
Abstract
In this research, quantitative structure property relationship (QSPR) regression along with multi criteria decision making (MCDM) approaches, including TOPSIS and Simple Additive Weighting (SAW), is deployed to rank the antihematological cancer drugs by structural and physicochemical attributes. Topological indices like zagreb, Randic and Atom Bond Connectivity (ABC) indices are calculated with the Maple program and chemspider was employed to obtain physicochemical descriptors such as boiling point; molar refraction; polarizability and molar volume which are both modeled using cubic and logarithm regression. The cubic model performed better than the logarithmic models, both in terms of correlation coefficients and predictive accuracy. In the present study, two compounds with more complexity of structure and greater connectivity, Carfilzomib and Zanubrutinib were ranked higher by QSPR and MCDM model considering the structural features compared to those with simpler molecular frameworks including Cyclophosphamide and Cytarabine which was placed lower. The above results suggest that compositing QSPR regression and MCDM is applicable towards computationally automatic, yet efficient process for screening drug candidates systematically at the beginning stages of hematologic cancer therapies.
Similar content being viewed by others
Introduction
Graph theory is the study of graphs, which are mathematical structures that model pairs of objects related by some set of relationships. A graph \(G\) is an ordering of a pair \(G = (V, E)\) where vertices are a set of vertices of the graph \(V\) and edges are a set of edges that connect pairs of vertices \(E .\) The degree of a vertex is one of the basic concepts in graph theory. The number of edges that meet at a vertex m, is called the degree of the vertex and is written as \(\beta _m.\) In the case of undirected simple graphs, this is equal to the number of vertices adjacent to v. The degree provides the basic input on the bonding between the vertices in a graph1.
Chemical graph theory is a branch of mathematical chemistry in which graphs are used to represent chemical compounds. Vertices are used to represent atoms, and chemical bonds are used to represent edges. Molecular graphs provide chemists with graph theoretic tools for examining the structural properties of chemical compounds. Chemical graph theory has been applied in numerous instances, such as for predicting the stability, reactivity, and physical properties of molecules2. It also plays a significant role in computer aided drug design and cheminformatics, where molecular structures are quantified.
Topological indices are quantitative characteristics of the graph theoretical structure of molecules with numerous applications in quantitative structure–activity relationship (QSAR) and quantitative structure–property relationship (QSPR) research. These properties are independent of the isomorphism of a graph and may prove valuable in modeling the biological, physical, and chemical properties of compounds. The Wiener index is a typical example, defined as the sum of the lengths of the shortest distances between any pair of vertices in a molecular graph. The Randic index, Zagreb indices, and Harary index are other popular topological indices, each describing a different aspect of the topology of the molecular graph3.
Ma et al.3 critically analysed the utility of topological indices in chemical graph theory, pointing to their practicability and limitations in the analysis of molecular structure. Their research calls for a rational understanding of these indices to arrive at precise predictive models. Khan et al.4 investigate degree based operators and topological descriptors in molecular graphs and prove their effectiveness in QSPR analysis of carbon derivatives. Their study emphasized the predictive capability of these descriptors for estimating chemical properties. Abubakar et al.5 carried out a QSPR study of antituberculosis drugs employing neighborhood degree-based topological indices coupled with support vector regression. Their results emphasized the utility of such indices in anticipating significant physical properties. Adnan et al.6 explored degree-based topological indices in the QSPR analysis, which proved to be feasible in describing the property of the antituberculosis drugs.
Iqbal et al.7 used QSPR modeling along with multicriteria decision analysis for the risk assessment of antiarrhythmic drugs. They combine predictive modeling with decision making frameworks to enhance drug selection strategies. Qin et al.8 using graph theoretic descriptors and machine learning to predict molecular properties of bladder cancer drugs. This study emphasizes the cooperation between graph theory and computational models in oncology drug discovery. Huang et al.9 studied application of QSPR analysis using XGBoost and regression based machine learning methods for glaucoma drugs. The results showed improved prediction performance for the physicochemical properties associated with therapeutic efficacy.
Qin et al.10 to study pulmonary cancer drugs using a QSPR approach based on topological modeling with Python. This study highlights the potential of computational topology in evaluating drug properties for cancer therapy. Qin et al.11 based a py value on topological indices predicting physicochemical properties of anti arrhythmia drugs. This model can be used as a computational framework for the systematic characterization of drugs. Wei et al.12 employed linear regression methodologies for the prediction of physical properties using QSPR on various drugs. Their findings may also support the knowledge that classic regression methods are effective in QSPR (Fig. 1).
Gutman and Polansky introduced the First and Second Zagreb Indices13 as:
Martínez et al.14 defined the Harmonic Index as follows:
The Forgotten Index was introduced by Furtula and Gutman15 as
The Shilpa Shanmukha Index was defined by Zhao et al.16 as follows:
The Atom Bond Connectivity Index was introduced by Estrada et al.17 as
The Randić index was defined by Randić et al.18as:
were
The Hyper Zagreb Index was introduced by Rajasekharaiah et al.20 as:
The Nirmala Index was defined by Kulli et al.21 as

