
Abstract
The article describes the successful synthesis and characterization of two copper complexes using the 6-phenyl-1,3,5-triazine-2,4-diamine ligand (L1). The complexes were characterized using IR, UV-vis, and X-ray spectroscopy, and their crystallographic data were determined using X-ray diffraction. Both complexes had each copper coordinated with four oxygen atoms from four acetate anions, which were bridged between the two copper atoms. The coordination sphere around each copper was in the form of a square pyramid, with an L1 ligands coordinated from nitrogen in the triazine ring in the axial position and four acetate anions coordinated from four oxygen atoms in equatorial positions. However, there were some differences between the two complexes, with complex 1 having no center of symmetry and complex 2 having a center of symmetry. The crystal networks of both complexes exhibited various non-covalent interactions, including intramolecular hydrogen bonding, intermolecular hydrogen bonding, and π-π interaction. Hirshfeld Surfaces Analysis was conducted to obtain more detailed information about these interactions, and the fingerprint results showed that hydrogen-hydrogen interactions contributed above 45% of the surfaces in both complexes. Overall, the study provides valuable insights into the synthesis and characterization of copper complexes using L1 ligands and highlights the importance of non-covalent interactions in crystal networks.
Similar content being viewed by others
Introduction
Copper(II) complexes have long fascinated coordination chemists due to their diverse structural chemistry and wide-ranging applications in catalysis, materials science, and molecular magnetism1,2,3,4,5,6,7,8,9,10. Among these, acetate-bridged copper systems occupy a particularly important niche, serving as archetypal examples of polynuclear coordination compounds while offering valuable insights into fundamental aspects of metal-ligand interactions11,12,13,14,15,16,17,18. The unique versatility of the acetate ligand - its ability to coordinate in various bridging modes while maintaining structural flexibility - makes these systems ideal for studying structure-property relationships in transition metal complexes .
The structural chemistry of copper acetate complexes is remarkably rich, with variations in coordination geometry, nuclearity, and supramolecular architecture arising from relatively minor changes in synthetic conditions or ligand environment19. The acetate ligand can serve as a bridging ligand in two ways. In the μ1,1-bridging mode, it coordinates to one metal ion through one of its oxygen atoms and to another metal ion through the other oxygen atom. Alternatively, in the μ1,2-bridging mode, it coordinates to both metal ions through both of its oxygen atoms. These different bridging modes have profound effects on the resulting complexes’ physical and chemical properties, influencing factors such as metal-metal distances, magnetic exchange interactions, and overall molecular topology20,21,22,23,24,25. Understanding these structural nuances is crucial for the rational design of copper-based materials with tailored properties. Recent advances in crystallographic techniques and computational methods have enabled more detailed investigations of these systems, revealing subtle correlations between ligand design, coordination geometry, and supramolecular organization. This study describes the synthesis and crystallographic characterization of two copper(II) complexes featuring acetate bridging ligands. The complexes were synthesized using a straightforward and efficient approach and characterized through spectroscopic and analytical techniques, including UV-Vis, IR, and X-ray crystallography. By analyzing their structural and spectroscopic properties, we aim to elucidate the influence of acetate bridging modes on coordination geometry and intermolecular interactions. Additionally, surface analysis was performed to explore the complexes’ solid-state interactions.
Experimental
Ligand preparation
6-phenyl-1,3,5-triazine-2,4-diamine ligand (L1)
The synthesis of L1 was carried out in two steps. Initially, a mixture of 4 g of cyanuric chloride and 25 ml of dry THF was combined in a flask and stirred under an argon gas atmosphere. Subsequently, a solution containing 0.84 g of phenyllithium in 25 ml of dry THF was added dropwise over a period of 20 min. The resulting mixture was stirred at room temperature for 3 h. Following this, 25 ml of water and 20 ml of toluene were introduced to the mixture and vigorously stirred until it separated into two distinct layers. The organic phase was separated and subjected to three washes with distilled water. The product was obtained through sedimentation and yielded 0.98 g, corresponding to an efficiency of 43.4%.
