
Abstract
The therapeutic potential of human umbilical cord-derived mesenchymal stem cells (hUC-MSCs) is significantly constrained by replicative senescence during in vitro expansion. To investigate this phenomenon, we established a long-term passaging model of hUC-MSCs and validated senescence-associated phenotypes. Through integrated transcriptomic and eccDNA profiling, we systematically analyzed three compartments—cell pellets (CP), cell culture media (CM), and extracellular vesicles (EVs)—at sequential timepoints (days 5, 22 and 43, post-seeding). Intriguingly, while eccDNA abundance in CP remained stable during senescence progression, CM and EVs exhibited higher eccDNA loads, respectively, compared to CP. Moreover, CM- and EVs-derived eccDNAs shared conserved size distribution patterns distinct from CP-associated eccDNAs. Furthermore, the compartment-specific eccDNA dynamics over time coincided with activation of growth-regulatory gene expression. We identified recurrent eccDNA species across all compartments, harboring genes linked to senescence-associated processes. Our study uncovers compartment-specific eccDNA dynamics during hUC-MSC aging and proposes their potential as biomarkers for senescence monitoring. These findings provide a foundation for developing strategies to mitigate senescence-related limitations in hUC-MSC clinical applications.
Similar content being viewed by others


Introduction
Human umbilical cord mesenchymal stem cells (hUC-MSCs) have emerged as promising candidates for regenerative medicine and tissue engineering due to their multipotency and immunomodulatory properties1,2,3,4. Human umbilical cord-derived mesenchymal stem cells (hUC-MSCs) have attracted considerable attention due to their low immunogenicity, ease of collection, and potent immunomodulatory properties. They are being actively investigated in regenerative medicine and have shown therapeutic potential in treating inflammatory diseases, autoimmune disorders, and tissue injuries. However, extended in vitro expansion of hUC-MSCs for therapeutic applications triggers replicative senescence, a process characterized by irreversible cell cycle arrest and functional deterioration, which critically compromises their clinical efficacy5,6,7. Notably, senescent hUC-MSCs exhibit impaired homing efficiency, diminished migratory capacity, and reduced immunosuppressive activity8. Furthermore, MSC senescence leads to the accumulation of DNA damage, heightening the risk of genomic instability5. These factors significantly impede the clinical applications of MSCs. Therefore, elucidating the molecular mechanisms underlying hUC-MSC senescence is critical not only for improving their therapeutic potential but also for enabling more effective senescence monitoring during in vitro expansion. Existing approaches for tracking cellular aging remain limited, highlighting the need for novel, biomarker-based strategies.
Extrachromosomal circular DNA (eccDNA) are covalently closed circular DNA molecules derived from chromosomal DNA, and have been implicated in genomic instability and cellular adaptation9,10. Their potential role in cellular senescence, especially in stem cells, remains largely unexplored. Recent advances in genomic research have highlighted the dual role of extrachromosomal circular DNA (eccDNA) as both a mediator of genomic instability and a dynamic regulator of cellular adaptation10,11. EccDNA comprises covalently closed, non-chromosomal DNA molecules ranging from hundreds of base pairs to megabases in length, generated through mechanisms such as chromothripsis, DNA repair errors, or retrotransposon excision12,13. While eccDNA has been implicated in tumor evolution, neurodegenerative disease, and adaptive responses to environmental stress11,14,15,16, its spatiotemporal dynamics and functional relevance during cellular senescence—particularly in the context of hUC-MSCs—remain entirely unexplored.
To address this knowledge gap, we established a replicative senescence model of hUC-MSCs through long-term serial passaging. Senescence phenotypes were validated through morphometric analysis of cell size and morphology (e.g., nuclear enlargement, loss of spindle shape), alongside viability assays and population doubling measurements. Furthermore, we performed high-resolution eccDNA sequencing to interrogate the correlation between eccDNA burden and senescence progression. Our study not only provides the first evidence linking eccDNA accumulation to hUC-MSC senescence but also proposes eccDNA landscape profiling as a sensitive tool for real-time monitoring of senescence status during therapeutic MSC preparation.