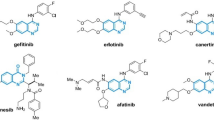
Chemical and molecular structures of hematologic cancer drugs for \(G_{i}, MG_{i}, i=1,2,3,...,11.\)
Bosutinib is used to treat chronic myeloid leukemia, especially in newly diagnosed patients or those resistant to other existing tyrosine kinase inhibitors. To assist in preventing the reproduction of cancer causing white blood cells, it attacks the unusual BCR ABL protein22. Carfilzomib is used in the treatment of multiple myeloma by inhibiting proteasomes, leading to cancer cell death. This was applied to patients who were inoculated before23. Cyclophosphamide A general alkylating agent is used in several cancers, such as lymphomas and breast cancer. It achieves its effect by destroying DNA and inhibiting cancer cell reproduction24. Cytarabine is primarily applied in acute myeloid leukemia. It suppresses DNA synthesis and is directed toward fast growing cancer cells25. Daunorubicin used to treat leukemia through its mechanism of action, which is the intercalating affinity of DNA and by blocking topoisomerase II. It also induces cancer cell death by breaking DNA26. Fludarabine it is mostly applied in chronic lymphocytic leukemia. It obstructs DNA cell division, inhibiting cancer cell growth27. Ibrutinib applied to B cell malignancies, such as CLL and mantle cell lymphoma. It blocks the signaling of cancer cells by inhibiting Bruton’s tyrosine kinase28. Lenalidomide treats multiple myeloma and myelodysplastic syndromes. It regulates the immune system and represses tumor growth29. Pomalidomide was applied to relapsed or intractable multiple myeloma. It boosts the immune system and prevents tumor angiogenesis30. Thalidomide is used to treat multiple myeloma and leprosy complications. It is immunomodulatory and anti angiogenic31. Zanubrutinib applied in mantle cell lymphoma and Waldenström macroglobulinemia. It selectively inactivates BTK, which interferes with B cell survival processes32.
Sun et al.33 emphasized the potential of memory T cells for adoptive T cell therapy based on their longevity and ability to provide long term immune surveillance in hematologic diseases. Their findings highlight approaches to maximize the potential of memory T cells to enhance therapeutic persistence and patient response. Zhou et al.34 in which various machine learning based computational methods were reviewed to predict drug target interaction and highlighted the role of state of the art models and algorithms for accelerating drug discovery pipelines. Their review illustrated the contribution of bioinformatics to reducing costs and increasing the accuracy of predicting treatments. Tang et al.35, which proposed a contrastive deep probabilistic matrix factorization model for drug disease association prediction. Their approach uses contrastive learning to exploit the non easy to observe relationships in biological data and outperforms contrastive learning by addressing the sparsity of data.
Li et al.36 proposed a dual ranking algorithm on multiplex networks to analyze heterogeneous and complex diseases. Their model effectively integrates multiple biological data sources, leading to better disease characterization and prediction accuracy in computational biology. Zhou et al.37 proposed a novel network embedding for predicting underground and terrorist communities. Leveraging graph representation learning methods, their approach contributed to the development of new associations in complex biomedical networks. Wang et al.38 mediated protection against LPS challenge in human PLEC 375 sirtuin 6 protects against LPS and induced inflammation. Their findings indicate that targeting sirtuin 6 could be a novel therapeutic approach for treating inflammatory pulmonary diseases.
Xianfang et al.39 utilized an indicator regularized non negative matrix factorization method for drug repositioning of coronavirus disease 2019. The authors identified potential antiviral agents using a cross system analysis of diverse biological and clinical datasets. Liu et al.40 proposed a matrix completion oriented ensemble approach with ridge regression for cancer drug response prediction. This greatly improved the robustness and prediction accuracy of numerous cancer associated datasets. Sheng et al.41 established an integrated molecular networking (IMN) approach for natural product dereplication. This process facilitates the discovery and characterization of novel natural products for pharmaceutical use. haematologic
Material and methods
This study selected 11 hematologic cancer drugs. They analyzed the chemical and molecular structures of bosutinib, carfilzomib, cyclophosphamide, cytarabine, daunorubicin, fludarabine, ibrutinib, lenalidomide, pomalidomide, thalidomide, The chemical structure is marked as \(G_1, G_2, G_3, G_4, G_5, G_6, G_7, G_8, G_9, G_{10}\) and \(G_{11}\) respectively and their respective molecular structures as \(MG_1, MG_2, MG_3, MG_4, MG_5, MG_6, MG_7, MG_8, MG_9, MG_{10}\) and \(MG_{11}.\) The legacy ChemSpider database was [https://legacy.chemspider.com] used to find such physicochemical properties of these drugs.
Table 1 indicates that various molecules of different drugs are partitioned by the connection (edges) among atoms of some specifics. A large majority of drugs possess numerous edges of the (2,3) type, that is, they tend to associate atoms having two and three bonds. The numbers in several edge classes were higher for carfilzomib and zanubrutinib, indicating that they have more complex structures. Drugs such as pomalidomide and thalidomide, on the other hand, contain fewer varieties of edges signaling the structures. This aids in visualizing the simple molecular structure and complexity of any given drug.
Result and discussion
Theorem 1
Let G be a graph and \(G_1\) denote the bosutinib; then the following axioms hold for the graph \(G_1: M_1(G_1) = 186,\) \(M_2(G_1) = 219,\) \(H(G_1) = 16.9,\) \(F(G_1) = 474,\) \(SS(G_1) = 41.400,\) \(ABC(G_1) = 27.662,\) \(RI(G_1) = 17.426,\) \(SC(G_1) = 18.073,\) \(GA(G_1) = 38.002;\) \(HZ(G_1) = 912;\) \(N(G_1) = 84.85.\)
Proof
Assume that bosutinib is denoted by \(G_1,\) and \(E_{s,t}\) is a set of edges linking vertices of degrees s and t within the graph. Between s and t vertices, frequencies \(|E_{s,t}|\) indicate the number of edges. \(|E_{1,2}| = 3\) indicates three edges linking vertices of degrees 1 and 2, and \(|E_{1,3}| = 3\) indicates three edges linking vertices of degrees 1 and 3. Likewise, \(|E_{2,2}| = 6,\) \(|E_{2,3}| = 21,\) \(|E_{3,3}| = 6.\) Then, Using the equation 1, 2, 3, 4, 5, 6, 7, 8, 9, and Table 1 we get
\(\square\)
Using a similar method, one can compute the topological indices of bosutinib. Furthermore, Python algorithm and maple is developed to compute the topological descriptor and its output is presented in Tables 2 and 3 represents their The physicochemical properties were collected from ChemSpider.
The molecular structures and complexities of drugs used to treat different hematologic cancers were studied by systematically calculating the topological indices of the drugs using Maple software. The Table 2 computed showed high level of variations in the compounds. Carfilzomib was rendered with the most consistent calculated indices and exhibited a highly branched and complex molecular design. In contrast, Cyclophosphamide and Cytarabine had the lowest values calculated in terms of less networked structures. These calculated graph-theoretical descriptors (including the Zagreb indices, Randic index, and GA index) provide valid quantitative information regarding the structural features, which in turn would serve as a strong QSPR modeling and rational drug design.
Table 3 shows physicochemical characteristics of hematologic cancers drugs mentioned as boiling point (BP), enthalpy of vaporization (EV), flash point (FP), molar refractivity (MR), Polarizability (P) and molar volume (MV). Carfilzomib had the highest points in various categories, including BP (975.6) and MV (619.6), indicating a larger and more complex machine. It is interesting to note that daunorubicin and fludarabine also have high physicochemical features, while lenalidomide and pomalidomide show average values. These variations explain the structural disparity of the drugs and can even affect the solubility, stability, and pharmacological activity of the drugs.
Cubic regression fits the relationship between variables using a third-degree polynomial equation. Cubic is appropriate when the data exhibit an inflection or double bends. (TI) denotes topological indices. where \(\alpha , \beta , \gamma ,\) and \(\delta\) are constants, and \(TI\) is the independent variable. This model is applicable when the value in the data has a nonlinear pattern with one or two bends or inflection points, that is, the line can bend back twice. It fits well even on complicated datasets where the trend increases and decreases, unlike the linear and quadratic models.
Logarithmic regression is used to model data where there is a rapid decrease or increase in the rate of change, followed by stabilization. It applies the natural logarithm of the independent variable. Where a and b are real numbers, x is any positive number, and log x is the natural logarithm of x.
It is applicable on the condition that \(x>0\) the logarithm is not defined when \(x=0\) or is a negative number. This model is good because it applies to processes that initiate rapidly and settle, such as population growth, learning rates, or returns on an investment. When the curve increases, it \(b>0\) goes below and drops as \(b<0.\) The variables \({M_1, M_2, H, F, SS, ABC, RI, SC, GA, HZ, N}\) are provided in this study as indices and used to define the independent variables of the model, which in this case are molecular or structural descriptors as predictors of the regression model. The dependent variables are boiling point, enthalpy of vaporization, flash point, molar refraction, molar volume, and polarizability, which are regarded as properties, and these are varied physical or chemical features that can be measured and are subject to variation brought about by the indices. The connection between the indices and properties was determined through regression analysis, and the quality of the models was determined using statistical parameters that included the correlation coefficient (R), which demonstrates the strength of the relationship; the coefficient of determination\((R ^2 ),\) which informs the proportion of the dependent variable that was explained by the independent variable; the standard error (SE), which shows the average level of error in the predicted values against the actual values; and the P-value, which shows whether a particular index contributed effectively to the model. These parameters assist in ascertaining the reliability and predictability of the regression models.
In the analysis, different physicochemical properties (such as boiling point, enthalpy of vaporization, flash point, molar refraction, molar volume, and polarizability) were analyzed as dependent variables and the first Zagreb index \(M_1(G)\) as independent. To capture the relationship between \(M_1(G)\) and each property, both cubic and logarithmic regression models were used. Such models assist in measuring the influence of the molecular structure described \(M_1(G)\) on the statistical behavior of compounds at the physical and chemical levels.
Table 4 indicates the superior performance of the cubic regression model when predicting six physicochemical properties from the \(M_1\) topological index. The higher and consistent R and \(R^2\) values and lower SE values from the cubic model attest to a better and more stable fit. Moreover, properties such as molar refraction and polarizability have \(R = 0.977\) and \(R^2 > 0.95,\) indicating a reliable connection with the \(M_1\) index. The cubic model is less susceptible to nonlinear relations, particularly those with inflection points or double curvature, which are prevalent in complex molecular behavior. Logarithmic models, on the other hand, can only be used in cases where the change is so rapid that it levels out over time. The values of \(R^2 > 0.95\) indicate that the cubic regression method fits better in explaining the relationship between the molecular structure and physicochemical properties.

