microbiota analysis has been reported in Supplementary data 11.
Digital PCR quantification
Digital PCR (dPCR) experiments were conducted on the Applied Biosystems QuantStudio Absolute Q Digital PCR System (ThermoFisher Scientific, USA). Each dPCR reaction comprised 1X dPCR Master Mix (ThermoFisher Scientific, A52490), 1X custom TaqMan dPCR assays specific for S. pyogenes and the 16 S rRNA gene (used as a control), and 1 µL of bacterial DNA sample, in a total volume of 10 µL. The DNA sample was then appropriately diluted to a concentration of 0.01 ng/ul with the aim of normalizing all the samples to the same amount of DNA prior to amplification in the instrument. The PCR reactions were then loaded onto the MAP16 plates (ThermoFisher Scientific, A52688, followed by the addition of 15 µL of Isolation Buffer (ThermoFisher Scientific, A52730) to the wells containing the PCR mix. All necessary steps for dPCR, including compartmentalization, thermal cycling, and data acquisition, were performed on the QuantStudio Absolute Q system according to the manufacturer’s instructions. The employment of specific probes for S. pyogenes (Ba07921919_s1) allowed the targeted detection of this pathogen, as well as Streptococcus spp. that differed from S. pyogenes (not S. pyogenes; Ba07922188_s1). The use of a 16 S rRNA gene probe (Ba04930791_s1) ensured the integrity and quality of the extracted bacterial DNA.
Data availability
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
References
Walker, M. J. et al. Disease manifestations and pathogenic mechanisms of group A Streptococcus. Clin. Microbiol. Rev. 27, 264–301. https://doi.org/10.1128/CMR.00101-13 (2014).
Maness, D. L., Martin, M. & Mitchell, G. Poststreptococcal illness: recognition and management. Am. Fam Physician. 97, 517–522 (2018).
Hahn, R. G., Knox, L. M. & Forman, T. A. Evaluation of poststreptococcal illness. Am. Fam Physician. 71, 1949–1954 (2005).
Sarno, L. D. et al. A scoping review of the management of acute mastoiditis in children: what is the best approach? Turk. J. Pediatr. 65, 906–918. https://doi.org/10.24953/turkjped.2023.320 (2023).
Vieira, A. et al. Rapid expansion and international spread of M1(UK) in the post-pandemic UK upsurge of Streptococcus pyogenes. Nat. Commun. 15, 3916. https://doi.org/10.1038/s41467-024-47929-7 (2024).
Lassoued, Y. et al. Unexpected increase in invasive group A Streptococcal infections in children after respiratory viruses outbreak in france: A 15-year time-series analysis. Open. Forum Infect. Dis. 10, ofad188. https://doi.org/10.1093/ofid/ofad188 (2023).
Rumke, L. W. et al. Nationwide upsurge in invasive disease in the context of longitudinal surveillance of carriage and invasive Streptococcus pyogenes 2009–2023, the netherlands: a molecular epidemiological study. J. Clin. Microbiol. 62, e0076624. https://doi.org/10.1128/jcm.00766-24 (2024).
Gouveia, C. et al. Sustained increase of paediatric invasive Streptococcus pyogenes infections dominated by M1(UK) and diverse emm12 isolates, portugal, September 2022 to May 2023. Euro. Surveill. 28. https://doi.org/10.2807/1560-7917.ES.2023.28.36.2300427 (2023).
Johannesen, T. B. et al. Increase in invasive group A Streptococcal infections and emergence of novel, rapidly expanding sub-lineage of the virulent Streptococcus pyogenes M1 clone, Denmark, 2023. Euro. Surveill. 28. https://doi.org/10.2807/1560-7917.ES.2023.28.26.2300291 (2023).
Davies, P. J. B. et al. Increase of severe pulmonary infections in adults caused by M1(UK) Streptococcus pyogenes, Central Scotland, UK. Emerg. Infect. Dis. 29, 1638–1642. https://doi.org/10.3201/eid2908.230569 (2023).