Results
Establishment and phenotypic validation of a replicative senescence model in hUC-MSCs
To systematically investigate the spatiotemporal dynamics of extrachromosomal circular DNA (eccDNA) during hUC-MSCs aging, we developed a long-term passaging model of human umbilical cord-derived MSCs (hUC-MSCs). Primary hUC-MSCs isolated from nine independent donors were subjected to continuous in vitro expansion over 43 days, spanning 12 passages (P1 to P12). Multi-compartment profiling was performed at critical senescence stages (P1, P6, P12) to capture eccDNA dynamics across intracellular (cell pellet, CP), secreted (conditioned media, CM), and vesicular (extracellular vesicles, EVs) fractions (Fig. 1A). Integrated Circle-seq and RNA-seq analyses were conducted to delineate eccDNA landscape evolution and transcriptomic reprogramming during senescence progression.

Characterization of replicative senescence features in hUC-MSCs. (A) The MSCs from the umbilical cords of nine puerperal volunteers were acquired and amplified in-vitro. Different methodologies are performed on three types of the cell contents. The cell pellets (CP), conditional cell culture media (CM) and extracellular vesicles (EVs) were sampled at passage 1 and passage 6, passage 12. Whole genome sequencing (WGS) was performed on CP samples from passage 1. RNA-Seq was performed on samples from all three timepoints of CP. And Circle-Seq was applied for eccDNA detection in samples of all three passages of CP, CM, and EVs. (B–D) The diameter, cell viability, and cumulative population doubling level of the nine MSCs cell lines during the long-term passage (****p < 0.0001, one-way ANOVA test).
Consistent with replicative senescence, prolonged culture induced progressive morphological deterioration, characterized by nuclear enlargement (P12 vs. P1: 1.8 ± 0.3-fold increase; p < 0.001) and loss of spindle-shaped morphology, accompanied by cellular flattening and irregular cytoplasmic spreading (Fig. 1B). Quantitative morphometric analysis revealed a significant increase in cell diameter at P6 (18.2 ± 2.1 μm) and P12 (23.5 ± 3.4 μm) compared to P1 (12.7 ± 1.6 μm; p < 0.01) (Fig. 1C). Proliferative capacity, assessed by cumulative population doubling level (CPDL), exhibited biphasic kinetics—rapid expansion during early passages (days 0–21) followed by marked attenuation from day 22 onward (slope reduction: 0.38 vs. 0.12; p < 0.001) (Fig. 1D). Concurrently, cell viability progressively declined post-P6, reaching 62.3 ± 5.7% of initial levels by P12 (Supplementary Fig. 1A).
Our data collectively demonstrate the successful establishment of a temporally resolved replicative senescence model that recapitulates hallmark features of hUC-MSC aging, including morphological degeneration, proliferative arrest, and functional decline.
Stable global eccDNA abundance and repeat composition during long-term culture of hUC-MSCs
To delineate the landscape of extrachromosomal circular DNA (eccDNA) during hUC-MSC senescence, we performed high-resolution Circle-Seq on cell pellets (CP) from P1, P6, and P12 cultures (Fig. 2A). To control for sequencing depth variability, eccDNA counts were normalized to eccDNAs per million mapped reads (EPM). Quantitative analysis revealed stable eccDNA abundance across passages, suggesting a conserved eccDNA load in CPs despite progressive senescence (not significant, FDR-adjusted Wilcoxon test, Fig. 2B). We next examined the genomic origins of eccDNAs by mapping them to protein-coding genes (eccGenes) and performing functional enrichment analysis to assess their potential biological roles. Using a length-normalized metric (eccDNA abundance = eccDNA count per gene/gene length), we identified 1,382 dynamically regulated eccGenes showing passage specific expression patterns (Fig. 2C). Notably, eccDNA-derived genomic elements, including 3’UTRs, 5’UTRs, introns, and exons exhibited temporal redistribution (not significant, FDR-adjusted Wilcoxon test, Fig. 2D). In contrast, repeat element composition remained invariant across passages (not significant, FDR-adjusted Wilcoxon test, Fig. 2E). Conclusively, we identified a highly abundant eccDNA profile in cell pellets of hUC-MSCs, and observed a relatively steady eccDNA number during consecutive culture.

