
Abstract
The link of epithelial–mesenchymal transition (EMT) program with metastasis is well documented. However, the role of ROS in EMT process is self-contradictory. DpdtpA was a dithiocarbamate derivative (an iron chelator) that exhibits excellent growth and wound healing inhibitory effects in colorectal cancer (CRC) cell lines, but the underlying mechanism of action was not fully determined. The present data in this study revealed that the ROS derived from autophagic degradation of ferritin were conducive to EMT repression. Genetically knockdown of NCOA4 promoted expression of mesenchymal characteristics, supporting that NCOA4 involved EMT regulation. In addition, DpdtpA treatment also caused depletion of Gpx4 and xCT, triggering ferroptosis as Erastin acted. The causal relationship analysis demonstrated that depletion of Gpx4 and xCT, (or ferroptosis) contributed to the EMT inhibition. Moreover, there seemed to be a feedback loop between Gpx4 and xCT, knockdown of Gpx4 led to upregulation of xCT, but knockdown of xCT brought about downregulation of Gpx4. Further study demonstrated that the depletion of Gpx4 and xCT was due to enhanced autophagy. In addition, PI3K/AKT/mTOR/pathway was showed involving EMT and ferroptosis regulation. In short, our data suggested that the status of EMT and ferroptosis largely was dominated by the continuous NCOA4-mediated ROS production.
Similar content being viewed by others

Introduction
Colorectal cancer (CRC) is a common adverse malignant tumor with high morbidity and mortality in the digestive system and is the leading cause of cancer-related death worldwide1. Surgical treatment and chemical treatment are still the main methods, although considerable progress has been made in the treatment of CRC. However, the side effects and drug resistance of chemotherapy can also lead to treatment failure; clearly, the development of more effective therapies is still urgently needed.
More than 90% of cancer-associated deaths are caused by metastatic disease rather than by the corresponding primary tumor2. The increased motility/invasiveness associated with the mesenchymal cell state has linked the epithelial-mesenchymal transition (EMT) program with metastasis3. The EMT is a biological process in which epithelial cells transform into mesenchymal surface cells, endowing the cells with invasiveness and infiltration4. These changes allow cancer cells to acquire high mobility and migrate from the primary site5,6. It is well documented that the transforming growth factor (TGF), receptor tyrosine kinase (RTK), Wnt, Notch, hedgehog, Hippo, cytokine, and nuclear receptor pathways are involved in EMT regulation7,8. In addition, reactive oxygen species (ROS) participate in the EMT process9, but their effects on the status of EMT are contradictory. Therefore, insight into the cellular and molecular mechanisms of EMT is needed to develop novel diagnostic and therapeutic strategies or inhibitors to prevent and treat metastasis.
NCOA4 mediates ferritin degradation in lysosomes through a process termed ferritinophagy10. The iron ions generated upon undergoing reduction can react with oxygen, producing hydroxyl radicals via the Fenton reaction. It has been demonstrated that some iron chelators act as ferritinophagy inducers; however, ferritinophagy does not necessarily result in the production of ROS, such as DFO11,12. Ferroptosis is an iron-dependent form of cell death characterized by the accumulation of peroxidated polyunsaturated fatty acids (PUFAs) that can be reduced by glutathione peroxidase 4 (Gpx4)-mediated glutathione (GSH)13. It is well documented that Gpx4 and xCT play crucial roles in ferroptosis induction: xCT expression is regulated by growth factor receptors14, CD44 variants15 and Smad316. Additionally, it is transcriptionally activated by nuclear factor erythroid 2-related factor 2 (Nrf2) and activating transcription factor 4 (ATF4) under conditions of oxidative stress or hypoxia17,18. The SLC7A11 catalytic subunit (called xCT) and SLC3A2 constitute system xc-, and the level of xCT is correlated with system xc- activity and tumor invasion19,20,21. Gpx4 is a selenoprotein, and a selenocysteine residue is located in its active site22. This protein acts as an antioxidant enzyme in reducing phospholipid hydroperoxides by using GSH in membranes, thus conferring cytoprotection against ferroptosis23. Cysteine is required for GSH synthesis, while cystine is transported into the cell through the system xc− transporter through the SLC7A11 subunit. Therefore, the cystine is transported into the cell via the systemic xc- transporter through the SLC7A11 subunit. Clearly, the Gpx4 and xc- systems modulate cellular redox homeostasis and determine ferroptosis fate24.
Ferroptosis has been shown to be involved in EMT regulation25; however, the role of ferroptosis in regulating EMT has been controversial. Yao et al. reported the promoting role of ferroptosis in lung adenocarcinoma EMT progression26. In contrast, ferritinophagy-mediated ferroptosis suppressed the EMT in gastric cancer and HepG2 cells in our previous studies27,28. These results indicated that there was a complex relationship between ferroptosis and EMT. Clarifying this relationship could help develop novel strategies for cancer therapies. In this study, we investigated the effect of the 2,2’-dipyridylhydrazone dithiocarbamate s-propionic acid ester (DpdtpA) on the EMT in CRC cells. The upregulation of epithelial features and downregulation of mesenchymal features indicated that DpdtpA could reverse TGF-β1-induced EMT, which involved the production of ROS. Further research also suggested the occurrence of ferroptosis accompanied by EMT inhibition. Correlation analysis showed that Gpx4 and xCT have their own effects on EMT status, and there was a feedback loop between them. Their levels are controlled by autophagic degradation through activation of LC3-II and beclin. In addition, the PI3K/AKT/mTOR complex played a crucial role in both EMT and ferroptosis induction. In summary, NCOA4-mediated ROS production may be a prime mover behind all the above mentioned events.
Materials and methods
General information
The HCT-116 cell line was obtained from HonorGene (Changsha, China). SW480 cells were purchased from Procell (Wuhan, China). Ferrostatin-1 (Fer-1), MTT, chloroquine (CQ), RPMI-1640, and other chemicals were purchased from Sigma‒Aldrich. 2,2’-Dipyridylhydrazone dithiocarbamate s-propionic acid ester (DpdtpA) was prepared by our laboratory29. Gpx4, xCT (SLC7A11), LC-3, beclin, vimentin, snail, and NCOA4 antibodies were obtained from Proteintech Group (Wuhan, China); antibodies against E-cadherin and secondary antibodies (fluorescence labeled for immunofluorescence) were purchased from Cell Signaling Technology (USA). Ferritin antibody for immunofluorescence was obtained from Santa Cruz Biotechnology (USA, Santa Cruz). The NCOA4 antibody for immunofluorescence was purchased from Atlas Antibody (Sweden). Secondary antibodies for western blotting were obtained from EarthOx, LLC (San Francisco, USA).
