
Abstract
The contraction-relaxation balance of trabecular meshwork (TM) cells is a critical physiological mechanism regulating aqueous humor outflow resistance. Both hypercontractility and impaired relaxation of TM cells can obstruct aqueous humor outflow. Such pathological changes not only directly compromise outflow function but may also reduce the clinical efficacy of ocular hypotensive therapies. However, the molecular regulatory mechanisms underlying TM cell contractility remain incompletely understood. Based on single-cell RNA sequencing data previously generated by our group, our analyses revealed abnormal expression patterns of tropomyosin family members (TPMs) in major TM cell subpopulations in primates with primary open-angle glaucoma (POAG). This study investigated the effects of TPM2 in TM cells on contractile-related molecular expression and biomechanical properties. The results showed that modulating TPM2 expression levels significantly affected the expression of Actin Alpha Cardiac Muscle 1 (ACTC1), Troponin T2, Cardiac Type (TNNT2), alpha-smooth muscle actin (α-SMA), and F-actin in human TM cells (HTMCs), and simultaneously altered the cellular elastic modulus. Thus, TPM2 may regulate aqueous humor outflow by modulating TM cell contractility, altering cytoskeletal architecture, and inducing changes in biomechanical properties.
Similar content being viewed by others

Introduction
The trabecular meshwork (TM), the primary aqueous humor outflow pathway in the eye, maintains intraocular pressure (IOP) homeostasis through active contractile regulation, which modulates outflow resistance by altering TM pore architecture1. Both contractile dysfunction in TM cells and cytoskeletal abnormalities can pathologically increase outflow resistance and are key aetiological factors in open-angle glaucoma2,3,4. However, the mechanisms underlying TM-mediated outflow dysfunction remain incompletely understood. Investigation of the regulatory mechanisms of TM cell contractility is essential to elucidate the pathogenesis of aqueous humor outflow dysfunction in this disease.
Our previous single-cell RNA sequencing (scRNA-seq) studies revealed that TM cells are predominantly composed of fibroblast and smooth muscle cell subpopulations with active contractile regulatory capacity5. In cynomolgus monkeys of primary open-angle glaucoma (POAG), tropomyosin family members (TPMs) exhibited significantly elevated expression in TM cells and may regulate contractility via the TPM-Ca²⁺-TnT/actin signaling axis. TPMs encode tropomyosins, which modulate contractility in muscle cells and cytoskeletal organization in non-muscle cells, thereby influencing their contractility6,7,8,9,10. Among TPMs, TPM2 plays a key regulatory role in the contraction and deformability of myofilaments11,12,13,14. These findings indicate that TPM2 plays a critical role in orchestrating TM contractile function.
Cytoskeletal remodeling modulates cellular migration and deformability. Biomechanical properties directly reflect cellular deformability and determine the extent of shape change during contractile activity15. In TM cells, deformability governs the dynamic adjustment of aqueous humor outflow pore dimensions, thereby critically influencing outflow function16. Recent studies on TM biomechanics using experimental and theoretical biomechanical approaches have revealed altered biomechanical properties in TM tissues and cells from glaucoma patients or high IOP animal models compared to normal controls, and these alterations correlate with impaired aqueous humor outflow17,18,19,20. Consequently, biomechanical abnormalities may both exacerbate contractile dysfunction of TM cells and contribute to the suboptimal IOP lowering efficacy of pilocarpine in the clinical setting.
This study investigated the regulatory role of TPM2 in TM contractility by modulating TPM2 expression levels in human trabecular meshwork cells (HTMCs). In addition, the study also simulated a high IOP environment through in vitro experiments to observe the effect of high pressure on TPM2 expression in TM cells. These findings should provide a theoretical basis for the development of novel IOP-lowering targeted therapies and advance therapeutic strategies for the treatment of glaucoma.
Methods
Cell culture
The primary human-derived trabecular meshwork cells (HTMCs) were supplied by Professor Xuanchu Duan’s group at the Aier Eye Institute of Central South University.
The primary HTMCs were cultured in Dulbecco’s modified Eagle’s medium/nutrient mixture F-12 (DMEM/F12) (Gibco, USA) containing 15% fetal bovine serum (FBS) (Absin, CHN), 1% penicillin-streptomycin (DMEM/F12) (Gibco, USA) and placed in a 37 °C, 5% CO₂ constant temperature and humidity incubator (Thermo Scientific) for routine culture. At 80–90% cell confluence, 1:3 passage21.