Graphical analysis of \(M_1\) based linear and cubic regression model.
Figure 2 shows the cubic and logarithmic models predicting six different physicochemical properties using the \(M_1\) topological index. The red dots represent the actual data, and the green and blue lines represent the cubic and logarithmic models, respectively. In most cases, the green cubic lines are closer to the red dots, indicating that they follow the real data more accurately. This is especially clear in the boiling point, flashpoint, and molar volume graphs. The cubic model also fits better for properties such as molar refraction and polarizability. Overall, the figure clearly shows that the cubic model provides a better and more reliable fit than the logarithmic model.
In this study, an array of physicochemical properties, such as the boiling point, enthalpy of vaporization, flash point, molar refraction, molar volume, and polarizability, were considered as dependent variables, whereas the Second Zagreb index, \(M_2(G),\) was considered as the independent variable. The relationship between \(M_2(G)\) and each property was tested using both cubic and logarithmic regression models. Such models provide details on the role of the structural characteristics which are encoded through \(M_2(G)\) affect the physical and chemical behaviors of molecules.
Table 5 presents a comparison of the predictions of various molecular properties using cubic and logarithmic algorithms based on the topological index \(M_2.\) The cubic model will always produce a superior fit with greater R and \(R^{2}\) counts and smaller SE, which implies that it is more precise in relation to the data. Although both models were statistically significant, the cubic model demonstrated more reliable and consistent results. This indicates that the cubic method is superior in explaining the associations between the selected properties and \(M_2\) relationships.

Graphical analysis of \(M_2\) based linear and cubic regression model.
Figure 3 demonstrates the correlation between \(M_2\) and other variables according to the cubic and logarithmic regressions. Actual data are plotted as red dots, whereas cubic and logarithmic fits are plotted as green and blue curves, respectively. Under normal circumstances, the cubic model is close to the data trend, particularly for nonlinear trends. A logarithmic model usually fits the data in a smoother, albeit less accurate, manner. This is more evident in variables such as BP and MV, in which sharp increases are better depicted in the cubic model. Comprehensively, the cubic regression proves to be the best in modeling the \(M_2\) based relations.
H(G) is treated as the independent variable in the given analysis, whereas the dependent variable is the various physicochemical properties that are likely to be dependent variables of boiling point, enthalpy of vaporization, flash point, molar refraction, molar volume, and polarizability. To examine their relationship, cubic and logarithmic regression models were used. These models provide a quantitative approach for investigating the effects of the topological structure embodied by H(G) on the measured molecular properties.
Table 6 presents a comparison of the performances of the cubic and logarithmic models in predicting various properties of H. The cubic model records greater values of R and \(R^2\) throughout the properties, implying that it presents more significant correlations and fits the information aptly. The logarithmic model does not explain as much of the variation, although in some cases, the standard errors are slightly lower. In terms of properties such as molar refraction and polarizability, the \(R^2\) values were perfected by the cubic models to the extent of 0.993, and the P-values were extremely low, indicating high significance. The boiling point, flash point, and molar volume are not better predicted using the cubic model. Therefore, the cubic model is a more reasonable and certain way of shaping the properties of H.

Graphical analysis of H based linear and cubic regression model.
Figure 4 shows that the cubic and logarithmic regression models are related to the variables of H. Red dots symbolize the real results, and the green and blue lines represent cubic and logarithmic fitting, respectively. A better fit may be obtained in the cubic model in general, particularly for variables such as BP, FP, and MV, where nonlinear trends can be observed. It is smoother than the logarithmic model; however, it overregulates the data in areas experiencing acute changes. The two models are not different in the case of variables such as MR and P. Overall, cubic regression provides a more effective grasp of the differences in data.
The independent variable in this study is the forgotten index F(G), and a range of disparate physicochemical properties, namely, the boiling point, enthalpy of vaporization, flash point, molar refraction, molar volume, and polarizability, are regarded as the dependent variables. Regression (both cubic and logarithmic) was used to determine the mathematical relations between those properties and F(G). The method of modeling aids in the inspection of how the molecular structure, measured with the help of F(G), influences the physical and chemical properties of compounds.
Table 7 confirms that the cubic model is stronger in forecasting the characteristics of F than the logarithmic model. It reaches higher correlation values (R) and coefficients of determination, \(R^{2}\) signifying a better relationship and accuracy of the model. The cubic model is inclined to provide fewer standard errors, which implies the accuracy of the predictions. Despite the accuracy in F-statistics between the logarithmic and overall models, the lower levels of \(R^{2}\) values demonstrate lower explanatory power. The lower P-values in both models are a constant reinstatement of statistical significance, but it is the cubic model that is a better and more reliable solution to use when it comes to modeling such properties.

Graphical analysis of F based linear and cubic regression model.
The correlation between variable F and the other output variables (BP, EV, FP, MR, P, and MV) was plotted with a cubic logarithmic regression model, as shown in Fig. 5. The red points denote the actual data points, the green curve indicates the cubic best fit, and the blue curve indicates the logarithmic best fit. The cubic model usually has a better fit, especially when the patterns of BP, FP, and MV tend to present more nonlinear patterns. Although the logarithmic model is smooth, it tends to differ from the actual trend at high values of F. In cases such as MR and P, both models performed similarly. Generally, a cubic regression model better approximates the data fluctuations in connection with F.
In this form a Shilpa Shanmukha index SS(G) is taken as the independent variable and the indices which are taken as independent variables are boiling point, enthalpy of vaporization, flash point, molar refraction, molar volume, polarizability which are taken as dependent variables. To test the correlation between SS(G) and these properties of molecules, cubic and logarithmic regressions were used. These models allow the determination of the contribution of structural differences symbolized as SS(G) to the major physicochemical properties of the compounds.
The statistical outcomes of the SS descriptor are shown in Table 8 for both the cubic and logarithmic models. In all the dependent variables, the cubic model demonstrates better results, with appreciable R and \(R^2,\) as well as lower standard errors. For example, an R value greater than 0.98 indicates a very strong fit, which is observed in the following parameters: molar refraction and polarizability in the cubic model. The p-values of each model were very small, and therefore, the models were statistically significant. Nonetheless, weaker correlations and larger prediction errors generally exist within the logarithmic model. The power of the cubic model is also supported by the F-statistic. In general, following Table 8 one can say that the cubic model will fit more and be more accurate and reliable in calculating physical properties using SS.
The graphical comparison of the cubic and logarithmic regression models is depicted in Fig. 6 which shows the regression models of different dependent variables (BP, EV, FP, MR, P, and MV) according to the independent variable SS. The actual points are marked by red dots, and the cubic and logarithmic regression fits are marked by green and blue lines, respectively. In the vast majority of subplots, the cubic model (green line) seems to better fit the data than the logarithmic model, reflecting the nonlinear tendencies best, particularly in pairs such as SS vs. BP and SS vs. MV. The smoother logarithmic model does not consider the abrupt shifts and curves of the data. Overall, the figure indicates that the cubic regression model can provide a more appropriate description of the relationships between SS and the other parameters.