Cell proliferation and viability assay
The effects of DpdtpA on the proliferation and viability of HCT-116 and SW480 cells were assessed using MTT assay. Cells were seeded in 96-well plates at a density of 5000 cells per well and treated for 48 h. Then, MTT solution (5 mg/ml) was added to each well, and cells were incubated for 4 h. After carefully removing the cell culture, 100 μl DMSO was added to each well to dissolve the formazan crystals. The absorbance was measured at 490 nm. The OD490 value indirectly reflects cell proliferation.
Cell migration and colony formation assay
The HCT-116 cells were seeded into 6-well plates and grown to 90% confluence. A sterile pipette tip was used to scratch the cell monolayer, and the suspended cells were removed by washing with PBS before adding medium containing different concentrations of DpdtpA (1/2 IC50). Wound healing was recorded at 0 h and 48 h using an Olympus microscope (model CKX53) to assess cell migration ability, which can effectively evaluate the impact of DpdtpA on cell migration by measuring the rate of cell movement into the scratched area. The colony formation assay was perfored based on our previous protocol30. Brieftly, the HCT-116 (or SW480) cells in the exponential phase were trypsinized and seeded in 6-well plates at the density of 500 cells/well. DpdtpA at dose of 1/20 or 1/10 IC50 with cell culture was added. Fourteen days later, colonies were fixed in 3.7% paraformaldehyde, stained with 0.1% crystal violet. Colonies containing 50 cells at least were counted under an inverse microscope (Nikon, Tokyo, Japan), and the clone numbers were analyzed subsequently.
ROS detection
A total of 3 × 105 HCT-116 cells were treated with agents (DpdtpA, etc.) for 24 h, followed by washing with PBS and trypsin digestion. Finally, the cells were stained with H2DCF-DA in serum-free culture medium and incubated for 30 min (Active Oxygen Detection Kit, Beyotime Biotechnology). The intracellular ROS concentration was assayed via a flow cytometer (Cytoflex, Beckman Coulter, USA).
Immunofluorescence analysis
The HCT-116 cells were grown in 6-well plates with coverslips overnight. To establish an EMT model, the cells were pretreated by 5 ng/ml TGF-β1 for 12–24 h. Following the treatments with DpdtpA (or NAC(150 μM)), the cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, blocked with 1% BSA, and incubated with either ferritin (H chain, Santa Cruz Biotechnology) or NCOA4 (alternative antibodies) (or vimentin and E-cadherin) primary antibody at 4 °C overnight based on the protocol described previously12. Next, the cells were further incubated with a fluorescence-labeled secondary antibody for 3 h at room temperature and further counterstained with DAPI after the secondary antibody was removed. Finally, the cells were visualized on a confocal laser scanning microscope (Nikon Eclipse Ts2, Japan), and representative cells were selected and photographed.
Knockdown of NCOA4, Gpx4 and xCT (SLC7A11)
NCOA4-specific siRNAs (siNCOA4, siG000008031A-1-5, siG000008031B-1-5), Gpx4 (siG161130095946-1-5, siG1611300953-1-5) and xCT (siG10118141107-5, siG10118141120-1-5) as well as negative control siRNA (siCON, siN0000001-1-5) were purchased from Ribo Biotechnology (Guangzhou, China). The procedure for knocking down NCOA4, Gpx4 and xCT was performed according to the manufacturer’s instructions. Briefly, HCT-116 (SW480) cells (1 × 106) were transfected with 100 pmol of siRNA using Lipofectamine™ Stem Transfection Reagent (Invitrogen, USA) for 12 h.
Western blotting analysis
HCT-116 (SW480) cells were hydrolyzed in lysis buffer on ice for 30 min. The supernatant was collected by centrifugation (14,000 × g) and stored at -80 °C. Following determination of the protein concentrations, 25 μg of protein was loaded on a 13% sodium dodecyl sulfate–polyacrylamide gel (SDS-PAGE), subjected to electrophoresis and transferred onto a PVDF membrane. Next, the investigated protein band was cropped and treated with 5% nonfat skim milk in TBS containing 0.1% Tween-20 for 2 h, the membrane was incubated at 4 °C overnight with the primary monoantibody at a dilution of 1:300 in TBST buffer. Finally, the membrane was incubated with a secondary antibody (1:2,000 in TBST) for 1 h at room temperature. The protein bands were visualized on a SYNGENE G: BOX Chemi XX9 (SYNGENE, UK). Grayscale analysis was performed with ImageJ.
Statistical analysis
The results are presented as the mean ± SEM. Comparisons between multiple groups were performed by one-way ANOVA with Dunnett post hoc correction. A P value < 0.05 was considered to indicate statistical significance.
Results
Antiproliferative and migratory effects of DpdtpA in HCT-116 and SW480 cells
Our previous studies revealed that the 2,2’-dipyridylhydrazone dithiocarbamate s-propionic acid ester (DpdtpA, insert in Fig. 1C) had excellent biological activity12,31. As a continuation of our work, we further determined the effect of DpdtpA on the proliferation of human colorectal carcinoma (CRC) cells. HCT-116 and SW480 cells were treated with various concentrations of the agent, and the cell viability, colony formation and wound healing ratio at the indicated time points were investigated. As shown in Fig. 1A–B, the cells underwent changes in morphology during DpdtpA treatment. Along with the occurrence of rounded cells, cell viability was markedly reduced in these two cell lines. The concentration-dependent growth inhibition data are presented in Fig. 1C. The IC50 values were 0.75 ± 0.22 μM for HCT-116 cells and 2.5 ± 0.34 μM for SW480 cells. To assess whether ROS are involved in growth inhibition, the ROS scavenger, N-acetyl-l-cysteine (NAC) was introduced into the assay. Clearly, NAC could partly affect the inhibitory effect of DpdtpA (Fig. 1D), indicating that ROS played a certain role in growth inhibition. Similarly, colony formation inhibition was observed in these two cell lines. Figure 1E showed that DpdtpA treatment resulted in smaller size in colonies in HCT-116 cell compared to that in control. Simialr scene was observed in SW480 cells (Fig. 1F). Those clearly indicated that DpdtpA had good proliferative inhibition in CRC cell lines. In addition, we wondered whether these changes might affect the migratory ability of HCT-116 and SW480 cells due to morphological changes. Thus, a wound healing assay was further carried out. Figure 1G–H showed that the decrease in wound closure was caused by DpdtpA administration. Quantitative analysis revealed that repair capacity of HCT-116 cell was significantly decreased (from 46.55 ± 2.57% to 19.35 ± 1.42%) after DpdtpA treatment. Similar attenuation was observed in SW480 cell (from 45.45 ± 2.83% to 20.63 ± 1.04%). Those strongly suggested that DpdtpA suppressed the proliferation and migration of CRC cells.