Lentiviral transduction
Lentiviruses for TPM2 overexpression, knockdown, and corresponding controls (dual-labeled with GFP and puromycin resistance) were constructed by Shanghai GENE Company (lentiviral vector sequences are listed in Table 1). Optimal transduction conditions were determined in preliminary experiments: HTMCs were transduced at multiplicities of infection (MOIs) of 10, 50, and 100, comparing standard transduction (Group M) versus enhancer-assisted protocols (Groups A and P). Cell viability and transduction efficiency were assessed by fluorescence microscopy at 72 h post-transduction. Based on these results, HTMCs were formally transduced under selected MOI and enhancer conditions. Culture medium was replaced 24 h post-transduction, followed by 4-day selection with 2.5 µg/mL puromycin initiated at 72 h to establish stable cell lines.
Stable transduction systems were prepared in the same way for the empty vector virus group, TPM2 knockdown group, and the TPM2 overexpression group.
Quantitative polymerase chain reaction (RT-qPCR)
Cells were harvested, and RNA was extracted using RNA Rapid Extraction Kit (ESScience, CHN), then PCR reaction was performed using Reverse Transcription Kit and RNA Amplification Kit (Sangon Biotech, CHN). Reaction procedures: pre-denaturation at 95 °C for 30 s; denaturation at 95 °C for 10 s; annealing at 60 °C for 30 s; extension at 72 °C for 30 s; repeat for 40 cycles, with GAPDH as the housekeeping gene and 2−ΔΔCT as the relative expression intensity (primer sequences are listed in Table 2).
Western blotting (WB)
Total protein was extracted using RIPA lysis buffer (Solarbio, CHN). Protein concentration was measured by BCA assay. Equal amounts of protein were separated by 10% SDS-PAGE, transferred to PVDF membranes, and blocked with 5% skimmed milk for 2 h at room temperature. Membranes were incubated overnight at 4 °C with the following primary antibodies: rabbit anti-TPM2 (1:2000), rabbit anti-α-SMA (1:5000), mouse anti-GAPDH (1:50,000), mouse anti-ACTC1 (1:5000), and mouse anti-F-actin (1:1000). After washing, membranes were incubated with HRP-conjugated goat anti-rabbit (1:10,000) or goat anti-mouse (1:5000) secondary antibodies for 1 h at room temperature. Proteins were visualized using an ECL system. For reprobing, membranes were stripped with rapid stripping buffer (Epizyme, CHN) for 20 min, re-blocked, and re-probed following the same procedure. GAPDH served as the loading control. Band intensities were quantified by grayscale analysis using ImageJ, and relative expression levels of target proteins were calculated. Experiments were performed in triplicate (antibody sources and catalog numbers are listed in Table 3).
Immunofluorescence
Cell slides were fixed with 4% PFA for 20 min, permeabilized with 0.5% Triton X-100 for 20 min, and blocked with 10% goat serum for 30 min. The slides were then incubated overnight at 4 °C with the following primary antibodies: rabbit anti-human TPM2 (1:250), rabbit anti-human α-SMA (1:1000), rabbit anti-human TNNT2 (1:250), mouse anti-human ACTC1 (1:250), and mouse anti-human F-actin (1:200). After washing, the slides were incubated with Coralite594-conjugated goat anti-rabbit (1:300) or goat anti-mouse (1:300) secondary antibody for 1 h at room temperature, protected from light. Finally, nuclei were stained with 10 µg/mL DAPI (Solarbio, CHN). Images were acquired using an inverted fluorescence microscope (Carl Zeiss AG, GER). Fluorescence intensity was analyzed using ImageJ software. All experiments were repeated three times.
Modulus of elasticity measurements
Cell Young’s modulus was determined using an atomic force microscope (AFM; JPK NanoWizard 4, Bruker, GER). Prior to measurements, the spring constant (k) of the AFM cantilever (CSG01, NanoAndMore GmbH) was calibrated in PBS via the thermal noise method. All measurements were performed in contact mode force spectroscopy. The setpoint was adjusted to limit the indentation depth to less than 1/10 of the sample height. Cells were plated on the AFM stage and visualized using an integrated inverted optical microscope. The cantilever was positioned above individual cells, and force-distance curves were acquired at a approach rate of 2 μm/s. For each experimental group, a minimum of 50 cells were measured. Young’s modulus was extracted by applying the Hertzian model to the indentation segment of the force-distance curves using the JPK SPM Data Processing software.

Effect of TPM2 on protein expression of contractile regulatory proteins in TM cells. (A-D) Overexpression of TPM2. (E-H) Knockdown of TPM2. *p < 0.05, **p < 0.01, ***p < 0.001.
The specific formula is E = 3 × (1-ν²)×Y/2×R. Where E is Young’s modulus, ν is Poisson’s ratio, Y is cell hardness and R is the radius of curvature of the probe.
Cytosolic pressurization
Cytosolic pressurization was applied using a cell compressor (Surforce Tech, CHN). Cell slides were placed into the device wells, which were then filled with basal medium. The compressor cover was secured by tightening the screws. A specific pressure value and duration were set via the control panel to initiate the pressurization process. Real-time pressure curves were monitored throughout the experiment (Fig. 1).
