
Abstract
This study provides insight into therapy for lung cancer, establishing p-Cresol (p-C) as an inhibitor of Pyruvate Dehydrogenase Kinase 3 (PDK3). PDK3 is critical in cancer metabolism by regulating the pyruvate dehydrogenase complex, shifting cellular energy production towards glycolysis, and promoting tumor growth and survival under hypoxic conditions. In this study, we have used computational and experimental approaches. Molecular docking reveals that p-C occupies PDK3’s binding pocket and forms interactions with key residues, especially Asp 287. Molecular dynamic simulation (MD) studies showed that p-C induced minimum alterations in PDK3, suggesting the structural stability of the PDK3-p-C complex. A fluorescence-based binding study demonstrated the binding of p-C to PDK3 with a binding constant of 3.8 × 108 M−1, indicating excellent binding affinity. Cell-based enzyme assay revealed significant inhibition of PDK3 by p-C, establishing it as a PDK3 inhibitor. Moreover, cellular assays also demonstrated significant inhibition of PDK3 activity and tumor progression. This study provides a promising therapeutic avenue for improving lung cancer treatment outcomes by targeting PDK3.
Similar content being viewed by others


Introduction
Cancer is the second most prevalent disease, which has challenged health care with high mortality rates1,2. According to a 2021 report released by the Centres for Disease Control and Prevention (CDC), cancer is the second leading cause of death in humans3. According to another report, more than 19 million new cancer cases were reported in the year 2020, which is also marked by 10 million deaths in the same year4,5,6. Still, these aren’t the peak cases we have witnessed to date. In the future, we are expected to record ~ 34 million new cases by the end of 20704,7. Those countries where the per-person income is still counted in the middle range and where the majority of the population cannot afford cancer cures are at a greater risk8,9. These alarming statistics are a hint for us to work and find a cure for this incurable and devastating disease. Due to this huge impact on the global human population, finding a cure for this disease is one of the most anticipated demands. Though efforts are made and new drugs come into the market in an attempt to provide a cure to treat cancer patients, no one has proven to be beneficial without any side effects10,11,12.
Cancer is a class of various grouped symptoms triggered by mutations in genomic DNA, failure of immune cells and proteins to limit risk cells that could turn into cancer and other environmental factors. It disturbs the cellular regulation processes, which, in standard cases, eliminate faulty cells. Pyruvate dehydrogenase kinases (PDKs) are protein kinases that have been found to show a role in cancer13. Thus, targeting them is an attractive strategy in cancer therapeutics. PDKs are critical modifiers of cellular machinery that drive cancer and play a central role in the metabolic processes that characterize many cancers. Driven by them, the pyruvate dehydrogenase complex (PDC) is an essential metabolic process to convert pyruvate into acetyl coenzyme A (acetyl-CoA)14,15. Under normoxic conditions, cells primarily use oxidative phosphorylation; however, PDKs mediated phosphorylation of PDC diverts pyruvate towards lactate formation, favouring glycolytic energy production even in aerobic conditions14,15.
Cancerous cells use this form of energy metabolism, a well-known factor for cancer. The Warburg effect provides cancerous cells with all the intermediates that aid their biosynthesis16. PDK3 is a unique protein with structural and regulatory features, which has helped it gain popularity because of its importance in cancer progression. Under hypoxic conditions, the expression of PDK3 is significantly elevated, a common feature of solid tumors. PDK3 comprises an N-terminal regulatory (R) domain that interacts with the lipoate moiety of E2, and a C-terminal kinase (K) domain responsible for catalysis17,18. PDC is inactivated because of PDK phosphorylating E1α at Ser264, Ser271, and Ser203. PDK3 thus also makes these cells resistant to apoptosis19,20,21. PDK3’s evidence in various cancers has been observed in many cancer types, viz. lung, breast, and ovarian cancer22. The other contribution of PDK3 in cancer is that it helps generate lactate, providing an environment relevant to tumor growth. Thus, the extracellular matrix gets acidified, which supports immune evasion and angiogenesis19,23. Due to its essential role in cancer, PDK3 is an important target for drug discovery that could be used for the prevention and treatment of cancer. PDK3 could be used to restore regular PDC activity and mitochondrial respiration and reduce the availability of glycolytic intermediates necessary for tumor growth19,24,25,26. Various PDK3 inhibitors are reported in pre-clinical stages19,27.
Cresols belong to the phenol family and have various industrial applications, such as their usage in producing resins, chemical disinfectants, and other chemical intermediate compounds. They are packed with antimicrobial and antiseptic properties, making them valuable in medical and industrial applications28. Recently, some published studies have reported that they could act as signaling molecules or bioactive agents and show interactions with enzymes in biochemical pathways29,30. Cresols are methyl phenols, and the position of the methyl group can vary, i.e., it can be at meta-, ortho, or para—position. Chemically, p-Cresol (p-C) is 4-methylphenol with the chemical formula: C₇H₈O and is a byproduct of microbial fermentation in the gut that can be detected in urine. Recent studies have shown the therapeutic potential of p-C in cancer. A recent study reported that p-C promoted bladder cancer cell invasion and migration involving the Ras and mTOR signaling pathways31. Another recent study investigated the effect of p-C on kidney and liver cancer cells and found that HepG2 human liver cancer cells’ malignant biological behavior was exacerbated by p-C32. Together, these studies highlight the importance of p-C in cancer therapeutics. Though to date, limited studies have investigated the effect of p-C in the field of cancer therapeutics, its potential role remains largely unexplored. Our study is the first to investigate the inhibitory effect of p-C on PDK3, highlighting its potential as a metabolic modulator in lung cancer therapeutics employing computational and experimental approaches. In this study, we used a combination of molecular docking, molecular dynamics simulations, fluorescence-based binding assays, and cell-based assays to establish p-C as an effective inhibitor of PDK3. These findings provide a platform to target cancer metabolism by modulating PDK3 function, offering a promising therapeutic strategy for lung cancer treatment.