Beres, S. B. et al. Increase in invasive Streptococcus pyogenes M1 infections with close evolutionary genetic relationship, Iceland and Scotland, 2022 to 2023. Euro. Surveill. 29 https://doi.org/10.2807/1560-7917.ES.2024.29.13.2400129 (2024).
Ramirez de Arellano, E. et al. Clinical, microbiological, and molecular characterization of pediatric invasive infections by Streptococcus pyogenes in Spain in a context of global outbreak. mSphere 9, e0072923. https://doi.org/10.1128/msphere.00729-23 (2024).
Nygaard, U., Holm, M., Rabie, H. & Rytter, M. The pattern of childhood infections during and after the COVID-19 pandemic. Lancet Child. Adolesc. Health. 8, 910–920. https://doi.org/10.1016/S2352-4642(24)00236-0 (2024).
De Maio, F., Fiori, B., Bianco, D. M., Sanguinetti, M. & Sali, M. Respiratory viruses in the pre and post-pandemic periods in an Italian Tertiary Hospital. Immun. Inflamm. Dis. 11, e909. https://doi.org/10.1002/iid3.909 (2023).
Di Sarno, L. et al. Antibiotic treatment for Streptococcal pharyngitis: time for a new approach? Acta Biomed. 94, e2023219. https://doi.org/10.23750/abm.v94i6.14928 (2023).
Shulman, S. T. et al. Clinical practice guideline for the diagnosis and management of group A Streptococcal pharyngitis: 2012 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 55, 1279–1282. https://doi.org/10.1093/cid/cis847 (2012).
Harberger, S., Goldin, J. & Graber, M. StatPearls (2025).
Coutinho, G., Sessa, D. M., Caretta-Barradas, A. & Altiner, S. Worldwide comparison of treatment guidelines for sore throat. Int. J. Clin. Pract. https://doi.org/10.1111/ijcp.13879 (2021).
Norton, L. & Myers, A. The treatment of Streptococcal tonsillitis/pharyngitis in young children. World J. Otorhinolaryngol. Head Neck Surg. 7, 161–165. https://doi.org/10.1016/j.wjorl.2021.05.005 (2021).
Massese, M. et al. Epidemiology of group A Streptococcal infection: are we ready for a new scenario? Lancet Microbe. https://doi.org/10.1016/S2666-5247(24)00071-5 (2024).
De Maio, F. et al. Reemergence of Streptococcus pyogenes infections in a large Italian Hospital: A Deja Vu from past years. J. Epidemiol. Glob Health. 14, 1778–1781. https://doi.org/10.1007/s44197-024-00316-3 (2024).
Ribet, D. & Cossart, P. How bacterial pathogens colonize their hosts and invade deeper tissues. Microbes Infect. 17, 173–183. https://doi.org/10.1016/j.micinf.2015.01.004 (2015).
Gao, Z., Kang, Y., Yu, J. & Ren, L. Human pharyngeal microbiome may play a protective role in respiratory tract infections. Genomics Proteom. Bioinf. 12, 144–150. https://doi.org/10.1016/j.gpb.2014.06.001 (2014).
Man, W. H., de Steenhuijsen Piters, W. A. & Bogaert, D. The microbiota of the respiratory tract: gatekeeper to respiratory health. Nat. Rev. Microbiol. 15, 259–270. https://doi.org/10.1038/nrmicro.2017.14 (2017).
Humphreys, G. J. & McBain, A. J. Antagonistic effects of Streptococcus and Lactobacillus probiotics in pharyngeal biofilms. Lett. Appl. Microbiol. 68, 303–312. https://doi.org/10.1111/lam.13133 (2019).
Xiong, D. et al. Enriched opportunistic pathogens revealed by metagenomic sequencing hint potential linkages between pharyngeal microbiota and COVID-19. Virol. Sin. 36, 924–933. https://doi.org/10.1007/s12250-021-00391-x (2021).