Graphical analysis of SS based linear and cubic regression model.
In present analysis, the Atom Bond Connectivity index ABC(G) is taken as an independent variable whereas such characteristic molecular properties as boiling point, enthalpy of vaporization, flash point, molar refraction, molar volume, and polarizability are considered dependent. Cubic and logarithmic regression models were applied to determine the effect of variations in ABC(G) on these characteristics. This model strategy reveals the extent to which the structure of molecules encapsulated by ABC(G) determines the physical and chemical properties of compounds.
Table 9 lists the statistical parameters of the ABC descriptor for both types of models. Similar to SS, the cubic model is better in most instances, particularly for molar refraction and polarizability, where R values are as high as 0.988. The standard errors are lower in the cubic model, which makes the prediction results more precise. Whereas the logarithmic model has acceptable results, the \(R^2\) values are slightly lower and the error values are greater. The stronger performance of the cubic model is also corroborated by the F-statistic and p-values. Based on Table 9, it has been realized that the Cubic model is a more viable alternative when modeling physical properties via ABC.

Graphical analysis of ABC based linear and cubic regression model.
The graphical analysis of the correlation between the ABC and other output variables in Fig. 7 is done with the help of the cubic and the logarithmic regression models. The points in red are observed, and the green and blue lines are related to the cubic and logarithmic fits, respectively. Overall, the cubic model is more informative of the data in the majority of the plots, as it approximates the non linear trends, especially in ABC vs. FP and ABC vs. MV. The logarithmic model is smoother in trend, but in some cases, it does not reproduce the curvature properly. Overall, the cubic regression model shows superiority over the existing relationship between ABC and the dependent variables.
The independent variable in this study was the Randic index RI(G) and the dependent variables were the boiling point, enthalpy of vaporization, flash point, molar refraction, polarizability, and molar volume. To understand the relationship between these molecular properties and RI(G), cubic and logarithmic regression models were used. This two modeling technique assists in interpreting how molecular branching into certain compounds, expressed by the quantity RI(G), affects the physical and chemical properties of the compounds.
Table 10 offers a comparison of the cubic and logarithmic models involving the RI descriptor to predict numerous physical properties. The cubic model has a better predictive performance because it records higher values of R and \(R^2,\) as well as lower standard errors. The results are statistically significant, as confirmed by the small p-values. The logarithmic model demonstrated worse results, an increased number of errors, and decreased accuracy. All properties are also in favor of the cubic model according to the F-statistics. The above highlights the fact that the cubic model is more effective and reliable when employing RI as the descriptor of property prediction.

Graphical analysis of RI based linear and cubic regression model.
In Fig. 8, the graphical tool compares the cubic and logarithmic regression models against the dependent variables plotted against the independent variable RI. The red dots denote real data, as opposed to cubic and logarithmic regression fits in green and blue lines, respectively. In the majority of plots, the cubic function provides a better representation of the nonlinear nature of the plot, especially in RI vs FP and RI vs MV, than the logarithmic function, which is smoother but less adaptively modeled. In other cases, including RI vs P and RI vs MR, both models worked equivalently, but the cubic model seemed marginally more successful in the level of variation brought to light. Overall, the cubic regression model is more efficient in representing complex relationships with RI.
As part of this analysis, the index of sum connectivity, was set as the independent variable, while several dependent variables were the boiling point, enthalpy of vaporization, flash point, molar refraction, polarizability, and molar volume. Cubic and logarithmic regression models were also used to reveal the mathematical correlations between such molecular properties and the level of SC(G). The present modeling emphasizes that the connectivity pattern in molecular graphs, which is represented through SC(G) affects the most important physicochemical parameters.
Table 11 indicates that the cubic model has a constant prediction power that surpasses that of the logarithmic model with the SC descriptor. It attains greater R and \(R^2\) values,which shows better fitting of data. The smaller standard errors in the cubic model indicate greater accuracy in the property estimation. The p-values were considerably low, and hence the findings are statistically important. The F-statistics also advocate the superiority of the cubic model in relationship capturing. Overall, SC is most applicable to a cubic model for predicting physical properties.

Graphical analysis of SC based linear and cubic regression model.
Figure 9 provides a graphic analysis comparing SC and six dependent variables (BP, EV, FP, MR, P, and MV) using both cubic and logarithmic regression models. The real data are marked with red dots, whereas the cubic and logarithmic fits are marked with green and blue lines, respectively. The cubic regression model (curved line) in the vast majority of plots seems to conform more to the data, providing that it is accurate in the non-linear trends (i.e., in the plots such as SC vs. BP, MR, and P), compared to the logarithmic model. The logarithmic model tends to reduce the variations and can fail in the region where the data are more curved. This indicates that the cubic model is perhaps more appropriate for use in more complex relationships when using SC based prediction.
The Geometric Arithmetic index GA(G) was considered as the independent variable, whereas the physicochemical properties such as boiling point, enthalpy of vaporization, flash point, molar refraction, polarizability, and molar volume were considered as the dependent variables in this analysis. To relate between both the GA(G) and each property, both cubic and logarithmic regression models were used. These models provide insight into the effect of the structural characteristics of the geometric arithmetic on the behavior of the molecular compounds.
Table 12 indicates that the cubic model is more advantageous than the logarithmic model in the application of the GA descriptor. Its R and \(R^2\) values are large, thus representing the data more precisely. The cubic model was also associated with fewer errors than the other model, indicating less imprecision in prediction. The strength of this model is indicated by greater F-statistics and lower p-values. Overall, the cubic model predicts properties with greater stability, accuracy, and reliability than the GA.

Graphical analysis of GA based linear and cubic regression model.
In Fig. 10 similar analysis was performed on GA as an independent variable against the same fixed six dependent variables (BP, EV, FP, MR, P, and MV). Similar to Fig. 10, the graph of the cubic regression model (green line) typically provides a better fit than the logarithmic model (blue line), especially when the data exhibit a curvy pattern, such as GA vs BP and GA vs MR. The cubic model is more responsive to changes in the data trends, whereas the logarithmic model is more generalized. In other cases, for example, GA vs. EV and GA vs. MV, the models differed more significantly, indicating the superiority of the cubic method in observing nonlinear trends. Altogether, the GA-based analysis proves the usefulness of cubic regression as a means of modeling complicated relationships among variables.
The hyper Zagreb index was used in this study as the independent and dependent variables, which are some physicochemical properties of the compound, including boiling point, enthalpy of vaporization, flash point, molar refraction, polarizability, and molar volume. Cubic and logarithmic regressions are used to evaluate the effect of these properties on HZ(G). These models provide insight into the degree squared based structure described by HZ(G) related to the molecular properties of the compounds.
Table 13 shows that the cubic model performs better than the logarithmic model when operating on the HZ descriptor. It has more R-values up to 0.955, \(R^2\)-values up to 0.912; that is why it is fit more to the data. The cubic model has a lower standard error, which indicates better prediction ability. F-statistics are more robust, and extremely small p-values, such as 0.0001, indicate that the results are statistically significant. The cubic model is more efficient and worthy of prediction with the HZ descriptor.