Materials and methods
Chemicals and reagents
Sigma-Aldrich Co. was the supplier of p-C. Himedia provided the additional chemicals needed to prepare the buffer (India). All buffers were prepared using MilliQ water from a Milli-Q® UF-Plus purification system. All the chemicals required in cell cultures, such as PDK3 antibody (MA5-27002), MTT [298-93-1; SRL], FBS, DMEM, and Trypsin-EDTA, were obtained from Gibco-life technologies, Thermo Fisher Scientific (USA). The source of H1299 was the National Centre for Cell Sciences in Pune, India (411007). Luria-Bertani Difco LB broth was purchased from Becton, Dickinson, and Company, Sparks, MD, USA. BIOMOL green reagent (BML-AK111-0250) was purchased from Enzo Lifescience.
Molecular docking
To analyze the interaction between the ligand p-C and the protein PDK3, molecular docking studies were performed using our proprietary docking software, InstaDock33,34. PyMod3 was used to further improve the crystal structure of PDK3 (PDB ID: 1Y8O), which we downloaded from RCSB, and PubChem provided the p-C structure. Furthermore, “PyMOL” and “Discovery Studio Visualizer” were used for all of the in-depth examination of the interaction between p-C and PDK335,36.
MD simulations
The native-PDK3 and p-C-PDK3 systems were simulated using GROMACS 2022.2 with the CHARMM36 force field. Protein termini were capped with suitable residues for stability. Systems were solvated in a TIP3P water box (10 Å) using 12 Å van der Waals cutoff and a 2 fs integration step. Minimization included 500 steps of steepest descent and 1000 conjugate gradient steps. We heated the systems from 0 to 300 K with 10 Å harmonic restraints, equilibrated them for 100 ps, and subjected them to a 100 ns production run to analyze trajectory changes. Further details are available in our recent publications37,38.
Principal component analysis (PCA) analysis
PCA was conducted on protein-ligand complexes using the default parameters in “GROMACS”39. The periodic conditions were eliminated by preprocessing the trajectory before performing PCA. To visualize the trajectory along the principal components (PCs), the ‘gmx an apron’ tool in GROMACS was utilized to compute the PC coordinates for each frame40. All other conditions were kept as mentioned in earlier studies.
Free energy landscape (FEL)
FEL is useful for exploring protein conformational changes across different energy states. The dynamics of biological systems can be analyzed by examining the steady states, represented by FEL minima, and the transient states, indicated by FEL barriers. This approach provides valuable insights into biomolecule recognition, aggregation, and folding processes. The plots were generated using SigmaPlot1541.
Protein expression and purification
PlasmID HMS library of human kinases (http://plasmid.med.harvard.edu/PLASMID) is the source of the PDK3 gene. A template was created using the amplified gene. Our research lab has already carried out the cloning. In the pET28b + expression vector, PDK3 amplicons containing a six-His tag were ligated. PDK3 cells were cultured overnight for revival as a primary culture. 1% of the primary culture was used to inoculate the secondary culture. The culture was induced with 1mM IPTG and incubated on a shaker for four hours at 37° C. The cells were then harvested by centrifugation and suspended in lysis buffer, sonicated, and then dissolved in a solution of 0.4% to 0.5% N-lauryl sarcosine prepared in 50 mM Tris and 100 mM NaCl buffer (pH 8.0) in order to separate the inclusion bodies (IBs). The protein eluted the column at a rate of 1 milliliter (ml) per five minutes. Any unbound protein was eliminated by washing the protein after the binding procedure was finished. After changing the washing buffer, which included a modest amount of imidazole, we performed the elution at different imidazole concentrations using our previous protocols.
Enzyme inhibition assay
Purified PDK3 (5 µM) protein was incubated with 200 µM of ATP for one hour at 37 °C. Following incubation, BIOMOL green reagent was added to stop the reaction and allowed to develop a green color for approximately 20 min. The protein incubated with different ligand concentrations was also incubated similarly, and all the samples were measured on a multiple reader OD at 620 nm. Protein kinase activity was defined as 100% when the protein’s OD620 was ligand-free. The protein’s percentage (%) activity impacted by the presence of increasing p-C concentrations was calculated. Three duplicates of each experiment were conducted.
Fluorescence binding assay
The protein PDK3 was subjected to a fluorescence-based quenching test with p-C acting as a ligand. The protein was stimulated at 280 nm to measure the fluorescence, and emission spectra were captured in the 300–400 nm region42,43. The quenching data was analysed using Modified Stern-Volmer (MSV) Eq. 143.
F0: “Maximum intensity of protein fluorescence”, F: “Maximum intensity of protein fluorescence in the presence ligands”; K: “Binding constant”; C: “Ligand concentration”; n: “number of binding sites”.
Maintenance of cells
H1299 was kept in an ideal condition at 37 °C and 5% CO2 after being obtained from the NCCS, Pune-411,007, India. The culture medium DMEM for H1299 cells included antibiotics (100 µg/mL streptomycin and 100 U/mL penicillin) and 10% Gibco heat-inactivated FBS. Further, the cells were plated at a density of 1 × 105 cells/well into 96-well culture plates. At passage numbers 20–22, 80%–85% of confluency, the cells received all treatments.
Cell proliferation assay
H1299, human non-small cell lung carcinoma, was used for cell-based studies and was obtained from the National Centre for Cell Science. The cancer cells were and were cultured in DMEM-supplemented with 10% fetal bovine serum (FBS). Approximately 5–6 × 103 cells were added to a 96 well plate, and cells adhered post-seeding (16–20 h) followed by compound. With DMSO, compound stock solutions were created. The treatments did not exceed a DMSO concentration of 0.2%. The MTT in a conc. 10 µL of 5 mg/ml was put in each well and left for 37 °C incubation with 5% CO2 for 2–4 h. After incubation, the 96 well plate was read with a micro-plate reader at 570 nm wavelength.