DpdtpA significantly inhibited both the proliferation and migration of CRC cells. (A) the effect of DpdtpA on the morphology of HCT-116 cells; (B) the effect of DpdtpA on the morphology of SW480 cells; (C) the growth inhibition of DpdtpA in HCT-116 and SW480 cell lines; (D) DpdtpA induced growth inhibition in CRC cells involving ROS production; (E) the effect of DpdtpA on colony formation of HCT-116 cells, the concentration as indicated in the figure; (F) the effect of DpdtpA on colony formation of SW480 cells; (G) the effect of DpdtpA on wound healing in HCT-116 cells; (H) the effect of DpdtpA on wound healing in SW480 cells. @@@, ### p < 0.01 vs control; one-way ANOVA with Dunnett post hoc correction.
ROS played a role in the EMT inhibition induced by DpdtpA in CRC cells
During epithelial wound healing, viable epithelial cells at the edge of the wound site undergo partial EMT to promote motility and movement as a coordinated group of cells to help reconstruct the epithelial cell sheet3. EMT is defined as a cellular process during which epithelial cells acquire mesenchymal phenotypes and behavior following the downregulation of epithelial features. Given that DpdtpA treatment leads to significant changes in morphology and impaired wound healing in CRC cells, it might exert its effect on EMT, as indicated by many studies revealing that EMT plays a critical role in cancer invasion and migration11. Thus, HCT-116 and SW480 cells were treated with DpdtpA at a 1/2 the half-maximal inhibitory concentration (IC50) for 24 h. Figure 2A–B showed that E-cadherin was markedly upregulated, while vimentin and snail were downregulated after DpdtpA exposure in HCT-116 cells, indicating that DpdtpA was able to inhibit EMT. Similarly, this repression of EMT was also observed in SW480 cells (Fig. 2C–D). In addition, immunofluorescence confocal microscopic analysis revealed that the trends in the expression of the EMT-related proteins E-cadherin and vimentin were similar to those observed via western blot analysis (Fig. 2E–F). These results demonstrated that DpdtpA inhibited the EMT process in CRC cells. In addition, since ROS are involved in growth inhibition, they may participate in EMT suppression32,33. To this end, the effect of NAC on the activity of DpdtpA was further investigated. As expected, DpdtpA-induced EMT inhibition was neutralized by NAC (Fig. 2G). To corroborate the effect of DpdtpA on EMT, an EMT model is needed. TGF-β1, a putative inducer of EMT was widely used by scholars34. Thus, the HCT-116 cells were pretreated with TGF-β1. Usually, when stimulated with TGF-β1, the cells undergo morphologic and phenotypic EMT-like changes, including cytoskeleton reorganization, mesenchymal marker up-regulation, and cadherin switching35,36. As expected, TGF-β1 treatment resulted in a significant increase in vimentin expression (Fig. 2H), while following treatment with DpdtpA (Fig. 2I), the regulatory effect of TGF-β1 on EMT-related proteins was significantly attenuated, confirming the above conclusion that DpdtpA was capable of repressing EMT.

The repressive effect of DpdtpA on EMT in CRC cells. (A) the changes in EMT-related proteins in HCT-116 cells; (B) quantitative analysis of EMT related proteins derived from (A), the results from two experiments; (C) the changes in EMT related proteins in SW480 cells; (D) quantitative analysis of EMT-related proteins derived from (C), the results from two experiments. (E–I) immunofluorescence confocal microscopic analysis of HCT-116 cells: (E) DMSO, (F) DpdtpA, (G) DpdtpA + NAC, (H) TGF-β1, and (I) DpdtpA + TGF-β1. @@@, ###, $$$ p < 0.01 vs control; one-way ANOVA with Dunnett post hoc correction.
Redox-active ferritinophagy had role in the ROS production
Our previous study revealed that DpdtpA was able to induce ferritinophagy in CT26 cells12, but this phenomenon might occur similarly in human colorectal cancer cells. As we know, ferritinophagy is a process that NCOA4 mediated ferritin degradation in lysosomes. To this end, the levels of NCOA4 and ferritin were investigated. As shown in Fig. 3A–B: DpdtpA treatment resulted in upregulation of NCOA4 and downregulation of ferritin in HCT-116 cell line, indicating that ferritinophagy indeed occurred. Similar changes in NCOA4 and ferritin were observed in SW480 cells (Fig. 3E–F). To determine whether ferritinophagy was responsible for the ROS production, the ROS scavenger NAC was used to assess the nature of ferritinophagy. Figure 3C–D showed that the addition of NAC significantly neutralized the regulatory effect of DpdtpA on ferritin and NCOA4, but enhanced autophagy-related proteins expression, suggesting that ferritinophagy was redox active and accompanied by autophagy (Fig. 3D). Similarly results were observed in SW480 cells (Fig. 3G–H), the levels of ferritin, NCOA4, LC3-II and beclin were significantly upregulated compare to that in DpdtpA treatment. In addition, immunofluorescence confocal microscopic analysis of HCT-116 cells further confirmed that ferritinophagy was redox active because NAC addition significantly decreased the NCOA4 expression (Fig. 3I–K). Next, analysis of ROS in different conditions via flow cytometry was conducted. DpdtpA treatment resulted in 30.76% increase in ROS abundance compared to that of control, addition of NAC significantly weaken the effect of the agent on ROS production (Fig. 3L), further supporting that the induced ferritinophagy was redox active. In addition, since autophagy occurs, does it have any impact on ROS? Thus, an autophagy inhibitor, 3-MA was introduced to the assay. Clearly autophagy inhibition indeed decreased the ROS production. Finally, ferritinophagy is a cellular event related to iron, the addition of iron chelator may also influene the ROS production. DFO, a widely used iron chelator, significantly attenuated the action of DpdtpA in ROS production (Fig. 3L), which was related to the decrease in free iron ion due to iron chelate formation. Those indicated that redox-active ferritinophagy was responsible for the ROS production.