Identification of the extrachromosomal circular DNA features in cell pellets of MSCs. (A) The workflow of eccDNA enrichment through DNA extraction, linear DNA removal by exonuclease V, Phi-29 rolling circle amplification, sequencing, and analysis by Circle-Map pipeline. (B) The eccDNA per million mappable reads of CP samples of passage 1, 6 and 12 (Wilcoxon test). (C) The differential eccDNA gene abundance between P1, P6 and P12. The X-axis and Y-axis represent sample id and gene id respectively. 1382 eccGenes presented differently between 3 groups by the Wilcox test. (D) Normalized ratio of eccDNA in different genomic elements. (E) Normalized mapping ratio of eccDNA reads aligned to repeats.
Distinct eccDNA profiles in cell pellets, media, and extracellular vesicles during hUC-MSC senescence
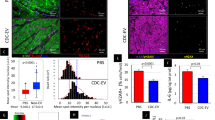
To delineate senescence-associated eccDNA secretion patterns, we performed comparative analysis of eccDNA landscapes in conditioned medium (CM) and extracellular vesicles (EVs) across P1, P6, and P12 (Fig. 3A). Size distribution profiling revealed conserved fragmentation patterns in CM and EVs, with predominant eccDNAs < 2,000 bp and shared peak sizes (~ 337, 583, 733, 937 bp) (Fig. 3B,C). In contrast to the stable eccDNA abundance observed in CPs, both CM and EVs exhibited progressive eccDNA depletion during senescence in P6 and P12 than in P1 (p < 0.01, FDR-adjusted Wilcoxon test, Fig. 3D,E, Supplementary Table 1). Notably, CM and EVs consistently harbored higher eccDNA loads than CPs throughout senescence (p < 0.01, FDR-adjusted Wilcoxon test, Fig. 3F). While equivalent eccDNA quantities were detected across compartments at P1, compartment-specific divergence emerged from P6 onward, with CPs maintaining baseline levels as CM/EVs declined (Fig. 3G, Supplementary Table 2, Supplementary Table 3). GC content analysis revealed compartment-specific patterns: CM-associated eccDNAs showed progressively lower GC content across passages (p < 0.01, p < 0.01 and p < 0.05, FDR-adjusted Wilcoxon test), while CP and EVs maintained stable GC profiles (Fig. 3H, Supplementary Table 4). Conclusively, dynamic size distribution, eccDNA number and GC content are observed during hUC-MSCs senescence and biopsy types.

Extracellular circular DNA identification from EVs and CM samples. (A) Technical pipeline of EVs extraction from the cell culture media. (B,C) The size distribution and the density of eccDNA size of P1, P6, P12 samples in EV, and CM respectively. (D,E) The comparison of eccDNA number in EVs and in CM of P1, P6 and P12 (Wilcoxon test). (F) The overall eccDNA number of CP, CM, EV samples from all timepoints are compared (Wilcoxon test). (G) EPM normalization scores under different conditions (CP, CM, EV) across three time points (p1, p6, p12). Statistical analysis was performed using Wilcoxon test. (H) The median GC values under different conditions (CM, CP, EV) across three time points (P1, P6, P12). Statistical significance between groups was assessed using Wilcoxon test.
Correlation between eccDNA profiles and growth-related gene expression
To elucidate the genomic determinants of eccDNA biogenesis, we mapped eccDNA reads to annotated genomic features. Comparative analysis revealed distinct compartmentalization patterns: CM and EVs exhibited enhanced eccDNA enrichment in gene-adjacent regions (2 kb upstream/downstream) and intronic sequences compared to CP (Fig. 4A, Supplementary Table 5). Conversely, CP eccDNAs showed preferential accumulation in 5’UTRs, exons, 3’UTRs, and CpG islands relative to secreted fractions (Fig. 4A). Repeat element profiling identified compartment-specific signatures, with CP eccDNAs demonstrating heightened representation of LINEs, satellite DNA, rRNA genes, tRNA loci, and small nuclear RNA clusters compared to CM/EVs (Fig. 4B, Supplementary Table 6). Integrating eccDNA mapping with transcriptomic data revealed bidirectional gene-eccDNA associations, 1,922 genes showed positive correlation with eccDNA generation frequency, and 1,220 genes exhibited negative correlation (Fig. 4C). Notably, key regulators including CPVL (carboxypeptidase, vitellogenic-like), KLF8 (Kruppel-like factor 8), TMEM258 (Transmembrane Protein 258), and NR0B1 (Nuclear Receptor Subfamily 0 Group B Member 1) demonstrated expression-eccDNA positive correlation, whereas ARG1 (arginase 1), GPA33 (glycoprotein A33), KDM5C (lysine demethylase 5C), MARCHF1 (membrane associated ring-CH-type finger 1), UTY (ubiquitously transcribed tetratricopeptide repeat containing, Y-linked), PAG1 (phosphoprotein associated with glycosphingolipid microdomains 1), and ARHGEF16 (Rho guanine nucleotide exchange factor 16) showed inverse relationships. However, only the 5’ UTR regions of GPA33 (glycoprotein A33) remained significantly affected after Benjamini—Hochberg FDR correction (Fig. 4D). Recurrent eccDNAs from MARCHF1, PAG1, CPVL, ARG1, and KLF8 were persistently detected across passages (Fig. 4E), with predominant origins in exons, introns, and 3’UTRs. Pathway analysis of eccDNA-associated genes identified enrichment in growth-regulatory processes, including: Actin cytoskeleton remodeling Ras/MAPK signaling Axon guidance Growth hormone signaling (Fig. 4F).