Immunofluorescence
Immunofluorescence is an extremely sensitive technique for IHC applications. 4% PFA is used to fix cells for 20 min at room temperature. After that, cells are washed three times using PBS (1X). Following a 30-minute treatment with 0.25% Triton X-100 in PBS (1X), the cells are rinsed at room temperature with PBS (1X). The treated well plate is washed three times with PBS (1X) to avoid unspecific antibody binding. The cells are then treated for 60 min with 3% BSA in PBS (1X). The blocking buffer can be eliminated by washing with PBS (1X) three times. The cells are then treated with primary antibodies against PDK3 (1:1000, Genetex, catalog #GTX132621) for an entire night at 4ᵒC. The cells undergo a 60-minute RT treatment with the secondary antibody the next day, three PBS (1X) washes, and DAPI mounting.
Statistical analysis
One-way analysis of variance (ANOVA) was used in the current investigation to compare the scope and treatment groups of TRAZ, followed by Tukey’s multiple comparison tests. GraphPad Prism 8.0 (GraphPad Software, San Diego, CA, USA) supported the statistical analysis. The nominal significance level was determined at p < 0.05, and values were presented as mean ± s.e.m.
Result and discussions
Molecular docking
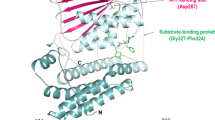
Molecular docking was carried out using InstaDock v1.2. A blind docking approach was applied by setting the grid box dimensions to 66 Å × 80 Å × 72 Å, with central coordinates positioned at X: −59.719 Å, Y: 10.72 Å, and Z: 79.185 Å. PDK3 is a protein kinase linked to various types of cancer progression by aiding its proliferation and metastasis. PDK3 has been targeted using multiple small molecules; we aim to target PDK3 with p-C as a therapeutic agent. The molecular docking studies showed that the ligand p-C interacts with PDK3 in the ATP-binding pocket, as shown in Fig. 1. The interaction involved key residues, including Ser252, Asp287, and Asn251. Asp287 is a catalytically critical residue in PDK3, which plays a key role in ATP binding as shown in Fig. 1B. Hydrophobic interactions were observed with residues Leu248, Leu344, and Ala354, suggesting strong ligand stabilization within the binding site. Hydrogen bonding with Ser252 and Asp287 enhances its binding affinity, while hydrophobic interactions with Leu248, Leu344, and Ala354 increase stability. Electrostatic interactions were minimal, with binding majorly driven by hydrophobic forces and hydrogen bonding. Figure 1D shows all the interactions in a 2-D representation. The calculated binding energy of p-C with PDK3 was − 5.8 kcal/mol, indicating a good binding affinity and suggesting its potential as a PDK3 inhibitor. These findings were further confirmed through molecular dynamics simulations and experimental validation.
The predicted inhibition constant (pKi) in molecular docking is a crucial parameter that estimates the potency of a ligand in inhibiting a target protein44,45. pKi was estimated as 4.25, which supports its moderate inhibitory potency. Ligand efficiency was calculated at 0.725, suggesting favorable binding efficiency relative to molecular size. Additionally, the torsional energy was recorded at 0.3113 kcal/mol, indicating minimal conformational strain upon binding. The molecular docking results show that the ligand binds efficiently to PDK3.

Structural features of PDK3 protein (1Y8O). (A) Docked p-C in the active site pocket of PDK3 in a cartoon representation. (B) Zoomed view of p-C interacting with PDK3. (C) Electrostatic surface view of p-C in the binding cavity of PDK3. (D) 2D figure of PDK3-p-C complex interactions.
Molecular dynamics simulations
Molecular dynamics (MD) simulations are essential to drug design and discovery tools. MD simulations predict how all the atoms in a molecular system, such as a protein, will move in a given time frame46. It is a sophisticated computational method for comprehending proteins’ stability, interaction mechanisms, and conformational dynamics in both free and complex states. Important MD parameters, including structural fluctuations and hydrogen bonds in PDK3 and PDK3 complex with p-C, were analysed.
Structural deviations
RMSD measures the overall structural stability of the protein-ligand complex over the simulation period. Figure 2A shows the RMSD plot of PDK3 in native form and in bound form with p-C. The value of RMSD for PDK3 was estimated at 0.32 nm and for the complex at 0.26 nm. These results were consistent across all three MD replicates, as shown in Figure S. Compared to the native-protein, the ligand-bound complexes showed slight variations, suggesting long-term stability. The systems stabilized the structures and achieved equilibrium over the 100 ns simulation. The ligand-induced stabilization is further supported by the PDK3-p-C complex’s narrower peak in Fig. 2C, which denotes a more constrained conformational space.

Structural deviations of PDK3 protein upon ligand binding; (A) RMSD of apo-PDK3 represented by black line and PDK3-p-C complex shown by pink line, (B) RMSF of apo-PDK3 and PDK3-p-C complex, (C) PDF of RMSD, (D) PDF of RMSF.
RMSF evaluates the flexibility of individual amino acid residues. Figure 2B highlights fluctuations across the PDK3 sequence, with peaks corresponding to loop regions or flexible domains. The unbound PDK3 (black) shows higher fluctuations at residues ~ 200 and ~ 300, suggesting flexible loop regions. However, in the PDK3-p-C complex (pink), the fluctuations are reduced, particularly around active site residues, implying that ligand binding stabilises these regions. The lower fluctuations at key catalytic residues, including Asp287 and Ser252, suggest that p-C may enhance the rigidity of the active site, which could influence enzyme activity. In Fig. 2D, the RMSF distribution shows that most residues experience reduced fluctuations upon ligand binding, aligning with the observed stabilization effects. As illustrated in Figure S1, these outcomes were consistent for all three MD replicates.