DpdtpA treatment resulted in the occurrence of both ferritinophagy and autophagy. (A) Western blotting analysis of ferritinophagy-related proteins in HCT-116 cells; (B) quantitative analysis derived from (A); (C) a redox activity of ferritinophagy and autophagy induced by the agent in HCT-116 cells; (D) quantitative analysis derived from (C); (E) western blotting analysis of ferritinophagy-related proteins in SW480 cells; (F) quantitative analysis derived from (E); (G) a redox activity of ferritinophagy and autophagy induced by the agent in SW480 cells; (H) quantitative analysis derived from (G); (I–K) immunofluorescence confocal microscopic analysis of ferritinophagy-related proteins in HCT-116 cells: (I) DMSO; (J) DpdtpA; (K) DpdtpA + NAC. (L) ROS production was partly from ferritinophagy in HCT-116 cells.
NCOA4 perpetrator was responsible for ferroptosis induction and EMT inhibition
Ferroptosis is an iron-dependent accumulation of reactive oxygen species (ROS) that lead to lipid peroxidation and cell death37. Generally, ferritinophagy leads to iron accumulation and, accordingly, triggers ferroptosis. Gpx4 depletion is a significant hallmark and indicator of ferroptosis38,39. As shown in Fig. 4A, along with ferritin downregulation, depletion of Gpx4 and xCT was observed in HCT-116 cells following DpdtpA exposure. The crucial changes in the expression of these proteins are presented in Fig. 4B. These results suggested that DpdtpA could induce ferroptosis. To validate this conclusion, the standard ferroptosis inducer, Erastin and the inhibitor ferrostatin-1 (Fer-1) were used in the present study. Figure 4C showed that DpdtpA treatment resulted in the depletion of ferritin, similar to the effect of Erastin, but this effect could be stopped by the addition of Fer-1, confirming that the mechanism of action of the agent involved ferroptosis induction. The quantitative analysis of those proteins was presented in Fig. 4D. Same protocol was applied to measure the changes of those protins in SW480 cell, similar results were obtained as that in HCT-116 cells (Fig. 4E–H). These results indicated that DpdtpA could induce ferroptosis in CRC cells. Next, the role of NCOA4 in ferroptosis induction was further investigated. As shown in Fig. 4I, in contrast to that of DpdtpA, genetic knockdown of NCOA4 resulted in an increase in Gpx4 and xCT, suggesting that there was a causal relationship between them in HCT-116 cells. It was anticipated that knockdown of NCOA4 would attenuate the regulatory effect of DpdtpA on Gpx4 and xCT. The significant alterations in ferritinophagy and ferroptosis-related proteins are presented in Fig. 4J. Similar changes were also observed in SW480 cells (Fig. 4K–L). In addition, since DpdtpA inhibits EMT and induces ferroptosis, the causal relationship between the two kind proteins requires to be further investigated. As a precautionary measure, Erastin and DpdtpA were used in parallel in the experimental design. As shown in Fig. 4M, DpdtpA acted as an Erastin, leading to EMT inhibition accompanied by depletion of Gpx4 and xCT, suggesting that ferroptosis was conducive to EMT inhibition. The results of the quantitative analysis of the indicated proteins are presented in Fig. 4N. Thus far, we inferred that NCOA4 was a perpetrator and responsible for all the cellular events mentioned above. Thus, the regulatory effects of NCOA4 on EMT and ferroptosis-related proteins were further assayed. As shown in Fig. 4O–P, downregulation of NCOA4 clearly counteracted the regulatory effect of DpdtpA on EMT and ferroptosis related proteins. Similar results were found in SW480 cells (Fig. 4Q–T). These findings suggested that NCOA4 was perpetrator and responsible for ferroptosis induction and EMT inhibition.

Ferritinophagy-mediated ferroptosis contributed to the EMT inhibition in CRC cells. (A–D) HCT-116 cells: (A) ferritinophagy along with downregulation of Gpx4 and xCT; (B) quantitative analysis derived from (A); (C) DpdtbA-induced ferroptosis; (D) quantitative analysis derived from (C). (E–H) SW480 cells: (E) ferritinophagy along with downregulation of Gpx4 and xCT; (F) quantitative analysis derived from (E); (G) DpdtpA-induced ferroptosis; (H) quantitative analysis derived from (G). (I) NCOA4 mediated ferroptosis induction in HCT-116 cells; (J) quantitative analysis derived from (I); (K) NCOA4 mediated ferroptosis induction in SW480 cells; (L) quantitative analysis derived from (K); (M–P): HCT-116 cells: (M) DpdtpA induced EMT inhibition as like Erastin; (N) quantitative analysis derived from (M); (O) NCOA4-mediated ferroptosis contributed to EMT inhibition; (P) quantitative analysis derived from (O). (Q–T): SW480 cells: (Q) DpdtpA induced EMT inhibition as like Erastin; (R) quantitative analysis derive from (Q); (S) NCOA4-mediated ferroptosis contributed to EMT inhibition; (T) quantitative analysis derived from (S). The results of the quantitative analysis were from two experiments. **, ##,$$,%%,&&,^^ p < 0.05 vs control; ***, ###,$$$,%%,&&&,^^^ p < 0.01 vs control; one-way ANOVA with Dunnett post hoc correction.
Gpx4 and xCT regulated the EMT
Since DpdtpA acts as an Erastin in ferroptosis induction, leading to the downregulation of both Gpx4 and xCT, the intrinsic role of these proteins in EMT must be determined. To this end, each of these genes was genetically knocked down via an interfering RNA technique. As shown in Fig. 5A, Gpx4 knockdown in HCT-116 cells led to an increase in epithelial characteristics (upregulated E-cadherin) and a decrease in mesenchymal characteristics (downregulated vimentin), which could be weakened by addition of Fer-1, suggesting that Gpx4 was involved in the EMT regulation, which was consistent with previous reports40. However, unexpectedly, Gpx4 knockdown led to the upregulation of xCT, which may be a stress response to Gpx4 depletion. The results of the quantitative analysis of the related proteins are presented in Fig. 5B. Similar results were obtained in SW480 cells (Fig. 5C–D). Next, the role of xCT in regulating EMT was further investigated in HCT-116 cells. As shown in Fig. 5E, similar to the downregulation of Gpx4, knocking down xCT also resulted in the downregulation of vimentin and snail; however, Gpx4 was also downregulated, which was consistent with that reported recently41. The results of the quantitative analysis of those proteins are shown in Fig. 5F. Similar changes in the expression of these genes were observed in SW480 cells (Fig. 5G–H). These findings suggested that there might be a feedback loop between Gpx4 and xCT.