Graphical analysis of HZ based linear and cubic regression model.
The graphical comparison of the regression models is illustrated in Fig. 11 based on the HZ as the independent variable and its effect on the six different output parameters, including BP, EV, FP, MR, P, and MV. The actual data points are presented as red dots, whereas the green and blue curves indicate the cubic and logarithmic regression models, respectively. The cubic model is more flexible to dynamic trends in the majority of the graphs, especially HZ vs. BP and HZ vs. MV, because it matches the data distribution. Conversely, the logarithmic model is prone to providing a less accurate but smoother curve, particularly in areas where the data change sharply. Overall, the cubic regression model appears to be more appropriate for describing nonlinear performance trend characteristics, which appear in the HZ-based trends.
In this section, the Nirmala index is considered as an independent variable and analyzed to determine its effect on the key physicochemical properties, namely boiling point, enthalpy of vaporization, flash point, molar refraction, polarizability, and molar volume, which are the dependent variables. Both cubic and logarithmic regression models are used to study the relationship between the number of properties and N(G). Such models are used to demonstrate that variations in N(G), a topological descriptor, are reflected in alterations in molecular behavior.
Table 14 shows that the cubic model has a more superior statistical performance than the logarithmic model when the N descriptor is utilized. The cubic model has larger correlation values, where R goes up to 0.985 and \(R^2\) up to 0.970, which is classified as a strong fit. It also experiences smaller standard errors, indicating greater accuracy in the estimations. The F statistics tend to be equal to or greater than and extremely small p-values, including\(1.11 \times 10^{-5},\)verify the statistical reliability. Overall, the cubic model is much more accurate and reliable than the logarithmic model.

Graphical analysis of N based linear and cubic regression model.
Figure 12 presents a graphical comparison of the cubic and logarithmic regression models under different parameters (BP, EV, FP, MR, P, and MV) against Nirmala (N). The dots on the graph are the observed data points, whereas the curves indicating the cubic and logarithmic models are indicated by the lines in green and blue, respectively. As an illustration, the cubic model can better describe the sharp increases present in the N vs. BP plot (and the N vs. MV plot) than the logarithmic model. These findings imply that a cubic regression model can produce a better fit in modeling the relationship that exists between the levels of nirmala and the parameters under study due to its adaptability in complicated drinking designs.
Multi criteria decision making (MCDM)
Multi Criteria Decision Making (MCDM) is a modern decision support tool that assists in the evaluation and ranking of alternatives in the presence of conflicting criteria. In drug discovery, MCDM techniques, such as the QSPR analogy, integrate and evaluate multiple physicochemical and pharmacological attributes of drugs. MCDM balances the weighing of differing criteria by normalizing property values, and in this way, MCDM discovers the most potential drug candidates. Some commonly used methods are TOPSIS (Technique for Order of Preference by Similarity to Ideal Solution) and SAW (Simple Additive Weighting). These methods provide a systematic approach for ranking molecules. Ultimately, MCDM aids in the optimization of drug candidates by multi criteria measurement and evaluation.
TOPSIS (technique for order preference by similarity to ideal solution)
Technique for Order Preference by Similarity to Ideal Solution (TOPSIS) is a widely used Multi Criteria Decision Making (MCDM) technique available for ordering a set of alternatives based on their similarity with an ideal solution. The general principle of TOPSIS is that the best alternative would be highly similar with the positive ideal solution, e.g., maximizing the desirable features and minimizing the undesirable ones, while simultaneously being as far away as possible from the negative ideal solution (doing exactly the opposite of the former) The TOPSIS method works by normalizing the decision set to eliminate scale variance, with proper weights illustrating the relative importance of each criterion and the computation of the distances of each alternative from the ideal and negative ideal solutions. The relative closeness of each alternative is calculated through these calculations, and therefore, a convenient ranking is obtained. Its simple nature and rational format have made TOPSIS popular in numerous applications, including supplier choice, performance appraisal, risk classification, and estimation. In the present problem, each alternative d is representative of an individual hematologic cancer drug, while each criterion c is representative of a topological index (such as the sum connectivity index, Nirmala index, etc.) applied for the characterization of the inherent drug molecule characteristics. The number of alternatives and criteria are indicated by the variables m and n, respectively. The raw value of the c th topological index for the d-th drug is denoted as \(x_{dc}.\) The normalized value \(r_{dc}\) reflects the scaled measure of \(x_{dc},\) eliminating the differences on scales of varying criteria. The weighted normalized value, denoted symbolically as \(v_{dc},\) is the product of the normalized value \(r_{dc}\) and the weight of criterion type c, denoted as \(w_c.\) Sets \(C_{\text {benefit}}\) and \(C_{\text {cost}}\) represent maximization and minimization criteria, respectively. The Euclidean distances at the ideal points of alternative i can be expressed as the distance from the ideal solution, \(S_i^+,\) and the distance from the negative ideal solution, \(S_i^-.\) The Relative Closeness Coefficient, \(C_i^*,\) reflects the relative closeness of each alternative to the ideal solution. In the entropy weight method, the ratio of the d-th alternative of the c-th criterion, the entropy measure of the dispersion of the c-th criterion being \(e_c,\) and the diversification degree of the c-th criterion being \(d_c,\) play important roles. The final entropy weight of the c-th criterion is denoted as \(w_c.\)
The steps involved in the TOPSIS method
Step 1: Compute the Proportion \(p_{dc}\) for Entropy Weighting:
Step 2: Calculate the Entropy\(e_c\) for Each Criterion:
Step 3: Compute the Degree of Diversification \(d_c\):
Step 4: Calculate the Entropy Weights \(w_c\):
These entropy weights would then be utilized through the TOPSIS or SAW method to objectively quantify the eye drug candidates.
Step 5: Normalize the Decision Matrix For each candidate drug, \(x_{dc}\) is the \(c^\text {th}\) topological index. The normalized value \(r_{dc}\) is calculated as follows:
Step 6: Compute the Weighted Normalized Decision Matrix:
For each criterion consideration, the criterion values were normalized and then multiplied by the corresponding entropy weights \(w_c\):
Step 7: Determine the Positive Ideal Solution \(A^+\) and the Negative Ideal Solution\(A^-\):
The positive and negative ideal solutions are defined as
Step 8: Calculate Separation Measures:
The Euclidean distance from each alternative to the ideal and anti-ideal solutions is computed as follows:
Step 9: Calculate the Relative Closeness to the Ideal Solution:
The relative closeness \(C_d^*\) of each alternative to the ideal solution is expressed as
Step 10: Rank the Alternatives:
The alternatives (drugs) are ranked in decreasing order of \(C_d^* .\) The drug with the highest value of \(C_d^*\) is considered the best choice based on the topological indices. Table 15 presents the numerical values of all drugs along with their corresponding topological indices.
The allocation of weights to the topological descriptors used in the analysis is presented in Table 16. Descriptors such as \(M_1\) (0.0624) and \(M_2\) (0.0609) are relatively given similar weights, reminding us that they are of similar significance in the decision making process. Descriptors such as H (0.0639) and SS (0.0632) are slightly weighted, suggesting that they have a modest impact on the evaluation of molecular properties. The other descriptors, which include F (0.0612) and N (0.0630), have relatively lesser weights and may therefore have a minimal impact on the analysis. This arrangement is important for converting normalized data into weighted scores, as in the case of TOPSIS or VIKOR.
Table 17 shows the normalized decision matrix of the chosen drugs against the selected topological indices, with all values on a common scale for an unbiased comparison. Carfilzomib scored a perfect 1.000 in all indices, a testament to its superior molecular attributes for more than a single topological measure. Bosutinib and zanubrutinib followed with high normalized values, specifically in \(M_1\) and \(M_2,\) as an indication of strong general molecular attributes. In contrast, cyclophosphamide ranked comparatively low in most indices, indicating that it has weaker topological properties than the other drugs. The compounds cytarabine, fludarabine, and ibrutinib ranked moderately, indicating a balance of molecular attributes with no exceptional attributes. Normalization makes the data comparable without any bias; hence, it is a good starting point for further research as well as multi criteria decision making.