Statistical methods
All data are presented as mean ± standard deviation and were analyzed using GraphPad Prism 10 software. Unpaired t-tests were employed for comparisons between two groups. For cellular Young’s modulus data, Cohen’s d values were calculated using Cohen’s formula along with their 95% confidence intervals to quantify the effect size of intergroup differences. Comparisons among multiple groups were performed using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. A p-value of less than 0.05 was considered statistically significant.
Results
Establishment of a stable transfection system for TPM2 overexpression and knockdown in cells
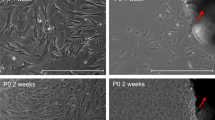
The transfection efficiency of the lentivirus was observed under an inverted fluorescence microscope (Carl Zeiss AG, GER) 72 h post-transfection. The optimal transfection conditions for HTMCs were determined to be a multiplicity of infection (MOI) of 10, using HitransG A as the transduction enhancer (Fig. 2).

Screening of lentiviral transfection conditions. (M) Conventional transfection group; (A) Co-transfection reagent group A; (P) Co-transfection reagent group P. A red circle identifies the selected optimal transfection condition based on transduction efficiency and cell viability. Scale bar = 200 μm.
Following transfection for 72 h under these optimal conditions, the cells were selected with 2.5 µg/mL puromycin (Solarbio, UK) for 4 days, with the medium changed daily, to establish stably transduced cell lines. Transfection efficiency was subsequently verified by RT-qPCR and Western blotting (WB).
The results showed that the TPM2 gene and protein expression levels were significantly higher in the OE-TPM2 group compared to the OE-NC group (p < 0.01) (Fig. 3A-C). The knockdown efficiency of three TPM2 knockdown lentiviral stable transduction systems was validated by WB and RT-qPCR. It was found that the knockdown efficiency in the shTPM2-1 (p < 0.05) and shTPM2-2 (p < 0.01) groups exceeded 50% compared to the shNC group (Fig. 3D-F). Among these, the shTPM2-2 group exhibited the most significant knockdown efficiency and was selected for subsequent TPM2 knockdown experiments.

Validation of lentiviral transfection efficiency. (A–B) WB analysis of TPM2 overexpression efficiency; (C) RT‒qPCR analysis of TPM2 overexpression; (D–E) WB analysis of TPM2 knockdown efficiency; (F) RT‒qPCR analysis of TPM2 knockdown. *p < 0.05, **p < 0.01.
Effect of overexpression of TPM2 in HTMC on proteins involved in contraction
After preparing the stable transfection system in the OE-NC and OE-TPM2 groups, we compared and analyzed the changes in the expression of contraction-related proteins and mRNAs in the two groups.
The effect of TPM2 on the expression levels of contractile regulatory proteins in myometrial-like TM cells was assessed by immunofluorescence (IF). The results demonstrated that TPM2 overexpression significantly increased the protein expression levels of ACTC1 (p < 0.05), TNNT2 (p < 0.01), and α-SMA (p < 0.05) in HTMCs (Fig. 4). Conversely, TPM2 knockdown markedly reduced the protein expression levels of ACTC1 (p < 0.05), TNNT2 (p < 0.001), and α-SMA (p < 0.05) (Fig. 5).

Effect of TPM2 overexpression on the expression of contractile regulatory proteins in TM cells. (A–C) Representative immunofluorescence images. (D–F) Quantification of average fluorescence intensity. *p < 0.05, **p < 0.01. Scale bar = 100 μm.

Effect of TPM2 knockdown on the expression of contractile regulatory proteins in TM cells. (A–C) Representative immunofluorescence images. (D–F) Quantification of average fluorescence intensity. *p < 0.05, ***p < 0.001. Scale bar = 100 μm.
RT-qPCR analysis demonstrated that TPM2 overexpression significantly upregulated the mRNA expression of ACTC1 (p < 0.01), TNNT2 (p < 0.05), and α-SMA (p < 0.01) in HTMCs, whereas its knockdown markedly downregulated these genes (ACTC1: p < 0.0001; TNNT2 and α-SMA: p < 0.01) (Fig. 6). Consistent with these findings, Western blot analysis showed that TPM2 overexpression increased the protein levels of ACTC1 (p < 0.05), TNNT2 (p < 0.001), and α-SMA (p < 0.01) (Fig. 7A-D). Conversely, TPM2 knockdown suppressed their expression (TNNT2 and α-SMA: p < 0.05; ACTC1: p < 0.01) (Fig. 7E-H).

Effect of TPM2 on mRNA expression of contractile regulatory molecules in TM cellss. (A-C) Overexpression of TPM2. (D-F) Knockdown of TPM2. *p < 0.05, **p < 0.01, ****p < 0.0001.