Gpx4 and xCT played roles in regulating EMT. (A) the effect of Gpx4 on EMT-related genes and xCT in HCT-116 cell; (B) quantitative analysis derived from (A), the results were from three experiments; (C) the effect of Gpx4 on EMT-related genes and xCT in SW480 cell; (D) quantitative analysis derived from (C), the results were from three experiments; (E) the effect of xCT on EMT-related genes and Gpx4 in HCT-116 cell; (F) quantitative analysis derived from (E), the results were from three experiments; (G) the effect of xCT on EMT-related genes and Gpx4 in SW480 cell; (H) quantitative analysis derived from (G), the results were from two experiments. @@,$$ p < 0.01 vs control; @@@, ###,$$$,%%%, ^^^ p < 0.01 vs control.
Lysosome and proteasome degradation played a role in the regulation of Gpx4 and xCT
Downregulated xCT/Gpx4 signaling contributes to ferroptosis induction. DpdtpA treatment resulted in ferritinophagy-mediated ferroptosis. The enhanced ROS production certainly triggered the occurrance of autophagy (Fig. 3C and G), thus, we speculated that the autophagy might have some role in controling the levels of Gpx4 and xCT. Whereupon, the effect of posttranslational modifications on ferroptosis related genes were investigated. It was reported that Gpx4 is regulated by autophagy42, and both Gpx4 and xCT are regulated by beclin43. In addition, protein synthesis controls many targets of genes in biosystems. The effect of the agent on protein synthesis was first determined. As shown in Fig. 6A, the protein synthesis inhibitor CHX significantly inhibited the expression of Gpx4 and xCT in HCT-116 cells; however, cotreatment with CHX and DpdtpA did not obviously decrease the expression of these genes, and the changes in Gpx4 and xCT expression under treatment with either CHX or both were almost parallel. The results of the quantitative analysis of those proteins are presented in Fig. 6B. Similar results were observed in SW480 cells (Fig. 6C–D). These results indicated that DpdtpA had no significant effect on protein synthesis. Next, the autophagy inhibitor chloroquine (CQ) was used to determine whether autophagy was involved in this regulatory effect. Figure 6E–F showed that CQ markedly attenuated the regulatory effect of DpdtpA on Gpx4 and xCT in HCT-116 cells, suggesting that their depletion was partly achieved by enhanced autophagy (activation of LC3-II and beclin, Fig. 3B and D). Similar results were obtained in SW480 cells. These findings suggested that autophagy played an important role in the regulation of Gpx4 and xCT. In addition, further research was conducted to investigate whether proteasome degradation was involved in the regulation of Gpx4 and xCT. Figure 6G–H showed that the proteasome inhibitor MG-132 had a certain regulatory effect on Gpx4, but had a lesser effect on xCT; however, it indeed attenuated the regulatory effect of DpdtpA on xCT. The regulatory effect of CQ and MG132 on DpdtpA were similar in SW480 cells (Fig. 6I–L).

The regulatory effect of DpdtpA on the expression of Gpx4 and xCT. (A) the regulatory effect of protein synthesis on Gpx4 and xCT in HCT-116 cells; (B) quantitative analysis derived from (A), the results were from two experiments; (C) the regulatory effect of protein synthesis on Gpx4 and xCT in SW480 cells; (D) quantitative analysis derived from (C), the results were from two experiments; (E) the effect of autophagy on the expression of Gpx4 and xCT in HCT-116 cells; (F) quantitative analysis derived from (E), the results were from three experiments; (G) the effect of the proteasome on the expression of Gpx4 and xCT in HCT-116 cells; (H) quantitative analysis derived from (G), the results were from three experiments; (I) the effect of autophagy on the expression of Gpx4 and xCT in SW480 cells; (J) quantitative analysis derived from (I), the results were from three experiments; (K) the effect of the proteasome on the expression of Gpx4 and xCT in SW480 cells; (L) quantitative analysis derived from (K), the results were from three experiments. @@,## p < 0.05 vs control; @@@, ### p < 0.01 vs control.
The PI3K/AKT/mTOR pathway was involved in the regulation of ferroptosis and EMT
It is well documented that the PI3K/AKT/mTOR pathway plays an important role in cell survival and proliferation44, and in tumor cells, the level of this pathway is greater than that in normal cells45,46. Considering that DpdtpA had an antiproliferative effect on CRC cells, we inferred that the PI3K/AKT/mTOR pathway may play a role in this antiproliferative effect. Thus, the expression levels of p-PI3K, AKT, phospho-AKT (a measure of AKT activation), and mTOR were determined via western blotting. As shown in Fig. 7A, DpdtpA treatment decreased the phosphorylation of p-PI3K, AKT and mTor, indicating that the mechanism of action of DpdtpA involved the PI3K/AKT/mTOR pathway. The quantitative analysis of the genes in this pathway was presented in Fig. 7B. The effect of the PI3K/AKT/mTOR pathway on the regulation of EMT and ferroptosis was further investigated. To this end, an agonist of this pathway, sc79, was used in the following assay. As shown in Fig. 7C, compared to DpdtpA, sc79 had the opposite effect on this pathway, as did both EMT and ferroptosis. The inhibitory effect of sc79 on proteins affected by DpdtpA was expected. Figure 7D showed the results of the quantitative analysis. Similarly, the same procedure was used for SW480 cells (Fig. 7E–H). These results clearly indicated that the PI3K/AKT/mTOR pathway played an important role in regulating EMT and ferroptosis.

The PI3K/AKT/mTOR pathway involved in the regulation of ferroptosis and EMT. (A) DpdtpA treatment resulted in inactivation of PI3K/AKT/mTOR in HCT-116 cell; (B) quantitative analysis derived from (A), the results were from three experiments; (C) the effect of PI3K/AKT/mTOR pathway on regulation of EMT and ferroptosis in HCT-116 cell; (D) quantitative analysis derived from (C), the results were from two experiments; (E) DpdtpA treatment resulted in inactivation of PI3K/AKT/mTOR in SW480 cell; (F) quantitative analysis derived from (E), the results were from three experiments; (G) the effect of PI3K/AKT/mTOR pathway on EMT-related and ferroptosis related genes in SW480 cell; (H) quantitative analysis derived from (G), the results were from two experiments. @@,##,$$,%%,^^,** p < 0.05 vs control, @@@, ###,$$$,%%%,^^^,&&&,***,!!! p < 0.01 vs control.