Hung, T. Y. et al. Antibiotics to eradicate Streptococcus pyogenes pharyngeal carriage in asymptomatic children and adults: A systematic review. J. Infect. 88, 106104. https://doi.org/10.1016/j.jinf.2024.01.003 (2024).
de Piters, S. Nasopharyngeal microbiota, host transcriptome, and disease severity in children with respiratory syncytial virus infection. Am. J. Respir Crit. Care Med. 194, 1104–1115. https://doi.org/10.1164/rccm.201602-0220OC (2016).
Del Chierico, F. et al. Changes in the pharyngeal and nasal microbiota in pediatric patients with adenotonsillar hypertrophy. Microbiol. Spectr. 12, e0072824. https://doi.org/10.1128/spectrum.00728-24 (2024).
Theodosiou, A. A., Dorey, R. B., Read, R. C. & Jones, C. E. The infant pharyngeal microbiomes: origin, impact and manipulation. Curr. Opin. Infect. Dis. 33, 548–555. https://doi.org/10.1097/QCO.0000000000000691 (2020).
Alcolea-Medina, A. et al. The ongoing Streptococcus pyogenes (Group A Streptococcus) outbreak in London, United Kingdom, in December 2022: a molecular epidemiology study. Clin. Microbiol. Infect. 29, 887–890. https://doi.org/10.1016/j.cmi.2023.03.001 (2023).
Turner, C. E. et al. Community outbreaks of group A Streptococcus revealed by genome sequencing. Sci. Rep. 7, 8554. https://doi.org/10.1038/s41598-017-08914-x (2017).
Thacharodi, A. et al. The burden of group A Streptococcus (GAS) infections: the challenge continues in the twenty-first century. iScience 28, 111677. https://doi.org/10.1016/j.isci.2024.111677 (2025).
Golden, A. R. et al. Invasive group A Streptococcus hypervirulent M1(UK) clone, Canada, 2018–2023. Emerg. Infect. Dis. 30, 2409–2413. https://doi.org/10.3201/eid3011.241068 (2024).
Valcarcel Salamanca, B. et al. Increase in invasive group A Streptococcal infections (iGAS) in children and older adults, Norway, 2022 to 2024. Euro. Surveill. 29. https://doi.org/10.2807/1560-7917.ES.2024.29.20.2400242 (2024).
Li, H. K. et al. Characterization of emergent toxigenic M1(UK) Streptococcus pyogenes and associated sublineages. Microb. Genom. 9. https://doi.org/10.1099/mgen.0.000994 (2023).
Lynskey, N. N. et al. Emergence of dominant toxigenic M1T1 Streptococcus pyogenes clone during increased Scarlet fever activity in England: a population-based molecular epidemiological study. Lancet Infect. Dis. 19, 1209–1218. https://doi.org/10.1016/S1473-3099(19)30446-3 (2019).
Toorop, M. M. A. et al. A core-genome multilocus sequence typing scheme for the detection of genetically related Streptococcus pyogenes clusters. J. Clin. Microbiol. 61, e0055823. https://doi.org/10.1128/jcm.00558-23 (2023).
Shelyakin, P. V., Bochkareva, O. O., Karan, A. A. & Gelfand, M. S. Micro-evolution of three Streptococcus species: selection, antigenic variation, and horizontal gene inflow. BMC Evol. Biol. 19, 83. https://doi.org/10.1186/s12862-019-1403-6 (2019).
Quinn-Bohmann, N. et al. Meta-analysis of the human upper respiratory tract Microbiome reveals robust taxonomic associations with health and disease. BMC Biol. 22, 93. https://doi.org/10.1186/s12915-024-01887-0 (2024).
Brouwer, S. et al. Pathogenesis, epidemiology and control of group A Streptococcus infection. Nat. Rev. Microbiol. 21, 431–447. https://doi.org/10.1038/s41579-023-00865-7 (2023).