Cell compression experimental setup. (A) Schematic diagram of the internal structure of the cell compressor. (B) Sealed state of the cell compressor after connection to the air pump and controller. (C) Controller interface for setting pressure and duration, with real-time pressure monitoring.
Western blot analysis confirmed that TPM2 overexpression in HTMCs resulted in a significant increase in F-actin expression (p < 0.05) and a notable decrease in G-actin expression (p < 0.01), leading to an elevated F-actin/G-actin ratio. This suggests that TPM2 overexpression promotes cytoskeletal polymerization in HTMCs (Fig. 8A–C). Conversely, TPM2 knockdown led to reduced F-actin expression (p < 0.05) and a significant increase in G-actin levels (p < 0.01), resulting in a decreased F-actin/G-actin ratio, which indicates enhanced cytoskeletal depolymerization following TPM2 knockdown (Fig. 8D–F).

WB analysis of TPM2 expression on TM cytoskeletal regulatory proteins. (A-C) Overexpression of TPM2. (D-F) Knockdown of TPM2. *p < 0.05, **p < 0.01.
Effect of overexpressing TPM2 in HTMC on Biomechanical properties
The AFM was corrected for the coefficient of elasticity with the set point set at 1nN and the zlength set at 3 μm. The Young’s modulus was measured by AFM for normal HTMC and was approximately 410.91 ± 298.86 Pa(Fig. 9).

AFM measurement of elastic modulus in HTMCs. (A) Schematic of the real-time measurement. (B) Force-displacement curve of an HTMC. (C) Statistical analysis of the elastic modulus in normal HTMCs. Scale bar = 100 μm.
AFM measurements on the stable HTMC transduction system revealed that the Young’s modulus of the OE-TPM2 group was approximately 1210.33 ± 494.12 Pa, while that of the OE-NC group was 411.43 ± 142.90 Pa. The Young’s modulus of the OE-TPM2 group was significantly higher than that of the OE-NC group, with a statistically significant difference (p < 0.0001; Cohen’s d = −2.20) (Fig. 10).

Effect of TPM2 overexpression on the elastic modulus of HTMCs. (A) Representative real-time measurement diagram. (B) Statistical analysis of HTMC elastic modulus. (C-D) Representative force-displacement curves. ****p < 0.0001. Scale bar = 100 μm.
Atomic force microscopy (AFM) measurements showed that the Young’s modulus of the shTPM2 group was approximately 743.26 ± 318.67 Pa, compared to 937.06 ± 412.79 Pa in the shNC group. The Young’s modulus of the shTPM2 group was significantly lower than that of the shNC group, and this difference was statistically significant (p < 0.05; Cohen’s d = 0.52) (Fig. 11).

Effect of TPM2 knockdown on the elastic modulus of HTMCs. (A) Representative real-time measurement. (B) Statistical analysis of the elastic modulus. (C-D) Representative force-displacement curves. *p < 0.05. Scale bar = 100 μm.
Effect of high pressure on TPM2 in HTMCs
Cell compression experiments were conducted on HTMCs from the OE-NC and OE-TPM2 groups at a pressure of 40 mmHg for 4 h. The compression significantly reduced the cell density of OE-TPM2 HTMCs, and the remaining cells exhibited irregular morphology (Fig. 12 A). Immunofluorescence analysis revealed that the nuclear fluorescent area fraction was significantly lower in the OE-TPM2 group compared to the OE-NC group (P < 0.001) (Fig. 12B). Similarly, the TPM2 fluorescent area fraction was significantly reduced in the OE-TPM2 group (P < 0.0001) (Fig. 12 C), while the average fluorescence intensity of TPM2 was considerably higher (P < 0.01) (Fig. 12D).

Analysis of HTMCs in the OE-NC and OE-TPM2 groups following high-pressure treatment. (A) Representative immunofluorescence images. (B, C) Quantification of the fluorescence-positive area. (D) Quantification of mean fluorescence intensity. Data are presented as mean ± SEM (n = 3). **P < 0.01, ****P < 0.0001. Scale bar = 100 μm.
Normal HTMCs were subjected to high-pressure treatment, with the pressure set at 40 mmHg, and the duration of action was 4 h and 8 h, respectively. The results showed that the expression of TPM2 decreased in HTMC after high-pressure treatment, and the decrease in TPM2 expression was more significant in the high-pressure group for 8 h (P<0.001) than in the high-pressure group for 4 h (P<0.01) (Fig. 13).

Expression of TPM2 in HTMCs following high-pressure treatment. (A) Representative immunofluorescence images. (B) Quantification of TPM2 mean fluorescence intensity. Data are presented as mean ± SEM (n = 3). **P < 0.01, ***P < 0.001. Scale bar = 100 μm.