Discussion
Colorectal cancer (CRC) is the leading cause of cancer-related death worldwide1. The prognosis of metastatic CRC is poor, with a 5-year survival rate of less than 20%47. Clearly, a new treatment is needed. Cancer cells have two significant characteristics: uncontrolled growth and infiltration. Obviously, a new agent with such inhibitory abilities is expected. DpdtpA is a dithiocarbamate derivative that was shown to have excellent biological activity in our previous study12. CRC cell proliferation and colony formation were inhibited (Fig. 1). In addition, DpdtpA also impaired wound healing. These findings prompted us to explore the underlying mechanism involved. It is well documented that dormant tumor cells of epithelial origin first acquire invasive potential to travel from the main tumor and enter the bloodstream48. During the invasive process, tumor cells must undergo epithelial-mesenchymal transition (EMT). Once tumor cells enter blood vessels, they will circulate. A portion of the circulating tumor cells at this stage may leak out of the blood vessels and begin to move to distant sites where they colonize49. To adapt to their new environment, tumor cells once again undergo mesenchymal-epithelial transition (MET). Clearly, reducing cancer-related mortality may be achieved by preventing cancer metastasis, while EMT inhibition in the early phases may be a good option50. Vimentin and E-cadherin are important markers of EMT. In cancer cells, E-cadherin is suppressed through the binding of transcriptional repressors (snail, slug, and zeb1) to the E-cadherin promoter51,52. Western and immunofluorescence analyses revealed that DpdtpA treatment caused significant downregulation of vimentin and upregulation of E-cadherin (Fig. 2), suggesting that the agent was able to inhibit EMT5,34. In addition, compelling evidence reveals reactive oxygen species (ROS) as crucial conspirators in EMT53, such as in TGF-β1-induced EMT6; However, it is unclear whether EMT is inhibited by ROS. The addition of NAC significantly attenuated the regulatory effect of DpdtpA on EMT-related proteins, suggesting that EMT inhibition was also achieved through ROS production (Fig. 2). This further strengthens the viewpoint that ROS play dual roles in regulating EMT, i.e., moderate ROS promote EMT, massive ROS reverse EMT54. In addition, the autophagy inhibitor 3-MA and DFO (iron chelator) both attenuated the regulatory effect of the agent on EMT-related proteins, indicating that EMT suppression might involve autophagy and iron ions. Thus, the source of ROS was further investigated. Mitochondria are crucial ROS sources due to the presence of the respiratory chain, but lysosomes and proteasomes may also be auxiliary sources. Since iron ions are involved, ferritin degradation in lysosomes or proteasomes might occur. The activation of autophagy-related proteins suggested that ferritinophagy occurred, and the downregulated ferritin and upregulated NCOA4 expression observed via western blotting and immunofluorescence assays supported the above conclusion (Fig. 3). This finding was consistent with previous findings6,12,27,28,55.
The regulatory effect of ferritinophagy on EMT is pluralistic. Generally, NCOA4-mediated ferritinophagy is correlated with ROS production; however, this process is highly dependent. Usually, ROS generated by downregulation of ferritin or upregulation of NCOA4 have redox activity, but are not iron chelators, such as DFO. Although DpdtpA acted as an iron chelator, its chelate was redox active (the iron chelate of DFO was redox inactive). In addition, a study showed that TGF-β1-induced EMT involved ROS production56; however, the ROS insult was periodic, while DpdtpA-induced EMT inhibition in CRC cells may be related to sustained insults from ROS45, strengthening the concept that NCOA4-mediated ROS production was a dominant driving force in the determination of EMT status.
Ferroptosis is an iron-dependent form of cell death57. Accumulating studies have demonstrated that the induction of ferroptosis in CRC successfully eliminates cancer cell resistance to other modes of cell death. Therefore, ferroptosis may be considered a new direction for the design of CRC therapy1. Ferroptosis may be caused by feritinophagy; however, ferritinophagy is not necessary for ferroptosis, and ROS may be a decisive factor. The depletion of Gpx4 and xCT triggers ferroptosis13,17, and DpdtpA downregulated Gpx4 and xCT like Erastin, indicating its ability to induce ferroptosis (Fig. 4). The genetic knockdown of NCOA4 protected cells from ferroptosis, suggesting that NCOA4-mediated ferritinophagy plays a crucial role in ferroptosis induction and EMT inhibition. Yao et al. reported a positive correlation between EMT and ferroptosis via single-cell RNA sequencing, and Gpx4 was found to strongly regulate EMT26. In addition, SLC7A11 inhibition was found to be a common cause of EMT and ferroptosis in A549 cells56. These results indicated that Gpx4 or xCT (SLC7A11) are involved in EMT regulation. In our study, genetic knockdown of either Gpx4 or xCT led to EMT inhibition in CRC cells (Fig. 5), which was consistent with previous report34. The contradictory conclusions about the role of ferroptosis in EMT may be related to the cell lines used. Importantly, we found that there was a feedback loop between Gpx4 and xCT: genetic depletion of Gpx4 led to redox stress, and in response to this stress, upregulation of xCT was necessary. As we know, GPX4 reduces intracellular lipid peroxides in the presence of GSH. Knockdown of GPX4 will increase oxidative stress (or ROS enhancement), and decrease GSH. This triggers biological system synthesizes GSH. To achieve this goal, it needs to provide cystine through xCT, triggering upregulation of xCT. Therefore, the upregulation of xCT was to cope with oxidative stress. However, when knockdown of xCT, similarly it also triggers ROS production, dowregulation of GSH, but it has little effect on the expression of Gpx4. Next, the mechanism of Gpx4 and xCT depletion was further investigated. Our data showed that autophagy-related proteins, such as beclin and LC3-II, influenced the levels of both Gpx4 and xCT (Fig. 6), which was consistent with previous findings45. Proteasome degradation of Gpx4 was also observed in our study (Fig. 6)58,59. In addition, the PI3K/AKT/mTOR pathway was found to play a role in proliferation, EMT and ferroptosis in CRC cells (Fig. 7). Similar results were found by others4,40.
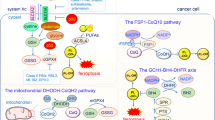
Taken together, these findings revealed that DpdtpA significantly regulated colony formation, proliferation, EMT and ferroptosis in CRC cells. Causality analysis revealed that ferritinophagy-mediated ferroptosis played a crucial role in EMT inhibition, while NCOA4-mediated continuous ROS production initiated these events and inactivates the PI3K/AKT/mTOR pathway (Fig. 8).