Woldan-Gradalska, P., Gradalski, W., Gunnarsson, R. K., Sundvall, P. D. & Rystedt, K. Is Streptococcus pyogenes a pathogen or passenger in uncomplicated acute sore throat? A systematic review and meta-analysis. Int. J. Infect. Dis. 145, 107100. https://doi.org/10.1016/j.ijid.2024.107100 (2024).
JJ, F. Streptococcus pyogenes. Basic Biology to Clinical Manifestations (2016).
Curatola, A. et al. Diagnosis and treatment of group A Streptococcal pharyngitis in children. Pediatr. Ann. 53, e234–e238. https://doi.org/10.3928/19382359-20240407-08 (2024).
Ramos Amador, J. T., Sanchez, B., Illan Ramos, M. & A. & Group A Streptococcus invasive infection in children: epidemiologic changes and implications. Rev. Esp. Quimioter. 36 (Suppl 1), 33–36. https://doi.org/10.37201/req/s01.09.2023 (2023).
Mohapatra, R. K., Kutikuppala, L. V. S., Mishra, S., Tuglo, L. S. & Dhama, K. Rising global incidence of invasive group A Streptococcus infection and Scarlet fever in the COVID-19 era - our knowledge thus Far. Int. J. Surg. 109, 639–640. https://doi.org/10.1097/JS9.0000000000000232 (2023).
Demczuk, W., Martin, I., Domingo, F. R., MacDonald, D. & Mulvey, M. R. Identification of Streptococcus pyogenes M1(UK) clone in Canada. Lancet Infect. Dis. 19, 1284–1285. https://doi.org/10.1016/S1473-3099(19)30622-X (2019).
Wolters, M. et al. Population of invasive group A Streptococci isolates from a German tertiary care center is dominated by the hypertoxigenic virulent M1(UK) genotype. Infection 52, 667–671. https://doi.org/10.1007/s15010-023-02137-1 (2024).
Peters, J. A. & Farhadloo, M. The effects of Non-Pharmaceutical interventions on COVID-19 cases, hospitalizations, and mortality: A systematic literature review and Meta-Analysis. AJPM Focus. 2, 100125. https://doi.org/10.1016/j.focus.2023.100125 (2023).
Y., T. The virulence factors and pathogenic mechanisms of Streptococcus pyogenes. J. Oral Biosci. 54, 96–100. https://doi.org/10.1016/j.job.2012.02.004 (2012).
Zhi, X. et al. Emerging invasive group A Streptococcus M1(UK) lineage detected by Allele-Specific PCR, england, 2020(1). Emerg. Infect. Dis. 29, 1007–1010. https://doi.org/10.3201/eid2905.221887 (2023).
Chiang-Ni, C. et al. Incidence and effects of acquisition of the Phage-Encoded Ssa superantigen gene in invasive group A Streptococcus. Front. Microbiol. 12, 685343. https://doi.org/10.3389/fmicb.2021.685343 (2021).
Richter, J. et al. Characterization of a novel CovS SNP identified in Australian group A Streptococcus isolates derived from the M1(UK) lineage. mBio 16, e0336624. https://doi.org/10.1128/mbio.03366-24 (2025).
Imohl, M., Fitzner, C., Perniciaro, S. & van der Linden, M. Epidemiology and distribution of 10 superantigens among invasive Streptococcus pyogenes disease in Germany from 2009 to 2014. PLoS One. 12, e0180757. https://doi.org/10.1371/journal.pone.0180757 (2017).
Mangioni, D. et al. Increase in invasive group A Streptococcal infections in milan, italy: a genomic and clinical characterization. Front. Microbiol. 14, 1287522. https://doi.org/10.3389/fmicb.2023.1287522 (2023).
Bonomo, C. et al. Molecular and clinical characterization of invasive Streptococcus pyogenes isolates: Insights from two Northern-Italy centers. Pathogens 14. https://doi.org/10.3390/pathogens14020152 (2025).