For the second step, 0.448 g of the product obtained from the first step was dissolved in 20 ml of acetonitrile at room temperature and placed in an ice bath. Next, 1 g of 28% ammonia aqueous solution was added dropwise and stirred vigorously. The reaction mixture was stirred for 60 min until it reached room temperature, and then refluxed for 4 h at 45 °C. After cooling, the resulting white precipitate was filtered and dried under vacuum (Fig. 1). The product weighed 0.221 g and had an efficiency of 59.09%. The melting point of the product was determined to be 210 °C.

Preparation of ligand L1.
Complex preparation
[Cu(L1)(µ-OAc)2]2 (1)
0.03 g (0.16 mmol) of L1 and 0.06 g (0.32 mmol) of copper (II) acetate mono hydrate were weighed and placed slowly at the bottom of a branched tube. Methanol was then added slowly until the volume reached one centimeter above the side branch of the tube. The tube was sealed and placed in a paraffin bath at a temperature of 60 °C. After two days, green crystals suitable for crystallization were obtained (Fig. 2a). The crystals were washed with acetone and ether, and then dried. The calculated analysis for C26H30Cu2N10O8 was C 42.33%, H 4.10%, N 18.99%, Cu 17.23%. The found analysis was C 42.57%, H 4.21%, N 19.22%, Cu 17.45%.
[Cu(L1)(µ-OAc)2]2.L1. 2CH3OH (2)
0.04 g (0.24 mmol) of L1 and 0.06 g (0.32 mmol) of copper chloride salt were weighed and carefully placed at the bottom of a container. Methanol was then slowly added to a volume of one centimeter above the side branch of the container. The container was sealed and placed in a paraffin bath at a temperature of 60 °C. After three days, suitable green crystals were obtained for crystallization (Fig. 2b). The crystals were washed with acetone and ether, and subsequently dried. The calculated analysis for C46 H48 Cu2 N20 O11 was C 46.66%, H 4.09%, N 23.66%, Cu 10.73%. The found analysis was C 47.24%, H 4.22%, N 24.34%, Cu 11.13%.

Preparation of (a) Complex 1 (b) Complex 2.
X-ray diffraction
The crystallographic data for both complexes are presented in Table 1, which was obtained through the characterization of the complex using X-ray diffraction.
Table 2 provides a compilation of the selective bond lengths for both complexes.
Table 3 displays the bond angles observed in the complexes.
IR spectroscopy
Figure S1 illustrates the IR spectrum of the complexes, which serves as confirmation of their synthesis. Table 4 provides a concise overview of the interpretation and peak positions of significant groups within the complex structures.
UV-Vis
A solution with a concentration of 2 × 10−5 M in methanol solvent was prepared from these complexes, and its UV-Vis spectrum was recorded in the range of 200–800 nm, as depicted in Fig. 3. In the UV-Vis spectrum of complex 1, absorption bands at λmax= 220 nm (ε = 92500 L mol−1 cm−1) and λmax= 245 nm (ε = 90500 L mol−1 cm−1) correspond to intra-ligand transitions. Similarly, in complex 2, the absorption bands at λmax= 219 nm (ε = 84000 L mol−1 cm−1) and λmax= 245 nm (ε = 81000 L mol−1 cm−1) are associated with intra-ligand transitions. The observed absorption bands in the UV region correspond to intra-ligand π→π* transitions, confirming the presence and intact electronic structure of the ligands in the complexes. This supports the successful coordination of the ligands and provides insight into their electronic environments, which is important for understanding the complexes’ chemical and photophysical properties.

UV-Vis Spectrum of (a) Complex 1 (b) Complex 2.
Hirshfeld analysis
To investigate the intermolecular interactions present within the crystals under investigation, we performed Hirshfeld surface analysis using CrystalExplorer2126. This analysis involved the use of Hirshfeld surface analyses, 2D fingerprint plots, and percentage contribution calculations. Specifically, Hirshfeld surfaces were employed in this study, with no properties involved in the calculation command.
Results and discussion
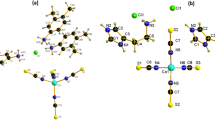
The X-ray diffraction analysis revealed that the complexes were crystallized in the solid state, with complex 1 adopting a monoclinic crystal system with space group P21/c, and complex 2 adopting a triclinic crystal system with space group P-1. The crystal structure of the complexes is depicted in Fig. 4.