Discussion
Glaucoma, the leading cause of irreversible blindness worldwide, is primarily caused by irreversible damage to the optic nerve due to abnormally elevated IOP. IOP regulation depends on the balance between aqueous humor production and outflow. As the primary pathway for aqueous humor drainage, the TM plays a critical role - any dysfunction or structural abnormality in the TM cells can increase outflow resistance and impair aqueous humor drainage. This pathological mechanism is the key etiology of open-angle glaucoma.
Building on our previous scRNA-seq study of TM cells in naturally occurring primate POAG, we demonstrated that TM cell subtypes are predominantly composed of fibrotic and smooth muscle-like populations. This finding indicates that TM cells exhibit dual contractile properties, characteristic of both muscle and non-muscle cells. Contractile capacity is regulated by actin–troponin–myosin interactions in muscle-like subtypes and by cycles of cytoskeletal disassembly and reassembly in non-muscle subtypes. Notably, the denser TM architecture observed in POAG cynomolgus monkeys suggests that excessive contraction or insufficient relaxation of TM cells may lead to constricted aqueous outflow pathways. Our results also identified significant changes in TPM expression in POAG TM cells, particularly enriched in the TPM-Ca²⁺-TnT/actin pathway, which controls cellular contractility. TPM2, a highly conserved member of the tropomyosin family, serves as a ubiquitous actin-binding protein involved in striated/smooth muscle contraction and non-muscle cytoskeletal organization6. Functional studies demonstrate the critical role of TPM2 in modulating contractility in human lung fibroblasts and myofilament dynamics in rabbit skeletal muscle cells11,12. In vascular smooth muscle, downregulation of TPM2 attenuates vascular contraction (18). Therefore, elevated TPM2 levels in POAG may be a key mechanism driving TM contractile dysfunction and subsequent aqueous outflow facility impairment.
In this study, TPM2 overexpression in HTMCs showed dual regulatory effects: upregulation of pro-contractile molecular signatures in muscular subtypes and enhancement of anti-relaxation markers in non-muscular subtypes. Increased expression of ACTC1 and TNNT2 indicates increased contractility of TM cells, suggesting potential hypercontractile states. Mechanistically, the tropomyosin (encoded by TPM), actin (encoded by ACTC), and troponin complexes (encoded by TNNT) form the core components of myofilaments. Muscle contraction relies on thick-thin filament interactions, and gene expression levels of these filament proteins correlate strongly with cellular contractility22,23. ACTC1 is critical for maintaining contractile function, as evidenced by contractile dysfunction following ACTC downregulation in cardiomyocytes24,25. TNNT2 mediates ACTC1 anchoring to myofilaments and initiates contraction, and its overexpression has been shown to ameliorate contractile dysfunction26,27. The increased ACTC1 and TNNT2 expression observed in this study likely reflects increased contractility of TM cells, with TM hypercontraction being a critical contributor to the structural narrowing of the TM tissue and consequent aqueous humor outflow abnormalities in POAG. In TPM2-overexpressing HTMCs, upregulated F-actin expression indicates increased cytoskeletal polymerization. Myofilament dynamics depend on actin polymerization-depolymerization cycles, with excess F-actin (polymerized actin filaments) increasing cellular stiffness and impairing the ability to relax28. This mechanism is consistent with reports showing that F-actin downregulation reduces stiffness in human vascular smooth muscle cells and that cytoskeletal disassembly reduces stiffness in endometrial cancer cells29,30. Increased F-actin expression in HTMC may therefore further increase resistance to aqueous humor outflow by causing increased cell stiffness and reduced diastolic capacity, leading to difficulty in opening the TM pore. In contrast, knockdown of TPM2 exhibited a completely opposite trend.
Given that cytoskeletal disassembly alters cellular deformability and motility, which limits the range of deformations of muscle and non-muscle cells during the execution of diastolic behaviors7,8,31, we introduced the biomechanical parameter elastic modulus to quantif1y TM contractility and its regulatory role in aqueous outflow resistance. Using AFM to analyze force-displacement curves in HTMCs, we demonstrated that TPM2 overexpression significantly increased cellular elastic modulus. Young’s modulus is controlled by fibrotic status and cytoskeletal organization, with fibroblast hyperactivation and proliferation driving tissue stiffening and modulus increase32,33,34. α-SMA, a key fibroblast marker, promotes TM stiffness via actin polymerization when overexpressed35,36. Previous studies have linked α-SMA upregulation in glaucomatous TM to ECM cross-linking and impaired aqueous humor outflow, which is mechanosensitive to TM stiffness18. Notably, TPM2 upregulation in HTMCs induced concomitant α-SMA overexpression. The coordinated up-regulation of α-SMA and F-actin correlates with a marked increase in elastic modulus, which together result in impaired deformability and relaxation of HTMCs. This biomechanical dysfunction likely underlies the limited clinical efficacy of micropulse laser therapy and cholinergic agents in reducing IOP, as these treatments do not address TM cytoskeletal stiffening and contractile dysregulation. Novel intraocular pressure-lowering agents such as ROCK inhibitors (e.g., netarsudil) can partially inhibit TM fibrosis and reduce cellular contraction, thereby promoting aqueous humor outflow37. Excessive TM fibrosis leads to reduced deformability and increased elastic modulus of the TM38,39. Therefore, knocking down TPM2 to reduce cellular elastic modulus and attenuate TM cell contraction may synergize with ROCK inhibitors in IOP-lowering therapy. The expression or functional status of TPM2 may also influence patient responsiveness to ROCK inhibitor treatment, suggesting its potential future use as a biomarker for predicting therapeutic efficacy or as a target for combination therapy.