The structural compactness of the protein in its native form and after binding to the ligand was studied and analyzed. A protein’s tertiary structure and general shape are directly indicated by the radius of gyration, denoted as Rg. It helps evaluate a protein’s compactness and folding characteristics. We computed the Rg value to assess the stability of PDK3 in its native form and after the binding of p-C, as shown in Fig. 3A. The average Rg calculated for native PDK3 and the protein-ligand complex was 2.22 nm and 2.23 nm, respectively. This increase in Rg values describes a less rigid structure during the simulation, although the variation was slight47. The PDF indicates very little change in the protein structure upon ligand binding, as shown in Fig. 3B. Table S1 summarizes the dynamic and structural parameters from the triplicate simulations.
SASA, often called the surface found around a protein by the molecule’s van der Waals contact surface and a hypothetical center of a solvent sphere, has been considered a crucial component in protein folding and stability studies. The SASA parameter can be used to examine the compactness treatment of backbone atoms. A lower SASA value indicates higher compactness, and higher values indicate less compactness48. We estimated the SASA to be over 100 ns, as shown in Fig. 3C and Fig. 3D. The SASA value of native-PDK3 was estimated to be 191 nm2, and PDK3, after binding with p-C, was found to be 192 nm2. In the PDK3 and p-C complex, a very slight increase in SASA was observed due to the expanded surface area of the protein due to the exposure of internal residues to the solvent-accessible surface. Triplicate MD runs showed highly reproducible Rg and SASA profiles, affirming structural compactness stability (Figure S2).

Structural compactness of PDK3 protein upon ligand binding; (A) Rg of apo-PDK3 (represented by black line) and PDK3-p-C complex (represented by pink line), (B) SASA of apo-PDK3 and PDK3-p-C complex, (C) PDF of Rg, (D) PDF of SASA.
Dynamics of hydrogen bonding
To assess the structural stability of free PDK3 and its complex with p-C, we analyzed hydrogen bonding interactions and other non-covalent contacts formed during the 100 ns molecular dynamics simulation. Specifically, hydrogen bonds were identified based on a widely accepted geometric criterion, where a hydrogen bond is considered present when the distance between the donor and acceptor atoms is ≤ 0.35 nm (3.5 Å. This cutoff allows for the detection of relevant and biologically meaningful interactions that contribute to the stabilization of the complexes. By tracking the number and persistence of such hydrogen bonds and other close contacts over the simulation time, we gained insights into the dynamic behavior and binding stability of each complex in comparison to the unbound protein. Throughout the 100 ns simulation, the number of intramolecular hydrogen bonds stayed constant, peaking at about 260–270 bonds, suggesting that p-C binding did not interfere with PDK3’s internal hydrogen bonding network (Fig. 4A). Transient but important interactions are indicated by the ligand’s frequent formation of one hydrogen bond and irregular formation of two bonds (Fig. 4C). Similar hydrogen bond trends were observed across all three replicate simulations (Figure S3). These hydrogen bonds enhance the overall binding stability and specificity of p-C for PDK3. The probability density function (PDF) analysis of intramolecular and intermolecular hydrogen bonds confirms these interactions’ stability (Fig. 4B and Fig. 4D).

2-D representation of (A) Intramolecular and (B) intermolecular hydrogen bond formation for apo PDK3 and PDK3 in complex with p-C over 100 ns of MD simulations. B and D represent PDF for the same.
Secondary structure analysis
Dynamic shifts in the secondary structure of residues drive conformational alterations within protein structures. It is essential to comprehend these changes in a protein’s secondary structure composition to understand its conformational behaviour and the polypeptide chain folding mechanism. We investigated the secondary structure dynamics of PDK3 to evaluate its stability both before and after binding with the ligand. According to our study, the structural elements of PDK3 exhibit relatively stable behaviour and achieve equilibrium throughout the simulation in both its native and p-C bound states (Fig. 5A & B). However, when PDK3 forms a complex with ligand, the secondary structure content of PDK3 is barely altered. The protein is usually structurally stable when there is no discernible change in content, indicating a more robust and ordered structure49,50. Table 1 shows various changes in the protein’s secondary structure upon ligand binding.

Secondary structure analysis (A) PDK3; (B) PDK3-p-C-complex.
Principle component analysis
To better understand how PDK3 behaves with and without p-C, we performed principal component analysis (PCA) to examine its conformational dynamics. Figure 6A shows the movement patterns of unbound PDK3, while Fig. 6B illustrates how the protein moves when bound to p-C. When PDK3 is unbound, the data points are tightly clustered, showing that the protein maintains a stable, rigid structure with minimal movement. However, once p-C binds, the data points spread out more, suggesting that the protein becomes more flexible and dynamic. This indicates that p-C binding triggers subtle structural rearrangements, altering how PDK3 moves and behaves. Multiple clusters in the ligand-bound state hint at distinct conformational states, meaning p-C may stabilize specific structural shifts. Overall, these findings indicate that while p-C does not destabilize PDK3, it influences its flexibility and, potentially, function.

PCA plot constructed by eigenvector 1 vs. eigenvector 2 for (A) apo-PDK3 and (B) for PDK3-p-C complex displaying the positive and negative residual movements.
Free energy landscape
The free energy landscape (FEL) analysis gives us a deeper insight into how stable PDK3 is in different states in its native-form and when bound to p-C. Figure 7A illustrates the FEL of unbound PDK3, while Fig. 7B shows the landscape after p-C binding. The color gradient, ranging from blue (low free energy) to red (high free energy), highlights the most stable conformational states. In Fig. 7A, we observe that unbound PDK3 has a well-defined, deep energy basin, meaning it tends to stay in a few stable conformations with minimal structural fluctuations, suggesting the rigidity of PDK3 in its native state. Figure 7B shows a spread-out energy landscape for the PDK3-p-C complex. The presence of multiple low-energy basins indicates that when the ligand binds, the protein gains more flexibility and can adopt a broader range of conformations. This aligns with the PCA results, reinforcing that p-C doesn’t destabilize PDK3 but influences its structural dynamics.