Minko, A. G. et al. Molecular genetic characterization of Streptococcus pyogenes strains isolated from patients with various manifestations of Streptococcal infection. Bull. Exp. Biol. Med. 175, 662–666. https://doi.org/10.1007/s10517-023-05922-y (2023).
Davies, M. R. et al. Atlas of group A Streptococcal vaccine candidates compiled using large-scale comparative genomics. Nat. Genet. 51, 1035–1043. https://doi.org/10.1038/s41588-019-0417-8 (2019).
Carroll, R. K. et al. Naturally occurring single amino acid replacements in a regulatory protein alter Streptococcal gene expression and virulence in mice. J. Clin. Invest. 121, 1956–1968. https://doi.org/10.1172/JCI45169 (2011).
Sumby, P., Whitney, A. R., Graviss, E. A., DeLeo, F. R. & Musser, J. M. Genome-wide analysis of group a Streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog. 2, e5. https://doi.org/10.1371/journal.ppat.0020005 (2006).
Jespersen, M. G., Hayes, A. J., Tong, S. Y. C. & Davies, M. R. Pangenome evaluation of gene essentiality in Streptococcus pyogenes. Microbiol. Spectr. 12, e0324023. https://doi.org/10.1128/spectrum.03240-23 (2024).
Khan, R. M. A., Anwar, S. & Pirzada, Z. A. Streptococcus pyogenes strains associated with invasive and non-invasive infections present possible links with Emm types and superantigens. Iran. J. Basic. Med. Sci. 23, 133–139. https://doi.org/10.22038/IJBMS.2019.38635.9164 (2020).
Smeesters, P. R. et al. Global Streptococcus pyogenes strain diversity, disease associations, and implications for vaccine development: a systematic review. Lancet Microbe. 5, e181–e193. https://doi.org/10.1016/S2666-5247(23)00318-X (2024).
Wu, Q., Jiang, Y., Lv, P. & Chen, M. Emergence of T4SS-type-ICE-carrying emm28 Streptococcus pyogenes causing invasive infection in shanghai, China. J. Glob Antimicrob. Resist. 41, 21–28. https://doi.org/10.1016/j.jgar.2024.12.005 (2025).
Reichardt, W., Muller-Alouf, H. & Kohler, W. Erythrogenic toxin type A (ETA): epidemiological analysis of gene distribution and protein formation in clinical Streptococcus pyogenes strains causing Scarlet fever and the Streptococcal toxic shock-like syndrome (TSLS). Zentralbl Bakteriol. 279, 283–293. https://doi.org/10.1016/s0934-8840(11)80406-8 (1993).
Beres, S. B. et al. Genome sequence of a serotype M3 strain of group A streptococcus: phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc. Natl. Acad. Sci. U S A. 99, 10078–10083. https://doi.org/10.1073/pnas.152298499 (2002).
Bianco, D. M. & De Maio, F. Applying the theory of broken windows to microbiome studies. NPJ Biofilms Microbiomes. 11, 89. https://doi.org/10.1038/s41522-025-00726-6 (2025).
Gogos, A. & Federle, M. J. Modeling Streptococcus pyogenes pharyngeal colonization in the mouse. Front. Cell. Infect. Microbiol. 9, 137. https://doi.org/10.3389/fcimb.2019.00137 (2019).
Calderaro, A. et al. Respiratory tract infections and laboratory diagnostic methods: A review with A focus on syndromic panel-based assays. Microorganisms 10. https://doi.org/10.3390/microorganisms10091856 (2022).
Guerra, S. & LaRock, C. Group A Streptococcus interactions with the host across time and space. Curr. Opin. Microbiol. 77, 102420. https://doi.org/10.1016/j.mib.2023.102420 (2024).
Herrera, A. L., Huber, V. C. & Chaussee, M. S. The association between invasive group A Streptococcal diseases and viral respiratory tract infections. Front. Microbiol. 7, 342. https://doi.org/10.3389/fmicb.2016.00342 (2016).