Additionally, we employed a cellular pressurization device to simulate elevate IOP conditions, investigating the impact of hydrostatic pressure on molecular expression levels associated with contractile function in normal HTMCs. While in vitro experiments are limited to simulating hydrostatic pressure effects, the induced mechanical stress from pressure elevation can indeed cause cellular deformation. This mechanical simulation effectively reflects TM cell responses to elevated pressure. Results demonstrated significant downregulation of TPM2 expression in normal HTMCs under acute high IOP conditions. This finding confirms TPM2’s critical role in modulating aqueous humor outflow resistance through TM contractility regulation. The observed TPM2 downregulation may represent a compensatory mechanism against acute pressure elevation to maintain aqueous humor homeostasis. However, under chronic high IOP conditions, pathological TPM2 upregulation due to dysregulated compensatory mechanisms may impair the TM’s ability to reduce outflow resistance through TPM2 downregulation, potentially contributing to the pathogenesis of POAG.
This study has two major limitations: First, although abnormal TPM2 expression in cynomolgus monkeys with POAG was enriched in pathways related to Ca²⁺-mediated relaxation and contraction, experimental validation of TPM2’s potential regulation of Ca²⁺ transients, relevant ion channels (e.g., TRPV4, Piezo1), or downstream RhoA/ROCK signaling pathways is still lacking. Follow-up studies will employ calcium imaging to monitor intracellular Ca²⁺ dynamics, assess functional activity of key ion channels, and examine changes in signaling molecules such as RhoA/ROCK to more comprehensively elucidate the mechanism of TPM2. Second, the cell pressurization device used to mimic hydrostatic pressure effects cannot fully replicate the intraocular high IOP mechanical environment. Moreover, further validation using glaucomatous TM cells has not yet been conducted. Therefore, the current setup only partially reflects the impact of high pressure on TPM2 expression in normal TM cells. Finally, this study only established and validated a mechanistic framework in an in vitro cell model. It did not explore the functional specificity of different TPM2 isoforms. Future research will clarify the specific functions and regulatory mechanisms of TPM2 isoforms and use adenoviral transfection techniques to modulate TPM2 expression in mice, further verifying its in vivo role through measurements of intraocular pressure and histological changes in the trabecular meshwork.
This study provides preliminary evidence that TPM2 expression in TM cells is mechanoresponsive to pressure stimuli. TPM2 overexpression induces increased TM contractility and limits cellular relaxation through cytoskeletal remodeling and altered biomechanical properties, ultimately leading to increased aqueous humor outflow resistance. The elevated TPM2 levels observed in POAG may critically contribute to aqueous humor outflow dysfunction. These findings position TPM2 as a novel therapeutic target for IOP-lowering strategies, which will be the focus of our future investigations.
Data availability
The data that support the findings of this study are available from the corresponding author, Xu Jia, upon reasonable request.
References
Wang, R. et al. Self-generating electricity system driven by aqueous humor flow and trabecular meshwork contraction motion activated BCKa for glaucoma intraocular pressure treatment. Mater. Horiz. https://doi.org/10.1039/d4mh01004c (2024).
Stamer, W. D. & Clark, A. F. The many faces of the trabecular meshwork cell. Exp. Eye Res. 158, 112–123. https://doi.org/10.1016/j.exer.2016.07.009 (2017).
Buffault, J., Labbe, A., Hamard, P., Brignole-Baudouin, F. & Baudouin, C. The trabecular meshwork: Structure, function and clinical implications. A review of the literature. J. Fr. Ophtalmol. 43, e217–e230. https://doi.org/10.1016/j.jfo.2020.05.002 (2020).
Buffault, J. et al. The dual effect of Rho-Kinase inhibition on trabecular meshwork cells cytoskeleton and extracellular matrix in an in vitro model of glaucoma. J. Clin. Med. 11 https://doi.org/10.3390/jcm11041001 (2022).
Jia, X. et al. Cell atlas of trabecular meshwork in glaucomatous non-human primates and DEGs related to tissue contract based on single-cell transcriptomics. iScience 26, 108024. https://doi.org/10.1016/j.isci.2023.108024 (2023).