Free energy landscape of (A) PDK3 and (B) PDK3-p-C complex.
Enzyme Inhibition assay
Protein kinases are enzymes that use ATP as a substrate to phosphorylate their target molecule. PDK3 shows ATPase activity, which is used to estimate the inhibitor potential of the ligands. The protein kinases hydrolyse ATP into ADP and free phosphate. The amount of phosphate is directly proportional to the activity of the protein. PDK3 protein (5 µM) was incubated with increasing ligand concentration (1–8 µM) of p-C and ATP (200 µM). BIOMOL Green provided a simple and convenient method for colorimetric phosphate quantitation, which was read calorimetrically at 620 nm. Protein without ligand was also incubated and considered to have the maximum activity of the protein, i.e., the highest release of phosphate and a high OD. Similarly, the OD was measured for all the ligand concentrations and plotted in terms of percentage, considering protein activity as 100% activity. The IC50 value calculated for p-C against PDK3 activity was 3.52 µM. Earlier published literature revealed ellagic acid and quercetin as PDK3 inhibitors, with IC50 values of 5 µM and 9.5 µM, respectively29,51. Figure 8 shows inhibition in the protein’s activity with increasing concentration of PDK3.

Enzyme inhibition assay of PDK3 with different concentrations of p-C.
Fluorescence binding assay
The in silico experiments showed that p-C binds in the active pocket of PDK3 to form a stable complex, stabilised by various bonds. The computational experiments were in line with the enzyme inhibition assay, which showed that p-C binds and inhibits the activity of PDK3. We further conducted a fluorescence quenching assay to complement the above experiments. The emission spectra of the protein with ligand ascending ligand concentration are shown in Fig. 9A. The quenching in fluorescence intensity was studied using the double log relation (modified Stern–Volmer equation) to assess the binding constant (K), as illustrated in Fig. 9B, as per our previous publications52,53. The binding constant for PDK3 and p-C was estimated as 3.8 × 108 M−1, which signifies a pronounced affinity between the protein and the ligand, highlighting the strength of their interaction.

Fluorescence quenching assay of PDK3 in the presence of the ligand p-C. (A) Fluorescence emission spectra, (B) Modified Stern–Volmer (MSV) of PDK3 fluorescence quenching against increasing ligand concentration.
Cell proliferation assay
MTT assay was performed to evaluate the cytotoxic effect of p-C on cancer cell proliferation. Cells were treated with increasing concentrations of p-C (5 µM, 10 µM, 20 µM, and 50 µM), and cell viability was measured by absorbance at 570 nm, as shown in Fig. 10. The inhibitors of PDK3, i.e., quercetin and ellagic acid, were used as a positive control, as the known IC50 of quercetin is 55.90 µM and 23.40 µM for the lung cancer cell line. The results indicate a significant, dose-dependent reduction in cell proliferation upon treatment. The control group exhibited significantly higher proliferation compared to all p-C-treated groups (5 µM, 10 µM, 20 µM, and 50 µM) (*p < 0.05, **p < 0.001). Additionally, all p-C -treated groups demonstrated a significant reduction in proliferation compared to the lowest concentration (5 µM) (###p < 0.001). Higher p-C concentrations (50 µM) further reduced proliferation compared to 10 µM and 20 µM treatments (^^^p < 0.001, $$$ p < 0.001). These findings suggest that p-C exerts a potent inhibitory effect on cancer cell proliferation in a dose-dependent manner. Data are presented as mean ± SEM, analyzed using Tukey’s multiple comparison test, with n = 3 independent experiments.

Cell proliferation assay: p-C exhibits a potent dose-dependent cytotoxic effect on cancer cell proliferation. The MTT assay was performed to evaluate the cytotoxic effect of p-C on cancer cell proliferation.
Live and dead assay
The fluorescence-based LIVE/DEAD™ Viability/Cytotoxicity Kit for mammalian cells distinguishes between live and dead cells according to their membranes’ integrity. It uses Ethidium Homodimer-1 (EthD-1) and Calcein-AM, two fluorescent dyes54. A non-fluorescent dye called Calcein-AM penetrates living cells and is hydrolysed into Calcein by intracellular esterase, which results in a green glow. Only living cells produce green fluorescence because intact membranes and esterase activity are characteristics of live cells. On the other hand, EthD-1 enters cells with damaged membranes, binds to nucleic acids, and releases red fluorescence. It is impermeable to intact membranes. The difference between living (green) and dead (red) cells can be easily distinguished according to this selective labelling55,56,57.
The cytotoxic effects of p-C on cancer cell viability were evaluated using live and dead cell staining. Cells were treated with p-C concentrations of 5 µM, 10 µM, 20 µM, 50 µM, and 100 µM. As the p-C concentration increased, a dose-dependent reduction in cell viability was observed. At higher concentrations (50 µM and 100 µM), there was a significant increase in dead cells, as indicated by red/orange fluorescence, alongside a corresponding decrease in viable cells, marked by green fluorescence, as shown in Fig. 11A and quantification is shown in Fig. 11B. The control group and the 5 µM p-C-treated group exhibited significantly higher proportions of live cells compared to all other p-C-treated groups (10 µM, 20 µM, 50 µM, and 100 µM). This trend underscores the potent cytotoxic effects of higher p-C-doses. Images were captured using a Zeiss microscope with a 20X magnification scale bar, confirming the dose-dependent cytotoxicity.