ORTEP representation of (a) Complex 1 (b) Complex 2 at the 50% probability level (Methanol and Hydrogens have been omitted for clarity).
Both complexes feature two copper(II) centers, each coordinated by four oxygen atoms from bridging acetate anions, with the acetate anions forming a paddle-wheel-like structure between the metal centers. In complex 1, which lacks a center of symmetry, the nitrogen atom of the neutral ligand 6-phenyl-1,3,5-triazine-4,2-diamine (L1) coordinates to the copper atoms with bond lengths of 2.275(3) Å and 2.240(3) Å, and the Cu···Cu distance is 2.7051(9) Å. The copper atoms in complex 1 are surrounded by four oxygen atoms from acetate anions with Cu-O bond lengths of 2.000(3) Å, 1.952(3) Å, 1.983(3) Å, and 1.956(2) Å. In contrast, complex 2 crystallizes in the centrosymmetric space group P-1, confirming an inversion center. The nitrogen atom of the L1 ligand occupies the axial position relative to the copper atoms, with a Cu-N bond length of 2.2444(16) Å, and the Cu···Cu distance is slightly shorter at 2.6738(4) Å. The four oxygen atoms from acetate anions coordinate each copper in complex 2 with Cu-O bond lengths of 1.9603(15) Å, 1.9701(15) Å, 1.9704(15) Å, and 1.9701(15) Å. The presence of two copper cations, four acetate anions with a negative charge, and neutral L1 ligands in both complexes indicates a +2 oxidation state for the copper centers.
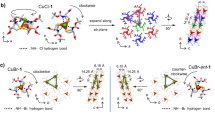
Complex 2 is distinguished from complex 1 by the presence of a free triazine ligand and two methanol molecules in its crystal lattice, which influence the hydrogen-bonding network’s orientation. The supramolecular architecture of complex 2, as shown in Fig. 5, is viewed along the a-axis and features centrosymmetric dimers formed due to the P-1 space group’s inversion symmetry. These dimers are primarily stabilized by strong intermolecular N-H···N hydrogen bonds between the L1 ligands (N14-H14···N2: 3.025(4) Å, 174±2°) and N-H···O hydrogen bonds between the L1 ligand’s amine groups and acetate oxygen atoms (N5-H5···O3: 2.883(4) Å, 150(2)°; N4-H4···O2: 2.968(4) Å, 148(2)°), as detailed in Fig. 5b. These dimers are interconnected through moderate-to-strong π-π interactions between phenyl and triazine rings of the L1 ligands (centroid separation: 3.776 Å), illustrated in Fig. 8, forming a two-dimensional layered network along the a-axis (Fig. 5). The free triazine ligands contribute to the hydrogen-bonding framework, but methanol-mediated hydrogen bonds were excluded due to positional disorder of the methanol molecules, observed during ambient-temperature data collection. Low-temperature crystallographic analysis is underway to resolve their precise positions. This combination of hydrogen bonds and π-π interactions drives the formation of the robust 2D supramolecular network in complex 2, significantly enhancing its structural stability.

Crystal network of (a) Complex 1(b) Complex 2.
Both complexes exhibit four oxygen atoms from four acetate anions coordinating each copper, with the acetate anions serving as a bridge between the two copper atoms. However, complex 1 lacks a center of symmetry, and the nitrogen atom of the neutral ligand 6-phenyl-1,3,5-triazine-4,2-diamine (L1) is attached to the copper atoms with bond lengths of 2.275(3) Å and 2.240(3) Å. In contrast, complex 2 has a center of symmetry, and the nitrogen atom of the neutral ligand 6-phenyl-1,3,5-triazine-4,2-diamine (L1) is in the axial position to the copper atoms, with a bond length of 2.2444(16) Å. In complex 1, the distance between the two metal centers is 2.7051(9) Å, with each surrounded by four oxygen atoms from four acetate anions. The bond length of copper 1 with oxygens is 2.000(3) Å, 1.952(3) Å, 1.983(3) Å, and 1.956(2) Å. The presence of two copper cations and four acetate anions with a negative charge, along with the neutrality of L1 ligands, indicates that the oxidation number of copper centers is +2. In contrast, complex 2 has a distance of 2.6738(4) Å between the two metal centers. Four oxygen atoms from four acetate anions are connected to the metal ion, with bond lengths of 1.9603(15) Å, 1.9701(15) Å, 1.9704(15) Å, and 1.9701(15) Å. Complex 2 differs from complex 1 due to the presence of a free triazine ligand and two methanol molecules in the crystal network. These additional components alter the orientation of the hydrogen bonds within the crystal network. It is important to note that the crystal information of complex 2 was collected at normal temperature, which means that the precise direction and location of the methanol molecules are not known. The figures provide an approximate representation of their location. This uncertainty arises from the strong vibration of the free methanol molecules within the crystal lattice and the possibility of some of them being displaced during the irradiation process. To address this issue, the compound will be analyzed at low temperature, which will enable the collection of crystallographic data for the compound at low temperature. As previously mentioned, the presence of two copper cations and four acetate anions with a negative charge, along with the neutrality of L1 ligands, indicates that the oxidation number of copper centers is +2.