Fiedler, T. et al. Protective mechanisms of respiratory tract Streptococci against Streptococcus pyogenes biofilm formation and epithelial cell infection. Appl. Environ. Microbiol. 79, 1265–1276. https://doi.org/10.1128/AEM.03350-12 (2013).
Wang, X. et al. The correlation of the intestinal with pharyngeal microbiota in early neonates. Front. Microbiol. 14, 1225352. https://doi.org/10.3389/fmicb.2023.1225352 (2023).
Monemo, P. et al. Pharyngeal carriage of beta-haemolytic Streptococcus species and seroprevalence of anti-streptococcal antibodies in children in Bouake, Cote d’Ivoire. Trop. Med. Infect. Dis. 5. https://doi.org/10.3390/tropicalmed5040177 (2020).
Guglielmetti, S. et al. Oral bacteria as potential probiotics for the pharyngeal mucosa. Appl. Environ. Microbiol. 76, 3948–3958. https://doi.org/10.1128/AEM.00109-10 (2010).
Soderholm, A. T., Barnett, T. C., Sweet, M. J. & Walker, M. J. Group A Streptococcal pharyngitis: immune responses involved in bacterial clearance and GAS-associated immunopathologies. J. Leukoc. Biol. 103, 193–213. https://doi.org/10.1189/jlb.4MR0617-227RR (2018).
Frost, H., Excler, J. L., Sriskandan, S. & Fulurija, A. Correlates of immunity to group A streptococcus: a pathway to vaccine development. NPJ Vaccines. 8, 1. https://doi.org/10.1038/s41541-022-00593-8 (2023).
Cohen, R. et al. Immune debt: recrudescence of disease and confirmation of a contested concept. Infect. Dis. Now. 53, 104638. https://doi.org/10.1016/j.idnow.2022.12.003 (2023).
Ye, Q. & Liu, H. Impact of non-pharmaceutical interventions during the COVID-19 pandemic on common childhood respiratory viruses - An epidemiological study based on hospital data. Microbes Infect. 24, 104911. https://doi.org/10.1016/j.micinf.2021.104911 (2022).
Kennis, M. et al. Seasonal variations and risk factors of Streptococcus pyogenes infection: a multicenter research network study. Ther. Adv. Infect. Dis. 9, 20499361221132101. https://doi.org/10.1177/20499361221132101 (2022).
Shieh, W. J. Human adenovirus infections in pediatric population - An update on clinico-pathologic correlation. Biomed. J. 45, 38–49. https://doi.org/10.1016/j.bj.2021.08.009 (2022).
Mank, V., Azhar, W. & Brown, K. StatPearls (2025).
Rosales, C. & Neutrophil A cell with many roles in inflammation or several cell types?? Front. Physiol. 9, 113. https://doi.org/10.3389/fphys.2018.00113 (2018).
Nakao, A. et al. Amoxicillin effect on bacterial load in group A Streptococcal pharyngitis: comparison of single and multiple daily dosage regimens. BMC Pediatr. 19, 205. https://doi.org/10.1186/s12887-019-1582-8 (2019).
Gualtieri, R. et al. Amoxicillin vs. placebo to reduce symptoms in children with group A Streptococcal pharyngitis: a randomized, multicenter, double-blind, non-inferiority trial. Eur. J. Pediatr. 183, 4773–4782. https://doi.org/10.1007/s00431-024-05705-1 (2024).
B Beres, S. et al. Integrative reverse genetic analysis identifies polymorphisms contributing to decreased antimicrobial agent susceptibility in Streptococcus pyogenes. mBio 13, e0361821. https://doi.org/10.1128/mbio.03618-21 (2022).
Yu, D., Zheng, Y. & Yang, Y. Is there emergence of beta-lactam antibiotic-resistant Streptococcus pyogenes in China? Infect. Drug Resist. 13, 2323–2327. https://doi.org/10.2147/IDR.S261975 (2020).