PDK3 expression in H1299 cells upon PDK3 treatment. p-C highlights the striking, dose-dependent impact of p-C on cancer cell viability. (A) p-C illustrates a potent, dose-dependent cytotoxic effect on cancer cell viability as assessed through live and dead staining. Cancer cells were exposed to p-C at concentrations of 5 µM, 10 µM, 20 µM, 50 µM, and 100 µM. The data reveal a dose-dependent increase in cell death, with higher concentrations of p-C (50 µM and 100 µM) resulting in a notable rise in dead cells and a corresponding decrease in viable cells. Both the control group and the lowest p-C concentration (5 µM) showed a significantly higher proportion of live cells compared to the other p-C -treated groups (10 µM, 20 µM, 50 µM, and 100 µM). Green fluorescence indicates live cells, while red/orange fluorescence marks dead cells. The images were captured using a Zeiss microscope, with a scale bar at 20X magnification. (B) fluorescence images showing live (green) and dead (red) cells after treatment. Quantification revealed that p-c (10, 20, and 50 µM) significantly reduced cancer cell viability compared to control (**p < 0.01 and *p < 0.05). Furthermore, p-c at 10 and 20 µM showed greater reduction in cancerous cells than the lower dose (5 µM) (##p < 0.01 and #p < 0.05). Data are expressed as mean ± SEM; n = 3; statistical analysis by Tukey’s multiple comparison test.
PDK3 is highly expressed in various cancers; therefore, the kinase has emerged as a therapeutic target for cancers22,58,59,60. PDK3 expression was studied in H1299 lung cancer cells before and after treatment with p-C. The in silico studies, inhibition assay, and binding assays show that p-C is a binding partner and an inhibitor of PDK3, and to validate this, we carried out immunofluorescence staining in cancer cells. The H1299 cells were incubated with PDK3-specific primary antibodies followed by secondary antibodies. H1299 cells without treatment were used as a control. With increasing the concentration of the ligand, the expression of PDK3 reduced gradually. Figure 12A shows a high expression of PDK3, which is significantly reduced at 10–20 µM concentration of the ligand; ligand at 5 µM and 10 µM did not show much effect. At very high doses, such as 50–100 µM, the PDK3 expression has been reduced dramatically when compared to the control group (Fig. 12A). The study overall presents the effect of p-C cancer cell lines. Higher doses above 50–100 µM are considered toxic, so we studied the concentration of p-C till 100 µM61,62. Images were captured using a Zeiss microscope at 20X magnification, with scale bars provided for reference and quantification is provided in Fig. 12B.

Immunofluorescence Staining of PDK3 Expression in Cancer Cells Treated with p-C). (A) Cancer cells were treated with increasing concentrations of p-C (5 µM, 10 µM, 20 µM, 50 µM, and 100 µM) and then subjected to immunofluorescence staining using an anti-PDK3 primary antibody, followed by a fluorescently labelled secondary antibody. The control group showed robust PDK3 expression, as indicated by strong green fluorescence throughout the cytoplasm. In contrast, treatment with higher concentrations of p-C (50 µM and 100 µM) resulted in a significant reduction in PDK3 expression, as evidenced by the decreased fluorescence intensity. At these higher doses, PDK3 expression was notably diminished, suggesting a dose-dependent inhibitory effect of p-C on PDK3. Cells treated with lower p-C concentrations (5 µM and 10 µM) displayed moderate PDK3 expression, but still lower than the control. Images were captured using a Zeiss microscope at 20X magnification. Scale bar: X µm. (B) fluorescence images showing PDK3 expression after treatment. Quantification demonstrated that p-C at 10, 20, 50, and 100 µM significantly reduced PDK3 expression compared to control (**p < 0.01 and *p < 0.05). Data are expressed as mean ± SEM; n = 3; statistical analysis by Tukey’s multiple comparison test.
Conclusions
Small molecule kinase inhibitors have gained popularity in recent years many of these compounds have FDA approval63. The development of kinase inhibitors has gained widespread attention due to their overexpression and mutation in various cancer types64,65. Kinases aid cancer survival by supporting the tumor microenvironment and aiding in metastasis and growth of cancer. PDK3, being a major kinase associated with tumor microenvironment and cancer progression, was targeted against lung cancer. We worked on the mechanistic insights of the binding of p-C with PDK3 using in silico and in vitro methods, including kinase inhibition and expression in H1299 lung cancer cells. All the studies complemented each other, and p-C was shown to be a binding and inhibiting agent of PDK3. The ligand also showed inhibitory effects against PDK3 in lung cancer cell lines, proving to be an effective therapy against cancer by inhibiting PDK3 activity. However, there are some limitations, as our results suggest that p-C may be a PDK3 inhibitor, which may have implications for addressing cancer metabolism in lung cancer. It is important to recognise a number of caveats. It is yet to be confirmed if p-C is relevant in a physiological tumor microenvironment because its inhibitory effect was first evaluated under carefully monitored in vitro and biochemical circumstances. There are issues with therapeutic selectivity, bioavailability, and safety because p-C is an endogenous microbial metabolite that has been shown to cause systemic toxicity at greater quantities. p-C’s structural optimisation was not included in the study; it might be required to increase its selectivity and potency against PDK3 in comparison to currently available synthetic inhibitors.
Data availability
All data supporting the findings of this study are available within the paper and supplementary material.
References
Filho, A. M. et al. The GLOBOCAN 2022 cancer estimates: data sources, methods, and a snapshot of the cancer burden worldwide. Int. J. Cancer. 156 (7), 1336–1346 (2025).
Boire, A. et al. Why do patients with cancer die? Nat. Rev. Cancer. 24 (8), 578–589 (2024).
Xu, J. et al. Mortality in the united States, 2021. NCHS Data Brief. (456), 1–8 (2022).
Kiri, S. & Ryba, T. Cancer, metastasis, and the epigenome. Mol. Cancer. 23 (1), 154 (2024).
Ferlay, J. et al. Cancer statistics for the year 2020: an overview. Int. J. Cancer. 149 (4), 778–789 (2021).
Torre, L. A. et al. Global cancer statistics, 2012. CA Cancer J. Clin. 65 (2), 87–108 (2015).
Soerjomataram, I. & Bray, F. Planning for tomorrow: global cancer incidence and the role of prevention 2020–2070. Nat. Rev. Clin. Oncol. 18 (10), 663–672 (2021).