In both complexes, the coordination sphere around each copper atom is arranged in a square pyramid configuration, with an L1 ligand coordinated from the nitrogen in the triazine ring in the axial position, and four acetate anions coordinated from four oxygen atoms in equatorial positions. The bond length between the L1 ligand and copper is longer than the other bonds, indicating a strong Jahn-Teller deviation in the direction of these ligands. The Jahn-Teller deviation is observed in copper (2+) octahedral complexes due to the d9 electron configuration. Figure 6 displays the coordination sphere around copper.

Coordination sphere of copper in (a) Complex 1 (b) Complex 2.
The crystal networks of both complexes exhibit a variety of non-covalent interactions, including intramolecular hydrogen bonding, intermolecular hydrogen bonding, and π-π interactions. Table 5 provides the relevant data regarding the hydrogen bonds, while Fig. 7 visually illustrates these interactions.
Both complexes exhibit well-defined hydrogen-bonding networks with D···A distances of 2.840(4)–3.025(4) Å and D-H···A angles between 148(2)–174(2)°, as summarized in Table 5, falling within conventional ranges for such interactions (2.5–3.2 Å for D···A, >120° for angles). The standard deviations (σ = 0.004–0.04 Å for distances) indicate measurable positional flexibility, particularly in N-H···O bonds. In complex 1, the supramolecular architecture is primarily driven by intramolecular N-H···O hydrogen bonds between the N-H groups of the 6-phenyl-1,3,5-triazine-4,2-diamine (L1) ligand and coordinated acetate oxygen atoms (N10-H4···O6: 2.948(4) Å, 156(2)°; N9-H7···O4: 2.972(4) Å, 159(2)°). These are complemented by intermolecular N-H···N interactions (N5-H3···O7: 2.840(4) Å, 156(2)°), which generate a one-dimensional polymer chain. This chain further connects four dimeric units into a two-dimensional hexagonal pattern, as illustrated in Fig. 7a. The phenyl rings of the L1 ligands are positioned with a centroid separation of 4.469 Å, which exceeds the typical threshold for significant π-π stacking (<4.0 Å), indicating that π-π interactions contribute negligibly to the stabilization of the supramolecular network. Instead, the robust hydrogen-bonding framework is the dominant factor in the structural stability of complex 1’s two-dimensional architecture.

Representation of intermolecular hydrogen bonds in (a) Complex 1 (b) Complex 2.
Complex 2 displays a more intricate scheme featuring:
-
• N-H···O bonds to acetate (N5-H5···O3: 2.883(4) Å, 150(2)°; N4-H4···O2: 2.968(4) Å, 148(2)°).
-
• A nearly linear N-H···N interaction (N14-H14···N2: 3.025(4) Å, 174±2°), the most geometrically constrained in the system.
-
• A moderate-to-strong π-π interaction (phenyl-triazine: 3.776 Å, Fig. 8) within the 3.3-3.8 Å stacking range.