Gateva, G. et al. Tropomyosin isoforms specify functionally distinct actin filament populations in vitro. Curr. Biol. 27, 705–713. https://doi.org/10.1016/j.cub.2017.01.018 (2017).
Meng, L. B. et al. TPM2 as a potential predictive biomarker for atherosclerosis. Aging (Albany NY). 11, 6960–6982. https://doi.org/10.18632/aging.102231 (2019).
Wang, H. W. et al. Ca(2+) metabolic disorder and abnormal expression of cardiac troponin involved in fluoride-induced cardiomyocyte damage. Chemosphere 201, 564–570. https://doi.org/10.1016/j.chemosphere.2018.03.053 (2018).
Gunning, P. W., Hardeman, E. C., Lappalainen, P. & Mulvihill, D. P. Tropomyosin - master regulator of actin filament function in the cytoskeleton. J. Cell. Sci. 128, 2965–2974. https://doi.org/10.1242/jcs.172502 (2015).
Prunotto, M. et al. Stable incorporation of alpha-smooth muscle actin into stress fibers is dependent on specific Tropomyosin isoforms. Cytoskeleton (Hoboken). 72, 257–267. https://doi.org/10.1002/cm.21230 (2015).
Bradbury, P. et al. Tropomyosin 2.1 collaborates with fibronectin to promote TGF-beta(1)-induced contraction of human lung fibroblasts. Respir Res. 22, 129. https://doi.org/10.1186/s12931-021-01730-y (2021).
Borovikov, Y. S. et al. The reason for a high Ca(2+)-sensitivity associated with Arg91Gly substitution in TPM2 gene is the abnormal behavior and high flexibility of Tropomyosin during the ATPase cycle. Biochem. Biophys. Res. Commun. 494, 681–686. https://doi.org/10.1016/j.bbrc.2017.10.161 (2017).
Kucukdogru, R. et al. Mechanochemical consequences of myopathy-linked mutations in Tpm2.2 on striated muscle contractility. FASEB J. 38, e23400. https://doi.org/10.1096/fj.202301604R (2024).
Kucukdogru, R. et al. Myopathy-linked mutations in TPM2 and their impact on troponin-mediated regulation of actomyosin contractility. FEBS J. https://doi.org/10.1111/febs.70180 (2025).
Wang, C., Xie, Y. & Wang, G. The elastic modulus and collagen of sclera increase during the early growth process. J. Mech. Behav. Biomed. Mater. 77, 566–571. https://doi.org/10.1016/j.jmbbm.2017.10.024 (2018).
Zhang, J. et al. Microstructure visualization of conventional outflow pathway and finite element modeling analysis of trabecular meshwork. Biomed. Eng. Online. 15, 162. https://doi.org/10.1186/s12938-016-0254-2 (2016).
Last, J. A. et al. Elastic modulus determination of normal and glaucomatous human trabecular meshwork. Invest. Ophthalmol. Vis. Sci. 52, 2147–2152. https://doi.org/10.1167/iovs.10-6342 (2011).
Raghunathan, V. K. et al. Glaucomatous cell derived matrices differentially modulate non-glaucomatous trabecular meshwork cellular behavior. Acta Biomater. 71, 444–459. https://doi.org/10.1016/j.actbio.2018.02.037 (2018).
Karimi, A. et al. Biomechanics of human trabecular meshwork in healthy and glaucoma eyes via dynamic schlemm’s Canal pressurization. Comput. Methods Programs Biomed. 221, 106921. https://doi.org/10.1016/j.cmpb.2022.106921 (2022).
Pant, A. D., Kagemann, L., Schuman, J. S., Sigal, I. A. & Amini, R. An imaged-based inverse finite element method to determine in-vivo mechanical properties of the human trabecular meshwork. J. Model. Ophthalmol. 1, 100–111 (2017).
Keller, K. E. et al. Consensus recommendations for trabecular meshwork cell isolation, characterization and culture. Exp. Eye Res. 171, 164–173. https://doi.org/10.1016/j.exer.2018.03.001 (2018).
Keyt, L. K. et al. Thin filament cardiomyopathies: A review of genetics, disease mechanisms, and emerging therapeutics. Front. Cardiovasc. Med. 9, 972301. https://doi.org/10.3389/fcvm.2022.972301 (2022).
McAdow, J. et al. A pathogenic mechanism associated with myopathies and structural birth defects involves TPM2-directed myogenesis. JCI Insight. 7 https://doi.org/10.1172/jci.insight.152466 (2022).
Frank, D. et al. Cardiac alpha-Actin (ACTC1) gene mutation causes Atrial-Septal defects associated with Late-Onset dilated cardiomyopathy. Circ. Genom Precis Med. 12, e002491. https://doi.org/10.1161/CIRCGEN.119.002491 (2019).