Luo, R. et al. Diagnosis and management of group a Streptococcal pharyngitis in the United States, 2011–2015. BMC Infect. Dis. 19, 193. https://doi.org/10.1186/s12879-019-3835-4 (2019).
Oliver, J. et al. Group A Streptococcus pharyngitis and pharyngeal carriage: A meta-analysis. PLoS Negl. Trop. Dis. 12, e0006335. https://doi.org/10.1371/journal.pntd.0006335 (2018).
Sauve, L., Forrester, A. M. & Top, K. A. Group A Streptococcal pharyngitis: A practical guide to diagnosis and treatment. Paediatr. Child. Health. 26, 319–320. https://doi.org/10.1093/pch/pxab025 (2021).
Goldstein, B., Giroir, B., Randolph, A. & International Consensus Conference on Pediatric, S. International pediatric sepsis consensus conference: Definitions for sepsis and organ dysfunction in pediatrics. Pediatr. Crit. Care Med. 6, 2–8 (2005). https://doi.org/10.1097/01.PCC.0000149131.72248.E6
di Simona, C. G. & Moro, M.L. Aggiornamento della Linea Guida della Regione Emilia-Romagna 442–447 (Medico e Bambino, 2015).
Kanagasabai, A. et al. Systematic review and meta-analysis of the accuracy of McIsaac and centor score in patients presenting to secondary care with pharyngitis. Clin. Microbiol. Infect. 30, 445–452. https://doi.org/10.1016/j.cmi.2023.12.025 (2024).
De Maio, F. et al. Profiling the gut microbiota to assess infection risk in Klebsiella pneumoniae-colonized patients. Gut Microbes. 17, 2468358. https://doi.org/10.1080/19490976.2025.2468358 (2025).
Posteraro, B. et al. Characterization of metallo beta-lactamase producing enterobacterales isolates with susceptibility to the aztreonam/avibactam combination. Antibiotics (Basel) 13. https://doi.org/10.3390/antibiotics13121221 (2024).
Posteraro, B. et al. In-depth characterization of multidrug-resistant NDM-1 and KPC-3 co-producing Klebsiella pneumoniae bloodstream isolates from Italian hospital patients. Microbiol. Spectr. 12, e0330523. https://doi.org/10.1128/spectrum.03305-23 (2024).
Jia, B. et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. https://doi.org/10.1093/nar/gkw1004 (2017).
Chen, L., Zheng, D., Liu, B., Yang, J. & Jin, Q. VFDB 2016: Hierarchical and refined dataset for big data analysis–10 years on. Nucleic Acids Res. 44 (D694-697). https://doi.org/10.1093/nar/gkv1239 (2016).
Junemann, S. et al. Updating benchtop sequencing performance comparison. Nat. Biotechnol. 31, 294–296. https://doi.org/10.1038/nbt.2522 (2013).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. https://doi.org/10.1093/bioinformatics/btu153 (2014).
Page, A. J. et al. Roary: rapid large-scale prokaryote Pan genome analysis. Bioinformatics 31, 3691–3693. https://doi.org/10.1093/bioinformatics/btv421 (2015).
Hadfield, J. et al. Phandango: an interactive viewer for bacterial population genomics. Bioinformatics 34, 292–293. https://doi.org/10.1093/bioinformatics/btx610 (2018).
Liu, M. et al. ICEberg 2.0: an updated database of bacterial integrative and conjugative elements. Nucleic Acids Res. 47, D660–D665. https://doi.org/10.1093/nar/gky1123 (2019).
Wishart, D. S. et al. PHASTEST: faster than PHASTER, better than PHAST. Nucleic Acids Res. 51, W443–W450. https://doi.org/10.1093/nar/gkad382 (2023).
De Pascale, G. et al. Lung microbiota composition, respiratory mechanics, and outcomes in COVID-19-related ARDS. Microbiol. Spectr. 12, e0357423. https://doi.org/10.1128/spectrum.03574-23 (2024).