Methanol-mediated hydrogen bonds were excluded due to positional disorder. The supramolecular architectures are primarily directed by these hydrogen bonds, with π-stacking providing secondary stabilization in complex 2. Complex (2) crystallizes in the centrosymmetric space group P-1, confirming the presence of an inversion center. Analysis of the crystal packing reveals the formation of centrosymmetric dimers, primarily stabilized by intermolecular N-H···N hydrogen bonds between the nitrogen atoms of the neutral 6-phenyl-1,3,5-triazine-4,2-diamine (L1) ligands (N14-H14···N2: 3.025(4) Å, 174±2°). These dimers are further reinforced by N-H···O hydrogen bonds between the L1 ligand’s amine groups and the oxygen atoms of acetate anions (N5-H5···O3: 2.883(4) Å, 150(2)°; N4-H4···O2: 2.968(4) Å, 148(2)°). A moderate-to-strong π-π interaction between phenyl and triazine rings (centroid separation: 3.776 Å) provides secondary stabilization, facilitating the connectivity of these dimers into a two-dimensional layered supramolecular network. The presence of free triazine ligands and methanol molecules in the crystal lattice was noted, but methanol-mediated hydrogen bonds were excluded due to positional disorder of the methanol molecules, as data were collected at ambient temperature. Ongoing low-temperature crystallographic analysis will provide further clarity on their precise positions. This hydrogen-bonding framework, complemented by π-π interactions, significantly contributes to the stability and orientation of the supramolecular architecture of complex 2, as illustrated in Figs. 7b and 8.

Intramolecular hydrogen bonds and π-π interaction in complex 2.
To gain a comprehensive and detailed understanding of the interactions and surfaces, we conducted Hirshfeld Surfaces Analysis. Figure 9 showcases the results of the Hirshfeld surfaces analysis for the complexes. In this analysis, the dnorm property was employed to highlight specific areas. Blue regions indicate that the contact distance between atoms inside and outside the surface is greater than the sum of their respective van der Waals radii. White areas represent a contact distance equal to the sum of the van der Waals radii, while small amounts of red areas indicate a contact distance between atoms inside and outside the surface that is less than the sum of their respective van der Waals radii27. The shape index map reveals complementary red and blue triangles on the Hirshfeld surfaces of the phenyl and triazine rings, confirming π-π stacking, while the curvedness map shows flat regions (low curvedness values) corresponding to the stacking interaction at 3.776 Å (Fig. 9).

Hirshfeld surfaces analysis of (a) complex 1 (b) complex 2.
In order to provide a more detailed understanding of the various interaction contributions in these complexes, we have included a comprehensive breakdown of the percentage contributions by atoms in the surface area in Figs. 10 and 11. The H(i)···H(e) interactions make a significant contribution to the surface, accounting for 49% in complex 1 and 46.6% in complex 2. The sum of section b in Figs. 10 and 11 represents the hydrogen bond contribution, which is 26% and 25.4% in complex 1 and 2, respectively. In the last part of Figs. 10 and 11, we have presented some of the significant carbon interactions in the complexes.

Surface contributions percentage of complex 1: (a) H-H interactions, (b) hydrogen bonds (c) carbon interactions (Include Reciprocal Contacts).

Surface contributions percentage of complex 2: (a) H-H interactions, (b) hydrogen bonds (c) carbon interactions (Include Reciprocal Contacts).
Conclusion
Two copper complexes of the ligand L1 (6-phenyl-1,3,5-triazine-2,4-diamine) were successfully synthesized and thoroughly characterized by spectroscopic methods (IR, UV-Vis) and single-crystal X-ray diffraction. The structural analysis revealed that both complexes feature a square pyramidal coordination geometry around copper, with coordination from a nitrogen atom of the L1 ligand and oxygen atoms from acetate anions. Complex 1 crystallizes in the monoclinic P2₁/c space group without a center of symmetry, while complex 2 crystallizes in the triclinic P-1 space group with a center of symmetry. Both complexes demonstrate significant non-covalent interactions, including hydrogen bonding and π–π stacking, which were further elucidated by Hirshfeld Surface Analysis. These findings contribute to the understanding of the coordination behavior of triazine-based ligands and provide insight into the design of copper complexes with specific structural and interaction features.
Data availability
Crystallographic data for the structure reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication CCDC-2452777 for C26H30Cu2N10O8 (1) and CCDC-2289690 for C44H48Cu2N20O8 (2). Copies of the data can be obtained by request from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (Fax: +441223336033, eMail: deposit@ccdc.cam.ac.uk.