Anandasabapathy, S. et al. Cancer burden in low-income and middle-income countries. Nat. Rev. Cancer. 24 (3), 167–170 (2024).
McLeod, M. et al. Quality indicators for evaluating cancer care in low-income and middle-income country settings: a multinational modified Delphi study. Lancet Oncol. 25 (2), e63–e72 (2024).
La’ah, A. S. & Chiou, S. H. Cutting-edge therapies for lung cancer. Cells 13 (5), 436 (2024).
Alsheikhly, A. S. & Alsheikhly, M. A. S. A Compr. Rev. Breast Cancer Latest Advancement Diagnosis Treatment (2025).
Kalofonou, F. et al. Monoclonal antibodies in metastatic Gastro-Esophageal cancers: an overview of the latest therapeutic advances. Int. J. Mol. Sci. 26 (3), 1090 (2025).
Sradhanjali, S. & Reddy, M. M. Inhibition of pyruvate dehydrogenase kinase as a therapeutic strategy against cancer. Curr. Top. Med. Chem. 18 (6), 444–453 (2018).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144 (5), 646–674 (2011).
Liu, J. et al. RRAD inhibits the Warburg effect through negative regulation of the NF-κB signaling. Oncotarget 6 (17), 14982–14992 (2015).
Iansante, V. et al. PARP14 promotes the Warburg effect in hepatocellular carcinoma by inhibiting JNK1-dependent PKM2 phosphorylation and activation. Nat. Commun. 6, 7882 (2015).
Kato, M. et al. Crystal structure of pyruvate dehydrogenase kinase 3 bound to lipoyl domain 2 of human pyruvate dehydrogenase complex. EMBO J. 24 (10), 1763–1774 (2005).
Kato, M. et al. Distinct structural mechanisms for Inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and radicicol. Structure 15 (8), 992–1004 (2007).
Khan, Z. F. et al. Exploring PDK3 Inhibition in lung cancer through drug repurposing for potential therapeutic interventions. Sci. Rep. 14 (1), 29672 (2024).
Goetze, K. et al. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol. 39 (2), 453–463 (2011).
Xie, J. et al. Beyond Warburg effect–dual metabolic nature of cancer cells. Sci. Rep. 4, 4927 (2014).
Anwar, S. et al. Targeting pyruvate dehydrogenase kinase signaling in the development of effective cancer therapy. Biochim. Et Biophys. Acta (BBA)-Reviews Cancer. 1876 (1), 188568 (2021).
Shestov, A. A. et al. Quantitative determinants of aerobic Glycolysis identify flux through the enzyme GAPDH as a limiting step. Elife 3, e03342 (2014).
Semenza, G. L. HIF-1: upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 20 (1), 51–56 (2010).
Denko, N. C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer. 8 (9), 705–713 (2008).
Chandramohan, S. et al. Investigation of ROS-mediated apoptotic and anti-metastatic effects of hydrastis Canadensis in lung adenocarcinoma cells: an in vitro and in Silico study. Tradit Med. Res. 10 (7), 42 (2025).
Liu, Z. et al. PDK3 drives colorectal carcinogenesis and immune evasion and is a therapeutic target for boosting immunotherapy. Am. J. Cancer Res. 14 (6), 3117 (2024).
Manning, G. et al. Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 27 (10), 514–520 (2002).
Dahiya, R. et al. Molecular interaction studies on ellagic acid for its anticancer potential targeting pyruvate dehydrogenase kinase 3. RSC Adv. 9 (40), 23302–23315 (2019).
Popov, K. M. et al. Primary structure of pyruvate dehydrogenase kinase establishes a new family of eukaryotic protein kinases. J. Biol. Chem. 268 (35), 26602–26606 (1993).
Hsu, Y. H., Huang, H. P. & Chang, H. R. The uremic toxin p-cresol promotes the invasion and migration on carcinoma cells via Ras and mTOR signaling. Toxicol. In Vitro. 58, 126–131 (2019).
Chen, X. et al. Effects of p-Cresol, a uremic toxin, on cancer cells. Translational Cancer Res. 12 (2), 367 (2023).
Mohammad, T., Mathur, Y. & Hassan, M. I. InstaDock: A single-click graphical user interface for molecular docking-based virtual high-throughput screening. Brief. Bioinform, 22(4), bbaa279 (2021).
Shafie, A. et al. Identification of phytoconstituents as potent inhibitors of casein Kinase-1 alpha using virtual screening and molecular dynamics simulations. Pharmaceutics 13(12), 2157 (2021).
DeLano, W. L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 40 (1), 82–92 (2002).
Biovia, D. S. Discovery studio visualizer. San Diego, CA, USA, 936. (2017).
Anwar, S. et al. Implications of tempol in pyruvate dehydrogenase kinase 3 targeted anticancer therapeutics: Computational, spectroscopic, and calorimetric studies. J. Mol. Liq. 350, 118581 (2022).
Jairajpuri, D. S. et al. Investigating the role of thymol as a promising inhibitor of pyruvate dehydrogenase kinase 3 for targeted cancer therapy. Int. J. Biol. Macromol. 259, 129314 (2024).
Berendsen, H. J., van der Spoel, D. & van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91 (1–3), 43–56 (1995).
Hess, B. et al. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4 (3), 435–447 (2008).
Maisuradze, G. G., Liwo, A. & Scheraga, H. A. Relation between free energy landscapes of proteins and dynamics. J. Chem. Theory Comput. 6 (2), 583–595 (2010).
Anwar, S. et al. Rosmarinic acid exhibits anticancer effects via MARK4 Inhibition. Sci. Rep. 10 (1), 1–13 (2020).
Shamsi, A. et al. MARK4 inhibited by ache inhibitors, donepezil and Rivastigmine tartrate: insights into alzheimer’s disease therapy. Biomolecules 10 (5), 789 (2020).