Heatmap of normalized data
The heatmap of the normalized data Fig. 13 provides an extended visualization of the normalized data for selected drugs (e.g., Bosutinib, Carfilzomib, Cyclophosphamide) for selected parameters ( \(M_1,\) \(M_2,\) H) through an offset color gradient from 0.0 to 1.0, where darker shades indicate higher levels. Carfilzomib stands out by maintaining consistently high levels at approximately 1.0 for all parameters, suggesting an intense and constant effect. In contrast, Cyclophosphamide (0.24-0.47) and cytarabine (0.32-0.37) show lower and less constant levels, indicating an inconsistent or weaker effect. The peak value of daunorubicin at 1.0 in the M2 parameter indicates a significant localized effect and represents an intriguing finding. This heatmap allows for an easy comparative analysis and shows distinct drug performance patterns among the tested parameters.
The alternate weighted normalized decision matrix is obtained by multiplying each of the normalized values calculated in Table 18 by their corresponding weights in Table 17; thus, Table 16 is obtained. This matrix is a good way to incorporate the specific molecular characteristics of the drugs and the relative significance of each of the topological descriptors. Therefore, it is subject to the emergence of a more complex and sophisticated dataset that can be employed in an advanced decision model. Among them, drugs with higher weights include carfilzomib, zanubrutinib, and daunorubicin, which raises their importance as descriptors, as they are not the only variables given their importance regarding their performance.

Heatmap of weighted normalized data.
Figure 14 shows the distribution of weighted physicochemical property scores for the different hematologic cancer drugs, including bosutinib, carfilzomib, and cyclophosphamide. The gradient from light green (lower, e.g., -0.06 for cyclophosphamide) to dark green (higher, e.g., 0.06 for bosutinib) highlights the relative performance over multiple indices, such as \(_{1},\) \(M_{2},\) and HZ. Notably, Carfilzomib occupied the maximum values (approximately 0.06) at all times, suggesting an overall good profile, whereas others, such as cyclophosphamide and cytarabine, showed comparatively low scores (approximately -0.05 to -0.06), indicating vulnerability in certain properties. This plot shows the drug performance variability and the merit of weighted normalization for identifying lead candidates for further optimization.
Bosutinib: The radar chart for bosutinib reveals a very balanced and even profile of values on several indices. This implies moderate variability and suggests a comparatively steady molecular structure with controllable entropy and structural diversity.
Carfilzomib: The radar chart of carfilzomib shows more variability and has more movement on the chart, suggesting a more complex molecular structure. The structure has moderate levels of diversity and entropy, suggesting a degree of stability combined with variability.
Cyclophosphamide: The radar chart for cyclophosphamide indicates relatively low variability with uniformly balanced values across indices. This indicates a steady molecular structure with medium entropy and a lower degree of structural diversity.
Cytarabine: The radar chart for cytarabine indicates a moderate degree of variability, suggesting the molecule has a balanced structure between stability and variability. The degree of entropy is quite high, indicating that the structure is more complex in relation to smaller molecules.
Daunorubicin: The radar chart for daunorubicin shows extensive variability, suggesting the more complex and heterogeneous structure at the molecular level. A higher degree of entropy suggests extensive structural diversification within the molecule.
Fludarabine: The radar chart for Fludarabine indicates balanced distribution of values and therefore low variability. The form of the structure is relatively stable with low entropy, and the degree of diversity in the structure of the molecule is moderate.
Ibrutinib: The radar chart for ibrutinib shows moderate variability, implying a stable but relatively complex molecular structure. This indicates moderate entropy, with a fair amount of diversity in the way it is configured.
Lenalidomide: As briefly indicated before, the radar chart in the case of lenalidomide indicates that the variability among the indices is low and even balanced implying simple molecular structure and limited entropy and structural variety.
Pomalidomide: The radar chart of pomalidomide highlighted a balanced set of values and least variability which mean that the molecular structure of the drug is relative stable. The unfavorable value of entropy and moderate number of structural diversity indicate a controlled and defined molecular structure that is complex.
Thalidomide: In the case of Thalidomide, variability in its radar chart indicates that the structure of this molecule is moderately stable and not very complex. It is somewhat complex in structure, as indicated by moderate levels of entropy and diversity.
Zanubrutinib: The radar chart of zanubrutinib distribution of variance is evenly spread over the indices, signifying that the structure of the molecule is relatively stable with moderate entropy and structural diversity. This implies a balanced molecule that is moderately complex in nature.

Radar charts illustrating the distribution of topological indices across various anticancer drugs.

Hierarchical clustering dendrogram of the analyzed drugs based on their topological and entropy-derived indices.
Figure 15 illustrates the topological and entropy based index diversity in a group of drugs under study, which indicates molecular architecture and complexity. Figure 16 presents a hierarchical clustering dendrogram that classifies drugs in accordance with weighted topological index profiles using Euclidean distance. The outline indicates two prominent clusters, each encompassing compounds with shared molecular characteristics. The first set included cyclophosphamide, cytarabine, lenalidomide, thalidomide, fludarabine, and pom They are quite simple in their topology with low values in their molecular indices, and this explains their proximity in The second set included carfilzomib, daunorubicin, bosutinib, ibrutinib, and zan, which are topologically more complex, with higher chemical diversity and entropy. There is especially tight correspondence between Ibrutinib and Zanubrutinib, which means the structures are highly similar In summary, clustering points to a clear stratification among topologically simpler and more complex drugs. This supports the topological index validity in revealing pertinent molecular similarities and defining strategies for classifying drugs.
The resulting TOPSIS based ranking of the drugs in terms of L+, L-, O_i, and Rank metrics is given in Table 19. The carfilzomib case was the first (with the highest O_i value of 0.996442), followed by daunorubicin and zanubrutinib, and the same trends as before in drug performance. These compounds exhibit good values for certain measurements, indicating that they are very strong and effective. Cyclophos was at the bottom because it had low values of L- and O_i; hence, the overall performance was poor. The scores favor the fact that drugs with more balanced metrics are more likely to succeed and prove the importance of these parameters in drug effectiveness assessment.