References
Beaudelot, J. et al. Photoactive copper complexes: properties and applications. Chem. Rev. 122(22), 16365–16609. https://doi.org/10.1021/acs.chemrev.2c00033 (2022).
Aflak, N. et al. Recent advances in Copper-Based solid heterogeneous catalysts for Azide–Alkyne cycloaddition reactions. Int. J. Mol. Sci. 23(4), 2383. https://doi.org/10.3390/ijms23042383 (2022).
Chang, F. et al. Copper-Based catalysts for electrochemical carbon dioxide reduction to multicarbon products. Electrochem. Energy Reviews. 5(3), 4. https://doi.org/10.1007/s41918-022-00139-5 (2022).
Tripathi, K. et al. Crafting copper complexes of variable nuclearity and coordination geometry through solvent tailoring: unveiling novel Structure, magnetic insight and computational marvels. J. Mol. Struct. 1321, 139817. https://doi.org/10.1016/j.molstruc.2024.139817 (2025).
Jäschke, A. et al. Structure and magnetic properties of some Copper(II) complexes supported by pendant Calix[4]Arene ligands. Eur. J. Inorg. Chem. 27(3). https://doi.org/10.1002/ejic.202300564 (2024).
Fomenko, I. S. et al. Copper(II) complexes with BIAN-Type ligands: synthesis and catalytic activity in oxidation of hydrocarbons and alcohols. Inorganica Chim. Acta. 565, 121990. https://doi.org/10.1016/j.ica.2024.121990 (2024).
Weng, Z. et al. Active sites of Copper-Complex catalytic materials for electrochemical carbon dioxide reduction. Nat. Commun. 9(1), 415. https://doi.org/10.1038/s41467-018-02819-7 (2018).
Castro, I. et al. Dinuclear Copper(II) complexes as testing ground for molecular magnetism theory. Polyhedron 169, 66–77. https://doi.org/10.1016/j.poly.2019.04.023 (2019).
Kumar, S., Sharma, R. P., Venugopalan, P. & Aree, T. Effect of differently substituted methoxybenzoates on the supramolecular assemblies of three [Cu(N-Hyden)2](o-/m-/p-Methoxybenzoate)2 complexes: Synthesis, spectroscopic characterization and single crystal structure determination. Polyhedron 133, 213–221. https://doi.org/10.1016/j.poly.2017.05.021 (2017).
Chauhan, C. et al. Structural studies and fabrication of mononuclear ionic Copper(II) complexes induced Polyvinylpyrrolidone films for optical band gap tunning. Inorg. Chem. Commun. 173, 113821. https://doi.org/10.1016/j.inoche.2024.113821 (2025).
Li, J. X., Du, Z. X., Zhang, L. L., Liu, D. L. & Pan, Q. Y. Doubly mononuclear cocrystal and Oxalato-Bridged binuclear copper compounds containing flexible 2-((3,5,6-Trichloropyridin-2-Yl)Oxy)Acetate tectons: Synthesis, crystal analysis and magnetic properties. Inorganica Chim. Acta. 512, 119890. https://doi.org/10.1016/j.ica.2020.119890 (2020).
Alter, M. et al. Photochemical origin of the darkening of copper acetate and resinate pigments in historical paintings. Inorg. Chem. 58(19), 13115–13128. https://doi.org/10.1021/acs.inorgchem.9b02007 (2019).
Ikram, M. et al. Spectral, Hirshfeld surface analysis and biological evaluation of a schiff base Copper(II) complex: towards a Copper(II) based human Anti-Glioblastoma agent. J. Mol. Struct. 1278, 134960. https://doi.org/10.1016/j.molstruc.2023.134960 (2023).
Sarakinou, K. M., Banti, C. N., Hatzidimitriou, A. G. & Hadjikakou, S. K. Utilization of metal complexes formed by Copper(II) acetate or Nitrate, for the Urea assay. Inorganica Chim. Acta. 517, 120203. https://doi.org/10.1016/j.ica.2020.120203 (2021).
Mujahid, M. et al. Structural and spectroscopic study of new Copper(II) and Zinc(II) complexes of coumarin oxyacetate ligands and determination of their antimicrobial activity. Molecules 28(11), 4560. https://doi.org/10.3390/molecules28114560 (2023).