De Pascale, G. et al. Staphylococcus aureus ventilator-associated pneumonia in patients with COVID-19: clinical features and potential inference with lung dysbiosis. Crit. Care. 25, 197. https://doi.org/10.1186/s13054-021-03623-4 (2021).
De Maio, F. et al. Profiling the urobiota in a pediatric population with neurogenic bladder secondary to spinal dysraphism. Int. J. Mol. Sci. 24. https://doi.org/10.3390/ijms24098261 (2023).
De Maio, F. et al. Nasopharyngeal microbiota profiling of SARS-CoV-2 infected patients. Biol. Proced. Online. 22, 18. https://doi.org/10.1186/s12575-020-00131-7 (2020).
De Maio, F. et al. Short-term gut microbiota’s shift after laparoscopic Roux-en-Y vs one anastomosis gastric bypass: results of a multicenter randomized control trial. Surg. Endosc. 38, 6643–6656. https://doi.org/10.1007/s00464-024-11154-6 (2024).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. https://doi.org/10.1038/s41587-019-0209-9 (2019).
Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 90. https://doi.org/10.1186/s40168-018-0470-z (2018).
Callahan, B. J. et al. DADA2: High-resolution sample inference from illumina amplicon data. Nat. Methods. 13, 581–583. https://doi.org/10.1038/nmeth.3869 (2016).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–596. https://doi.org/10.1093/nar/gks1219 (2013).
McMurdie, P. J. & Holmes, S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 8, e61217. https://doi.org/10.1371/journal.pone.0061217 (2013).
Xu, S. et al. MicrobiotaProcess: A comprehensive R package for deep mining microbiome. Innov. (Camb). 4, 100388. https://doi.org/10.1016/j.xinn.2023.100388 (2023).
P., D. Vegan, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930 (2003).
Shetty, S. A., Hugenholtz, F., Lahti, L., Smidt, H. & de Vos, W. M. Intestinal Microbiome landscaping: insight in community assemblage and implications for microbial modulation strategies. FEMS Microbiol. Rev. 41, 182–199. https://doi.org/10.1093/femsre/fuw045 (2017).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. https://doi.org/10.1186/s13059-014-0550-8 (2014).
Peschel, S., Muller, C. L., von Mutius, E., Boulesteix, A. L. & Depner, M. NetCoMi: Network construction and comparison for Microbiome data in R. Brief. Bioinform. 22 https://doi.org/10.1093/bib/bbaa290 (2021).
Bach, L. L., Ram, A., Ijaz, U. Z., Evans, T. J. & Lindstrom, J. A longitudinal study of the human oropharynx microbiota over time reveals a common core and significant variations with self-reported disease. Front. Microbiol. 11, 573969. https://doi.org/10.3389/fmicb.2020.573969 (2020).
Acknowledgements
We acknowledge the contribution of the Microbiota Analysis & Microbial WGS Research Core Facility - GSTeP of the Fondazione Policlinico Universitario “A. Gemelli” IRCCS to the sample processing and analysis.
Funding
M.S. acknowledges EU funding within the MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases (Project no. PE00000007, INF-ACT).
Author information
Authors and Affiliations
Contributions
This study was designed by FDM MLS and AG. Sample and collection were performed by FDM, RR, MLS, AG and LDS. The whole genome sequencing and 16s rRNA sequencing were carried out by FDM, RR and GS. Data analysis and figure preparation were performed by FDM and RR. FDM and RR wrote the initial draft. FDM, RR, MLS, AG, GS, LDS, BF, BP, AC and MS revised the final manuscript. All authors participated in the discussion and interpretation of the results. All authors have read and agreed to the published version of the manuscript. All authors had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
De Maio, F., Rosato, R., La Sorda, M. et al. Elucidating the role of group A Streptococcus genomics and pharyngeal microbiota in acute paediatric pharyngitis. Sci Rep 15, 33617 (2025). https://doi.org/10.1038/s41598-025-19099-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-19099-z