Ranking of drugs based on TOPSIS score.
Figure 17 displays the drug rankings obtained using the TOPSIS methodology, which computes the relative proximity of each candidate to the ideal solution for all criteria. Carfilzomib ranked first with a near 1.0 measure for relative closeness, indicating perfect performance in all descriptors considered. This was followed in the second to fourth ranks by Daunorubicin, Bosutinib, and Zanubrutinib, all of which had high scores and favorable multi criteria profiles. Cyclophosphamide and Cytarabine occupy the lowest ranks with low scores for closeness, indicating poor proximity to the ideal solution and are in agreement with earlier SAW based rankings. This consistency in all the ranking models reaffirms the internal stability and strength of the multi criteria decision making approach applied in this study.
Evaluation of hematologic cancer drugs using the SAW technique
The Simple Additive Weighting (SAW) method is a straightforward yet effective method for ranking alternatives in Multi Criteria Decision Making (MCDM). In this study, we ranked hematologic cancer drugs according to various topological indices calculated from their structures at the molecular level. Each drug is considered an alternative, and each index is a criterion reflecting the molecular complexity, connectivity, or information content. The first step in the process is to normalize the values in the decision matrix. Benefit criteria (for which higher values are advantageous) and cost criteria (for which lower values are advantageous) are treated using different normalization functions. Each criterion, after normalization, was weighted in proportion to its importance. These weights were determined objectively using the entropy function. The SAW score of each drug was calculated as the sum of its normalized weighted values. Hematologic cancer drugs with high SAW scores are promising. This measure is popular because of its ease of computation, transparency, and handling of heterogeneous criteria.
Step 1: Normalize the Decision Matrix
Step 2: Compute the SAW Score
Step 3: Rank the Alternatives
The structures of the chemicals are ordered in descending order for \(V_d ,\) and greater values indicate good overall performance for all weighted topological indices.
Table 20 shows the normalized quantities of some of the topological descriptors included in the SAW based screening of potential candidates for the bladder. These descriptors\(M_1,\) \(M_2,\) H, F, SS, ABC, RI, SC, GA, HZ, and Ncapture various structural, connectivity, and complexity related aspects of each molecule. Normalization ensures that all descriptors are dimensionless and directly comparable to facilitate a fair multi criteria evaluation. Carfilzomib always attained the maximum attainable quantity (1.0) in all descriptors, indicating flawless structural features across all criteria, making it the top candidate in the raw descriptor space. Daunorubicin and zanubrutinib exhibited bright profiles with high normalized quantities in most descriptors, particularly in the entropy based and degree sensitive descriptors H, SS, GA, and HZ. Cyclophosphamide and Cytarabine exhibit relatively poor scores, and in many cases, particularly in F, ABC, and GA, this indicates that their topology is simpler and more dispersed.
In Table 21, the weighted normalized values of several drugs on various parameters (\(M_1,\) \(M_2,\) H, F, SS, ABC, RI, SC, GA, HZ, and N) are provided. Table 21 displays diversified values for all drugs, of which the highest values are shown by carfilzomib for most of the parameters, particularly M2 (0.06094), H (0.06397), and ABC (0.06387). This suggests that the performance of carfilzomib may be better than that of the other drugs for such parameters. Similarly, Cyclophosphamide displayed the lowest values, particularly for the parameters of \(M_1\) (0.01538) and \(M_2\) (0.01487), suggesting that it has a weak performance compared to other drugs. Drugs such as Bosutinib, Fludarabine, and Ibrutinib showed intermediate values for most parameters, which may indicate a balanced performance for some parameters. These parameters reflect the diversified performance or efficacy attributes of the aforementioned drugs, which depict their disparate performance under diverse conditions. Overall, Carfilzomib appears to be the best performing medication based on such calculations, but the performance of some medications, such as Zanubrutinib and Daunorubicin, appears prominent on some parameters.

Ranking of drugs based on TOPSIS score.
The radar chart in Fig. 18 provides a comparative picture of the performance of various drugs on various parameters. Carfilzomib has the best performance, as it has the highest values across, particularly \(M_2\) and H, which indicate its best performance. Daunorubicin and bosutinib also performed well, but not as exceptionally as arfilzomib. Cyclophosphamide and Cytarabine have the lowest values, especially on \(M_1\) and \(M_2,\) indicating that they are less effective. Zanubrutinib, Ibrutinib, and Fludarabine exhibited normal performance, as they were better than the others on some parameters but did not score well on all parameters. Overall, the chart confirms that the most effective drug from this dataset is carfilzomib.
Table 22 presents the SAW scores calculated using the Simple Additive Weighting (SAW) method, a multi criteria decision making technique that employs normalized descriptor values weighted by entropy. The SAW score reflects the overall performance of each drug based on topological or structural descriptors. Carfilzomib, daunorubicin, and bosutinib emerged as the top three candidates, suggesting a strong global performance that may be linked to favorable molecular complexity and connectivity. These results align with observations from complementary decision making frameworks, such as TOPSIS and clustering. In contrast, cyclophosphamide, cytarabine, and lenalidomide ranked lowest, corresponding to simpler molecular structures and lower entropy values. The SAW based ranking supports previous findings by providing a transparent and reliable aggregation of weighted criteria, reinforcing its utility in drug prioritization.