Seguel, G. V., Rivas, B. L. & Paredes, C. Study of the interactions between copper(II) acetate monohydrate and orotic acid and orotate ligands. J. Chil. Chem. Soc. 55(3), 355–358. https://doi.org/10.4067/S0717-97072010000300018. (2010).
Kumar, U., Thomas, J. & Thirupathi, N. Factors dictating the Nuclearity/Aggregation and acetate coordination modes of Lutidine-Coordinated Zinc(II) acetate complexes. Inorg. Chem. 49(1), 62–72. https://doi.org/10.1021/ic901100z (2010).
Levchenkov, S. I. et al. Structure of the planar Acetate-Bridged binuclear Copper(II) complex based on 1,3-Bis(3-Formyl-5-Tert-Butylsalicylidenimino)Propanol-2. Russ. J. Coord. Chem. 43(10), 630–634. https://doi.org/10.1134/S1070328417100049 (2017).
Abedi, M. et al. Structure, magnetic properties and DFT study of a dinuclear Copper(II) macrocyclic complex containing acetate groups. Inorganica Chim. Acta. 520, 120310. https://doi.org/10.1016/j.ica.2021.120310 (2021).
Huang, P. J. & Miyasaka, H. Canting angle dependence of Single-Chain magnet behaviour in Chirality-Introduced antiferromagnetic chains of Acetate-Bridged Manganese(<scp>iii</scp>) Salen-Type complexes. Dalton Trans. 49(46), 16970–16978. https://doi.org/10.1039/D0DT03615C (2020).
Sheng, Y. et al. Structures, and magnetic properties of Acetate-Bridged lanthanide complexes based on a tripodal oxygen ligand. Front. Chem. 10. https://doi.org/10.3389/fchem.2022.1021358 (2022).
Dominelli, B. et al. Dinuclear zwitterionic Silver(<scp>i</scp>) and Gold(<scp>i</scp>) complexes bearing 2,2-Acetate-Bridged Bisimidazolylidene ligands. Dalton Trans. 48(37), 14036–14043. https://doi.org/10.1039/C9DT03035B (2019).
Cherkashina, N. V. et al. )–Pd(II) complex: Synthesis, Structure, and formation of bimetallic PtPd2 nanoparticles. Russ. J. Coord. Chem. 45(4), 253–265. https://doi.org/10.1134/S107032841904002X (2019). The First Heterometallic Acetate-Bridged Pt(II.
Zhang, J., Dou, L., Hu, Z. F. & Dong, W. K. The investigation of two Bis(Phenoxo)-Acetate-Bridged binuclear Nickel(II) complexes driven by different alcohol solvents. J. Coord. Chem. 76(5–6), 596–611. https://doi.org/10.1080/00958972.2023.2201656 (2023).
Mondal, I., Chatterjee, S. & Chattopadhyay, S. An acetate bridged centrosymmetric Zinc(II) complex with a tetradentate reduced schiff base ligand: Synthesis, characterization and ability to sense nitroaromatics by turn off fluorescence response. Polyhedron 190, 114735. https://doi.org/10.1016/j.poly.2020.114735 (2020).
Spackman, P. R. et al. CrystalExplorer : A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 54(3), 1006–1011. https://doi.org/10.1107/S1600576721002910 (2021).
McKinnon, J. J., Jayatilaka, D. & Spackman, M. A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 37, 3814. https://doi.org/10.1039/b704980c (2007).
Acknowledgements
We thank the University of Qom for their assistance with this project.
Author information
Authors and Affiliations
Contributions
Babak Mirtamizdoust: Conceptualization, Synthesis, Investigation, Writing—original draftAmirhossein Karamad: characterization, Writing the article, Supervision, Writing—review & editingFaeze Mojtabazade: Student, SynthesisHassan Hosseini-Monfared : SupervisorRahman Bikas: Supervisor, Investigation, Writing—review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mirtamizdoust, B., Karamad, A., Mojtabazade, F. et al. Synthesis, characterization, and hydrogen bonding analysis of copper complexes with acetate bridging: insights into interactions in crystal networks. Sci Rep 15, 40084 (2025). https://doi.org/10.1038/s41598-025-23955-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-23955-3