Unuma, K., Aki, T., Nagano, S., Watanabe, R. & Uemura, K. The down-regulation of cardiac contractile proteins underlies myocardial depression during sepsis and is mitigated by carbon monoxide. Biochem. Biophys. Res. Commun. 495, 1668–1674. https://doi.org/10.1016/j.bbrc.2017.12.020 (2018).
Wei, B., Jin, J. P. & TNNT3. Isoform genes, regulation, and structure-function relationships. Gene 582. TNNT1, 1–13. https://doi.org/10.1016/j.gene.2016.01.006 (2016).
Xie, S. Y. et al. USP28 serves as a key suppressor of mitochondrial morphofunctional defects and cardiac dysfunction in the diabetic heart. Circulation 149, 684–706. https://doi.org/10.1161/CIRCULATIONAHA.123.065603 (2024).
Luo, Q., Kuang, D., Zhang, B. & Song, G. Cell stiffness determined by atomic force microscopy and its correlation with cell motility. Biochim. Biophys. Acta. 1860, 1953–1960. https://doi.org/10.1016/j.bbagen.2016.06.010 (2016).
Khoukaz, H. B. et al. PAI-1 regulates the cytoskeleton and intrinsic stiffness of vascular smooth muscle cells. Arterioscler. Thromb. Vasc Biol. 44, 2191–2203. https://doi.org/10.1161/ATVBAHA.124.320938 (2024).
Salker, M. S. et al. LeftyA decreases actin polymerization and stiffness in human endometrial cancer cells. Sci. Rep. 6, 29370. https://doi.org/10.1038/srep29370 (2016).
Sarna, M. et al. Undifferentiated bronchial fibroblasts derived from asthmatic patients display higher elastic modulus than their non-asthmatic counterparts. PLoS One. 10, e0116840. https://doi.org/10.1371/journal.pone.0116840 (2015).
Janbandhu, V. et al. Hif-1a suppresses ROS-induced proliferation of cardiac fibroblasts following myocardial infarction. Cell Stem Cell 29, 281–297 e212, (2022). https://doi.org/10.1016/j.stem.2021.10.009
Link, P. A. et al. Combined control of the fibroblast contractile program by YAP and TAZ. Am. J. Physiol. Lung Cell. Mol. Physiol. 322, L23–L32. https://doi.org/10.1152/ajplung.00210.2021 (2022).
Quinn, K. P. et al. Optical metrics of the extracellular matrix predict compositional and mechanical changes after myocardial infarction. Sci. Rep. 6, 35823. https://doi.org/10.1038/srep35823 (2016).
Peng, M. et al. Cross-linked actin networks (CLANs) affect stiffness and/or actin dynamics in Transgenic transformed and primary human trabecular meshwork cells. Exp. Eye Res. 220, 109097. https://doi.org/10.1016/j.exer.2022.109097 (2022).
Watanabe, M., Ida, Y., Ohguro, H., Ota, C. & Hikage, F. Establishment of appropriate glaucoma models using dexamethasone or TGFbeta2 treated three-dimension (3D) cultured human trabecular meshwork (HTM) cells. Sci. Rep. 11, 19369. https://doi.org/10.1038/s41598-021-98766-3 (2021).
Batra, M. et al. A new ophthalmic drug in the treatment of chronic primary open angle glaucoma and ocular hypertension. Eur. J. Ophthalmol. 31, 2237–2244. https://doi.org/10.1177/11206721211008783 (2021). Netarsudil.
Lin, Y. C., Sung, Y. K., Jiang, X., Peters-Golden, M. & Nicolls, M. R. Simultaneously targeting myofibroblast contractility and extracellular matrix Cross-Linking as a therapeutic concept in airway fibrosis. Am. J. Transpl. 17, 1229–1241. https://doi.org/10.1111/ajt.14103 (2017).
Petrou, C. L. et al. Clickable decellularized extracellular matrix as a new tool for Building hybrid-hydrogels to model chronic fibrotic diseases in vitro. J. Mater. Chem. B. 8, 6814–6826. https://doi.org/10.1039/d0tb00613k (2020).
Funding
National Natural Science Foundation of China, 81960175; National Natural Science Foundation of China, 82360207; Guizhou Provincial Basic Research Program(Natural Science), zk[2025] General Program 472.
Author information
Authors and Affiliations
Contributions
BYS and XJ designed the study. BYS, YG, and YJX analysed the data and designed tables and fgures. RH and QYL interpreted the data. XJ helped supervise the project, helped with data interpretation and revised the manuscript. BYS wrote the manuscript. XJ supervised the project. All authors discussed the results and contributed to the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shen, B., Gong, Y., Xu, Y. et al. TPM2 regulates contractility and biomechanical properties in trabecular meshwork cells. Sci Rep 15, 41108 (2025). https://doi.org/10.1038/s41598-025-24993-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-24993-7