Ranking of drugs based on TOPSIS score.
The pie chart in Fig. 19 shows the share of Simple Additive Weighting (SAW) scores of various drugs. The highest share was for carfilzomib (16.3 %), indicating that it had the best performance among all the drugs considered. Daunorubicin had the next highest share, 13.0%, indicating its strong efficacy. Bosutinib and zanubrutinib share similar 11.8% and 11.7% market share, respectively, which means that they perform well but just slightly less than the first two drugs. Other drugs such as ibrutinib, fludarabine, and pomalidomide each take about 6–7% of the market, which means that they have moderate performance. Cyclophosphamide and cytarabine take the lowest share, 4.4% and 5.5%, respectively, which means that they have the poorest performance from the considered analysis. The chart shows the relative performance of the drugs based on the SAW score, which reaffirms that the best performing drug remains carfilzomib. Carfilzomib and Zanubrutinib obtained the best scores in the MCDM analysis because their molecular graphs were structurally complex and had high scores in various topological charts. This indicates that structural complexity can increase the potential for drug molecular target interactions. It is also worth mentioning that molecular complexity is a performance correlator in structural models, but not necessarily a higher therapeutic effect, and should be considered together with clinical evidence.
Notably, Carfilzomib exhibited consistently high normalized scores over all topological indices, indicating structural thermostability and flexibility to interact with different molecular targets. Zanubrutinib, although slightly inferior in its overall values, performed well regarding indices associated with branching and information entropy that indicates of moderate, balanced topology known to enhance the binding selectivity and stability. On the other hand, drugs like Cyclophosphamide and Cytarabine had more compact molecular structures with unconnected structure having lower descriptor values leading to weaker orderings in both TOPSIS as well SAW approaches. These results indicate that the structural complexity, as measured by topological indices, is a prominent factor in virtual drug screening prioritization. Although a higher degree of topological complexity does not necessarily lead to better clinical efficacy, it indirectly implies wider possibilities for favorable physicochemical behavior and molecular action indicating the applicability of QSPR and MCDM methods for preclinical drug exploration.
Conclusion
The present study shows that employment of quantitative structure property relationship (QSPR) regression in conjunction with multi criteria decision making (MCDM) methodologies like TOPSIS and Simple Additive Weighting (SAW) can be a good strategy for ranking hematologic cancer drugs. The model integrates set of topological indices that includes Zagreb, Randic, Hyper Zagreb and Atom bond connectivity with significant physico chemical descriptors thus encompassing structural as well as property based features of drug assessment. Our results consistently demonstrated the top positions of Carfilzomib and Zanubrutinib, which is in accordance with their relative molecular complexity, connectivity counts and descriptor values. By contrast, drugs of a less complex structure (such as Cyclophosphamide and Cytarabine) appeared more toward the bottom in the ranking. The proportions in SAW and TOPSIS are relative computational scores, not actual results of clinical efficacies or market shares. The continued dominance of cubic regression over logarithmic models also affirms the capacity of such QSPR methods to discern non linear structure property relationships. Collectively, these results emphasize the potential of QSPR MCDM integration as a decision making assistance in early drug discovery for systematic prioritization of candidates prior to experimental testing. This method could be extended in future analyses to larger drug libraries, and adapted by inclusion of biological or clinical outcome data for better relationship between complexity and therapeutic outcome.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Wilson, R. J. Introduction to graph theory (Pearson Education India, 1979).
García Domenech, R., Gálvez, J., de Julián Ortiz, J. V. & Pogliani, L. Some new trends in chemical graph theory. Chem. Rev. 108(3), 1127–1169 (2008).
Ma, Y. et al. The usefulness of topological indices. Inf. Sci. 606, 143–151 (2022).
Khan, A. R. et al. On degree based operators and topological descriptors of molecular graphs and their applications to QSPR analysis of carbon derivatives. Sci. Rep. 14(1), 21543 (2024).
Abubakar, M. S., Aremu, K. O., Aphane, M., & Amusa, L. B. (2024). A QSPR analysis of physical properties of antituberculosis drugs using neighbourhood degree based topological indices and support vector regression. Heliyon 10(7).
Adnan, M., Bokhary, S. A. U. H., Abbas, G. & Iqbal, T. Degree-based topological indices and QSPR analysis of antituberculosis drugs. J. Chem. 2022(1), 5748626 (2022).
Iqbal, S., Iqbal, H., Tarar, M. A., Hanif, M. F. & Fiidow, O. A. Evaluation of antiarrhythmic drug through QSPR modeling and multi criteria decision analysis. Sci. Rep. 15(1), 29216 (2025).
Qin, H., Hashem, A. F., Hanif, M. F. & Fiidow, O. A. Graph theoretic and machine learning approaches in molecular property prediction of bladder cancer therapeutics. Sci. Rep. 15(1), 28025 (2025).
Huang, L., Alhulwah, K. H., Hanif, M. F., Siddiqui, M. K. & Ikram, A. S. On QSPR analysis of glaucoma drugs using machine learning with XGBoost and regression models. Comput. Biol. Med. 187, 109731 (2025).
Qin, H. et al. On QSPR analysis of pulmonary cancer drugs using python driven topological modeling. Sci. Rep. 15(1), 3965 (2025).
Qin, H. et al. A python approach for prediction of physicochemical properties of anti arrhythmia drugs using topological descriptors. Sci. Rep. 15(1), 1742 (2025).
Wei, J., Hanif, M. F., Mahmood, H., Siddiqui, M. K. & Hussain, M. QSPR analysis of diverse drugs using linear regression for predicting physical properties. Polycyclic Aromat. Compd. 44(7), 4850–4870 (2024).
Gutman, I., and Polansky, O. E. Mathematical concepts in organic chemistry (Springer Science and Business Media, 2012).
Mendez-Bermudez, C. T., Rodrıguez, J. A. & Sigarreta, J. M. Computational and analytical studies of the harmonic index models. MATCH Commun. Math. Comput. Chem 85, 395–426 (2021).
Furtula, B. & Gutman, I. A forgotten topological index. J. Math. Chem. 53(4), 1184–1190 (2015).
Zhao, W., Shanmukha, M. C., Usha, A., Farahani, M. R. & Shilpa, K. C. Computing SS index of certain dendrimers. J. Math. 2021(1), 7483508 (2021).
Estrada, E., Torres, L., Rodriguez, L., and Gutman, I. (1998). An atom bond connectivity index: modelling the enthalpy of formation of alkanes.
Randic, M. Characterization of molecular branching. J. Am. Chem. Soc. 97(23), 6609–6615 (1975).
Vujošević, S., Popivoda, G., Vukićević, ŽK., Furtula, B. & Škrekovski, R. Arithmetic–geometric index and its relations with geometric–arithmetic index. Appl. Math. Comput. 391, 125706 (2021).
Rajasekharaiah, G. V. & Murthy, U. P. Hyper Zagreb indices of graphs and its applications. J. Algebra Combin. Discrete Struct. Appl. 8(1), 9–22 (2021).
Kulli, V. R. Nirmala index. Int. J. Math. Trends Technol. IJMTT 67, 98–156 (2021).
Kantarjian, H. M., Jabbour, E. J., Lipton, J. H., Castagnetti, F. & Brümmendorf, T. H. A review of the therapeutic role of bosutinib in chronic myeloid leukemia. Clin. Lymphoma Myeloma Leuk. 24(5), 285–297 (2024).
Georgoulis, V., Haidich, A. B., Bougioukas, K. I. & Hatzimichael, E. Efficacy and safety of carfilzomib for the treatment of multiple myeloma: An overview of systematic reviews. Crit. Rev. Oncol. Hematol. 180, 103842 (2022).
Quan, X. Y. et al. Revisited cyclophosphamide in the treatment of lupus nephritis. Biomed. Res. Int. 2022(1), 8345737 (2022).
Hunault, M. et al. Intermediate dose cytarabine as postinduction AML therapy. NEJM Evidence 4(7), 2400326 (2025).
Pourmadadi, M. et al. Nanoparticles loaded with Daunorubicin as an advanced tool for cancer therapy. Eur. J. Med. Chem. 258, 115547 (2023).
Fabrizio, V. A. et al. Optimal fludarabine lymphodepletion is associated with improved outcomes after CAR T cell therapy. Blood Adv. 6(7), 1961–1968 (2022).
Brown, J. R. et al. Zanubrutinib or ibrutinib in relapsed or refractory chronic lymphocytic leukemia. N. Engl. J. Med. 388(4), 319–332 (2023).
Jan, M., Sperling, A. S. & Ebert, B. L. Cancer therapies based on targeted protein degradation lessons learned with lenalidomide. Nat. Rev. Clin. Oncol. 18(7), 401–417 (2021).
Dimopoulos, M. A. et al. Daratumumab plus pomalidomide and dexamethasone versus pomalidomide and dexamethasone alone in previously treated multiple myeloma (APOLLO): an open label, randomised, phase 3 trial. Lancet Oncol. 22(6), 801–812 (2021).
Yamamoto, J., Ito, T., Yamaguchi, Y. & Handa, H. Discovery of CRBN as a target of thalidomide: A breakthrough for progress in the development of protein degraders. Chem. Soc. Rev. 51(15), 6234–6250 (2022).
Tam, C. S., Muñoz, J. L., Seymour, J. F. & Opat, S. Zanubrutinib: Past, present, and future. Blood Cancer J. 13(1), 141 (2023).
Sun, D. et al. Unlocking the full potential of memory T cells in adoptive T cell therapy for hematologic malignancies. Int. Immunopharmacol. 144, 113392 (2025).
Zhou, L. et al. Revealing drug target interactions with computational models and algorithms. Molecules 24(9), 1714 (2019).
Tang, X. et al. CDPMF DDA: Contrastive deep probabilistic matrix factorization for drug disease association prediction. BMC Bioinform. 26(1), 5 (2025).
Li, X., Xiang, J., Wu, F. & Li, M. A dual ranking algorithm based on the multiplex network for heterogeneous complex disease analysis. IEEE/ACM Trans. Comput. Biol. Bioinf. 19(4), 1993–2002 (2022).
Zhou, R. et al. NEDD: A network embedding based method for predicting drug disease associations. BMC Bioinform. 21(13), 387 (2020).
Wang, J. et al. SIRT6 protects against lipopolysaccharide induced inflammation in human pulmonary lung microvascular endothelial cells. Inflammation 47(1), 323–332 (2024).
Xianfang, T., Cai, L., Meng, Y., Xu, J., Lu, C., et al. Indicator regularized non negative matrix factorization method based drug repurposing for COVID 19. Front. Immunol. 11. (2021)
Liu, C. et al. An improved anticancer drug response prediction based on an ensemble method integrating matrix completion and ridge regression. Mole. Therapy Nucl. Acids 21, 676–686 (2020).
Sheng, Y., Wang, J., Liu, S. & Jiang, Y. IMN4NPD: An integrated molecular networking workflow for natural product dereplication. Anal. Chem. 96(7), 2990–2997 (2024).
Funding
This study received no funding to support this article.
Author information
Authors and Affiliations
Contributions
Lina Huang:review and editing, MATLAB. Saba Hanif: data analysis, Computation, and verification of calculations. Muhammad Kamran Siddiqui:Investigation, Organization, maple, and funding resources. Muhammad Faisal Hanif: supervised the project and methodology, coordinated it, and wrote the starting adumbrate of the paper. Muhammad Farhan Hanif: Software review and validation. Mohamed Abubakar Fiidow: Formal analysis of experiments, validation, securing funding, and software improvement. The ultimate report of the paper is reviewed and approved by all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, L., Hanif, S., Siddiqui, M.K. et al. Multi criterion decision making analysis of hematologic cancer drugs via topological indices and physicochemical properties. Sci Rep 15, 38707 (2025). https://doi.org/10.1038/s41598-025-23474-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-23474-1