NCOA4-mediated ROS production contributed to the autophagic degradation of Gpx4 and xCT, EMT inhibition and inactivation of PI3K/AKT/mTor in the mechanism of action of DpdtpA in CRC cells.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper.
Abbreviations
- ROS:
-
Reactive oxygen species
- NCOA4:
-
Nuclear receptor coactivator 4
- DpdtpA:
-
2,2’-Dipyridylhydrazone dithiocarbamate s-propionic acid ester
- NAC:
-
N-acetyl-L-cysteine
- CQ:
-
Chloroquine
- CHX:
-
Cycloheximide
- Fer-1:
-
Ferrostatin-1
- ECL:
-
Electrochemiluminescence
- SEM:
-
Standard error of the mean
- ANOVA:
-
Analysis of variance
- siRNA:
-
Small interfering RNA
- PI3K:
-
Phosphoinositide-3 kinase
- AKT:
-
Protein kinase B
- mTOR:
-
Mammalian target of rapamycin
- MTT:
-
3-(45)-Dimethylthiahiazo (-z-y1)-35-dicytetrazoliumromide
References
Wang, Y. et al. Ferroptosis in colorectal cancer: Potential mechanisms and effective therapeutic targets. Biomed. Pharmacother. 153, 113524 (2022).
Lambert, A. W., Pattabiraman, D. R. & Weinberg, R. A. Emerging biological principles of metastasis. Cell 168, 670–691 (2017).
Zhang, Y. & Weinberg, R. A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 12, 361–373 (2018).
Yuan, R. et al. Cucurbitacin B inhibits TGF-β1-induced epithelial–mesenchymal transition (EMT) in NSCLC through regulating ROS and PI3K/Akt/mTOR pathways. Chin. Med. 17, 24 (2022).
Serrano-Gomez, S. J., Maziveyi, M. & Alahari, S. K. Regulation of epithelialmes-enchymal transition through epigenetic and posttranslational modifcations. Mol. Cancer 15, 18 (2016).
Xu, J., Lamouille, S. & Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 19, 156–172 (2009).
Moustakas, A. & Heldin, C. H. Signaling networks guiding epithelial–mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 98, 1512–1520 (2007).
Derynck, R., Muthusamy, B. P. & Saeteurn, K. Y. Signaling pathway cooperation in TGF-induced epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 31, 56–66 (2014).
Wang, Z., Li, Y. & Sarkar, F. H. Signaling mechanism(s) of reactive oxygen species in epithelial-mesenchymal Transition reminiscent of cancer stem cells in tumor progression. Curr. Stem Cell Res. Ther. 5, 74–80 (2010).
Mancias, J. D., Wang, X., Gygi, S. P., Harper, J. W. & Kimmelman, A. C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109 (2014).
Huang, T. F. et al. Growth inhibition of a novel iron chelator, DpdtC, against hepatoma carcinoma cell lines partly attributed to ferritinophagy-mediated lysosomal ROS generation. Oxid. Med. Cell Longev. 2018, 4928703 (2018).
Sun, Y. J. et al. Ferritinophagic flux activation in CT26 cells contributed to EMT inhibition induced by a novel iron chelator. DpdtpA. Oxid. Med. Cell Longev. 2019, 8753413 (2019).
Hirschhorn, T. & Stockwell, B. R. The development of the concept of ferroptosis. Free Radic. Biol. Med. 133, 130–143 (2019).
Tsuchihashi, K. et al. The EGF receptor promotes the malignant potential of glioma by regulating amino acid transport system xc(-). Cancer Res. 76, 2954–2963 (2016).
Ishimoto, T. et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 19, 387–400 (2011).
Kim, D. H., Kim, W. D., Kim, S. K., Moon, D. H. & Lee, S. J. TGF-β1-mediated repression of SLC7A11 drives vulnerability to GPX4 inhibition in hepatocellular carcinoma cells. Cell Death Dis. 11, 406 (2020).
Sasaki, H. et al. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 277, 44765–44771 (2002).
Sato, H. et al. Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. Biochem. Biophys. Res. Commun. 325, 109–116 (2004).
Guo, W. et al. Disruption of xCT inhibits cell growth via the ROS/autophagy pathway in hepatocellular carcinoma. Cancer Lett. 312, 55–61 (2011).
Sugano, K. et al. Expression of xCT as a predictor of disease recurrence in patients with colorectal cancer. Anticancer Res. 35, 677–682 (2015).
Takeuchi, S. et al. Increased xCT expression correlates with tumor invasion and outcome in patients with glioblastomas. Neurosurgery 72, 33–41 (2013).
Carlisle, A. E. et al. Selenium detoxification is required for cancer-cell survival. Nat. Metab. 2, 603–611 (2020).
Yang, W. S. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014).
Zhang, Z. et al. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 25, 101047 (2019).
Ling, H., Xiao, H., Luo, T., Lin, H. & Deng, J. Role of ferroptosis in regulating the epithelial–mesenchymal transition in pulmonary fibrosis. Biomedicines 11, 163 (2023).
Yao, J. et al. Single-cell RNA-seq reveals the promoting role of ferroptosis tendency during lung adenocarcinoma EMT progression. Front. Cell Dev. Biol. 9, 822315 (2022).
Li, H. et al. Ferritinophagic flux was a driving force in determination of status of EMT, ferroptosis, and NDRG1 activation in action of mechanism of 2-pyridylhydrazone dithiocarbamate s acetic acid. J. Oncol. 2021, 3015710 (2021).
Guan, D. et al. Ferritinophagymediated ferroptosis and activation of Keap1/Nrf2/HO-1 pathway were conducive to EMT inhibition of gastric cancer cells in action of 2,2’-di-pyridineketone hydrazone dithiocarbamate butyric acid ester. Oxid. Med. Cell Longev. 2022, 3920664 (2022).
Wang, T. T. et al. Copper ion attenuated the antiproliferative activity of di-2-pyridylhydrazone dithiocarbamate derivative; however, there was a lack of correlation between ROS generation and antiproliferative activity. Molecules 21, 1088 (2016).
Li, Y. L. et al. p53-Mediated PI3K/AKT/mTOR pathway played a role in PtoxDpt-induced EMT inhibition in liver cancer cell lines. Oxid. Med. Cell Longev. 2019, 2531493 (2019).
Wang, L. et al. DpdtpA, a multimetal ion chelator, attenuates tau phosphorylation and microglial inflammatory response via regulating the PI3K/AKT/GSK-3β signal pathways. Neuroscience 526, 196–203 (2023).