Yu Tanchuk, V. et al. A new scoring function for molecular Docking based on Autodock and Autodock Vina. Curr. Drug Discov. Technol. 12 (3), 170–178 (2015).
Govinda, K., Hassan, M. M. & Sirimulla, S. KinasepKipred: a predictive model for estimating ligand-kinase inhibitor constant (pKi). BioRxiv. 798561 (2019).
Karplus, M. & McCammon, J. A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 9 (9), 646–652 (2002).
Jiang, Z. et al. Effects of an electric field on the conformational transition of the protein: a molecular dynamics simulation study. Polymers 11 (2), 282 (2019).
Ali, S. hassan M, Islam A, Ahmad F.. A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded States. Curr. Protein Pept. Sci. 15(5): 456–476. (2014).
Shukla, R. & Tripathi, T. Molecular dynamics simulation of protein and protein–ligand complexes. Computer-aided drug design, 133–161 (2020).
Morris, C. J. & Corte, D. D. Using molecular Docking and molecular dynamics to investigate protein-ligand interactions. Mod. Phys. Lett. B. 35 (08), 2130002 (2021).
Dahiya, R. et al. Investigation of inhibitory potential of Quercetin to the pyruvate dehydrogenase kinase 3: towards implications in anticancer therapy. Int. J. Biol. Macromol. 136, 1076–1085 (2019).
Parveen, I. et al. Synthesis, Estrogen receptor binding affinity and molecular Docking of pyrimidine-piperazine-chromene and-quinoline conjugates. Bioorg. Med. Chem. Lett. 27 (18), 4493–4499 (2017).
Queen, A. et al. Biological evaluation of p-toluene sulphonylhydrazone as carbonic anhydrase IX inhibitors: an approach to fight hypoxia-induced tumors. Int. J. Biol. Macromol. 106, 840–850 (2018).
Kim, M. J. et al. Transcriptome dynamics of alternative splicing events revealed early phase of apoptosis induced by Methylparaben in H1299 human lung carcinoma cells. Arch. Toxicol. 94, 127–140 (2020).
Sena-Lopes, Â. et al. Cell viability analysis of Toxocara Cati larvae with LIVE/DEAD® Viability/Cytotoxicity kit. Exp. Parasitol. 212, 107871 (2020).
Kim, J. Y. et al. Comparative study of cytotoxic signaling pathways in H1299 cells exposed to alternative bisphenols: BPA, BPF, and BPS. Toxicol. Res. 13 (6), tfae200 (2024).
Shen, J. et al. Magnolol inhibits the growth of non-small cell lung cancer via inhibiting microtubule polymerization. Cell. Physiol. Biochem. 42 (5), 1789–1801 (2017).
Lu, C. W. et al. Overexpression of pyruvate dehydrogenase kinase 3 increases drug resistance and early recurrence in colon cancer. Am. J. Pathol. 179 (3), 1405–1414 (2011).
Cui, L. et al. Overexpression of PDK2 and PDK3 reflects poor prognosis in acute myeloid leukemia. Cancer Gene Ther. 27 (1), 15–21 (2020).
Xu, J. et al. Metabolic enzyme PDK3 forms a positive feedback loop with transcription factor HSF1 to drive chemoresistance. Theranostics 9 (10), 2999 (2019).
Ying, Y. et al. A uremic solute, P-cresol, inhibits the proliferation of endothelial progenitor cells via the p38 pathway. Circ. J. 75 (9), 2252–2259 (2011).
London, J. A. et al. Synthesis and toxicity profile in 293 human embryonic kidney cells of the β D-glucuronide derivatives of ortho-, meta-and para-cresol. Carbohydr. Res. 499, 108225 (2021).
Li, J. et al. Kinase inhibitors and kinase-targeted cancer therapies: recent advances and future perspectives. Int. J. Mol. Sci. 25 (10), 5489 (2024).
Tomuleasa, C. et al. Therapeutic advances of targeting receptor tyrosine kinases in cancer. Signal. Transduct. Target. Therapy. 9 (1), 201 (2024).
Jaradat, S. K. et al. Targeting receptor tyrosine kinases as a novel strategy for the treatment of triple-negative breast cancer. Technol. Cancer Res. Treat. 23, 15330338241234780 (2024).
Acknowledgements
Mohd Shahnawaz Khan extend his appreciation to Ongoing Research Funding Program (ORF-2025-352), King Saud University, Riyadh, Saudi Arabia for funding this research. Dr. Akram Ashames is grateful to Ajman University, UAE for supporting the publication through IRG (2024-IRG-PH-8).
Author information
Authors and Affiliations
Contributions
Conceptualization, Akram Ashames and Saleha Anwar; Data curation, Anas Shamsi, Nayab, Pinky; Investigation, Akram, Saleha, Suhel Parvez, Khuzin Dinislam and Mohd Shahnawaz Khan; Methodology, Saleha Anwar, Nayab, Pinky and Moyad Shahwan; Resources, Md. Imtaiyaz Hassan, Moyad Shahwan and Suhel Parvez; Software, Anas Shamsi, Saleha Anwar and Mohd Shahnawaz Khan; Supervision, Akram Ashames and Anas Shamsi; Validation, Saleha Anwar, Pinky, Md. Imtaiyaz Hassan and Anas Shamsi; Visualization, Akram, Khuzin Dinislam, Saleha and Anas Shamsi; Writing – original draft, Saleha Anwar, Anas Shamsi, Mohd Shahnawaz Khan and Akram Ashames; Writing – review & editing, Moyad Shahwan, Md. Imtaiyaz Hassan, Khuzin Dinislam and Suhel Parvez.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ashames, A., Anwar, S., Sulaimani, M.N. et al. Targeting cancer metabolism through p-cresol mediated Inhibition of pyruvate dehydrogenase kinase 3 in lung cancer. Sci Rep 15, 39561 (2025). https://doi.org/10.1038/s41598-025-25246-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-25246-3