Li, L. L. et al. Ferritinophagy-mediated ROS production contributed to proliferation inhibition, apoptosis and ferroptosis induction in action of mechanism of 2-pyridylhydrazone dithiocarbamate acetate. Oxid. Med. Cell Longev. 2021, 5594059 (2021).
Lu, Q. et al. ROS induces epithelial–mesenchymal transition via the TGF-β1/PI3K/Akt/mTOR pathway in diabetic nephropathy. Exp. Ther. Med. 17, 835–846 (2019).
Tang, Z. et al. TIPE2 inhibits the migration and invasion of epithelial ovarian cancer cells by targeting Smad2 to reverse TGF-β1-induced EMT. FASEB J. 38, e70045 (2024).
Maeda, M., Johnson, K. R. & Wheelock, M. J. Cadherin switching: Essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition. J. Cell Sci. 118, 873–887 (2005).
Sarrió, D. et al. Epithelial-mesenchymal transition in breast cancer relates to the basallike phenotype. Cancer Res. 68, 989–997 (2008).
Latunde-Dada, G. O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta 1861, 1893–1900 (2017).
Wenzel, S. E. et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 171, 628-641.e26 (2017).
Yang, W. S. & Stockwell, B. R. Ferroptosis: Death by lipid peroxidation. Trends Mol. Med. 26, 165–176 (2016).
Cai, J. Y. et al. Fatostatin induces ferroptosis through inhibition of the AKT/mTORC1/GPX4 signaling pathway in glioblastoma. Cell Death Dis. 14, 211 (2023).
Lee, N. et al. xCT-driven expression of GPX4 determines sensitivity of breast cancer cells to ferroptosis inducers. Antioxidants 10, 317 (2021).
Xue, Q. et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy 19, 1982–1996 (2023).
Tan, Y. et al. HucMSC-derived exosomes delivered BECN1 induces ferroptosis of hepatic stellate cells by regulating the xCT/GPX4 axis. Cell Death Dis. 13, 319 (2022).
Fumarola, C., Bonelli, M. A., Petronini, P. G. & Alfieri, R. R. Targeting PI3K/AKT/mTOR pathway in non-small cell lung cancer. Biochem. Pharmacol. 90, 197–207 (2014).
Laplante, M. & Sabatini, D. M. mTOR signaling in growth control and disease. Cell 149, 274–293 (2012).
Strimpakos, A. S., Karapanagiotou, E. M., Saif, M. W. & Syrigos, K. N. The role of mTOR in the management of solid tumors: An overview. Cancer Treat. Rev. 35, 148–159 (2009).
Han, Z. et al. The survival prediction of advanced colorectal cancer received neoadjuvant therapy-a study of SEER database. World J. Surg. Oncol. 22(1), 175 (2024).
Biller, L. H. & Schrag, D. Diagnosis and treatment of metastatic colorectal cancer: A review. JAMA 325, 669–685 (2021).
Jeong, K. et al. FAK inhibition reduces metastasis of α4 integrin-expressing melanoma to lymph nodes by targeting lymphatic VCAM-1 expression. Biochem. Biophys. Res. Commun. 509, 1034–1040 (2019).
Sainz, B. Jr., Carron, E., Vallespinós, M. & Machado, H. L. Cancer stem cells and macrophages: Implications in tumor biology and therapeutic strategies. Mediators Inflamm. 2016, 9012369 (2016).
Aokage, K. et al. Dynamic molecular changes associated with epithelial–mesenchymal transition and subsequent mesenchymal-epithelial transition in the early phase of metastatic tumor formation. Int. J. Cancer. 128, 1585–1595 (2011).
Du, B. & Shim, J. S. Targeting epithelial-mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules 21, 965 (2016).
Lee, J. Y. & Kong, G. Roles and epigenetic regulation of epithelial–mesenchymal transition and its transcription factors in cancer initiation and progression. Cell Mol. Life Sci. 73, 4643–4660 (2016).
Giannoni, E., Parri, M. & Chiarugi, P. EMT and oxidative stress: A bidirectional interplay affecting tumor malignancy. Antioxid. Redox Sign. 16, 1248–1263 (2012).
Wang, R. et al. Ferritinophagy-mediated apoptosis and paraptosis induction involved MAPK and PI3K/AKT pathway in mechanism of an iron chelator. Biochem. Pharmacol. 218, 115874 (2023).
Sun, L. et al. Lipid peroxidation, GSH depletion, and SLC7A11 inhibition are common causes of EMT and ferroptosis in A549 cells, but different in specific mechanisms. DNA Cell Biol. 40, 172–183 (2021).
Salovska, B. & Liu, Y. Posttranslational modification and phenotype. Proteomics 23, e2200535 (2023).
Zhu, Z. et al. FBXO31 sensitizes cancer stem cells-like cells to cisplatin by promoting ferroptosis and facilitating proteasomal degradation of GPX4 in cholangiocarcinoma. Liver Int. 42, 2871–2888 (2022).
Li, D. et al. CST1 inhibits ferroptosis and promotes gastric cancer metastasis by regulating GPX4 protein stability via OTUB1. Oncogene 42, 83–98 (2023).
Acknowledgements
This work was supported by the Natural Science Foundation of Henan Province (No. 252300421392), the Key Science and Technology Projects of the Henan Provincial Department of Science and Technology (232102310361; 232102310241), the Key Research Project Funding Program of Higher Educational Institutions of Henan Province (24A350013), the Doctoral Initiation Fund of Xinxiang Medical University (505505), University of Sanya of Talent Introduction Project (USYRC25-17), Hainan Province Higher Education Scientific Research Project (Hnky2025ZC-15).
Author information
Authors and Affiliations
Contributions
Y.F., Q.Z. and Y.S. performed the experiments; they contributed equally to this work. C.L. and Y.Y. conceived and designed the experiments. Y.L., B.H. (2022 undergraduate in clinical medicine), C.L. and W.Z. analyzed the data and prepared the figures. C.L. prepared and wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fu, Y., Zhao, Q., Li, Y. et al. NCOA4-mediated ferroptosis conducive to EMT inhibition in colorectal cancer cells was achieved through the PI3K/AKT/mTOR pathway. Sci Rep 15, 39542 (2025). https://doi.org/10.1038/s41598-025-23180-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-23180-y
Keywords
This article is cited by
-
Morusinol exerts anti-glioma activity by targeting the PI3K/AKT/mTOR signaling pathway
Naunyn-Schmiedeberg's Archives of Pharmacology (2026)