The genomic function analysis of eccDNA in long term passage of hUC-MSCs. (A) Normalized ratio of eccDNA in different genomic elements. (B) Normalized mapping ratio of eccDNA reads aligned to repeats. (C) The number of genes associated with eccDNA across different genomic regions: Gene2KU, 5’UTR, Exon, Intron, 3’UTR, and Gene2KD. Positive bars (green) indicate enriched regions, while negative bars (brown) represent underrepresented ones. Values on each bar provide a quantitative comparison across these elements. (D) The distribution of genes associated with different genomic regions, including Gene2KU, 5’UTR, Exon, Intron, 3’UTR, and Gene2KD. The x-axis shows the p-value (− log10), and the y-axis represents the adjusted p-value (padj, − log10). Genes with significant expression changes are highlighted, with thresholds set at p-value = 0.01 and padj = 0.01 (marked by blue lines). Points are color-coded based on their genomic region, with labeled genes representing notable candidates within their respective categories. (E) The distribution of recurrent genes on eccDNAs across P1, P6, and P12. Each bar is segmented by sample count within the respective group, with colors representing the different time points (black for P1, brown for P6, and blue for P12). (F) The enriched biological pathways based on the gene set analysis of recurrent presented eccDNAs. Pathways include the regulation of the actin cytoskeleton, Ras signaling, axon guidance, growth hormone synthesis, secretion, and action, T cell receptor signaling, mRNA surveillance, Fc epsilon RI signaling, biosynthesis of nucleotide sugars, and D-amino acid metabolism. The x-axis indicates the count of genes associated with each pathway, showing the relative enrichment across these biological processes.
Shared eccDNAs show similar size distributions across compartments
To identify eccDNAs persistently present across intracellular and extracellular compartments, we analyzed shared eccDNA populations at progressive senescence stages (P1, P6, P12) under two sequence overlap thresholds (90% and 100%). At 90% overlap criteria, 5,977, 283, and 174 eccDNAs were common to CP, CM, and EVs at P1, P6, and P12, respectively (Fig. 5A). Size distribution analysis further revealed that eccDNAs shared across all three compartments predominantly occupied smaller size ranges compared to those present in any two compartments, which exhibited similar size profiles (Fig. 5B). Stringent sequence matching reduced the number of eccDNAs detected across all three compartments, with most shared eccDNAs showing a smaller size range.

Recurrent eccDNA is identified in CP, CM, EVs at P1, P6, P12. (A) The recurrently presented eccDNA in passage 1(P1), passage 6 (P6), passage 12 (P12) in CP, CM, EVs. The black bar represents eccDNA number in P1, blue bar represents eccDNA number in P6, and the yellow bar represents eccDNA number in P12. (B) The comparison of length distribution of eccDNA in P1, P6, P12. The brown line represents the interaction of CP and CM, blue line represents the interaction of CP and EV, and the yellow line represents the interaction of CP, EV and CM.
Discussion
Our study delineates the dynamic landscape of extrachromosomal circular DNA (eccDNA) during replicative senescence of human umbilical cord mesenchymal stem cells (hUC-MSCs), providing the first evidence of extracellular vesicle (EV)-mediated eccDNA trafficking. As a clinically relevant cell source, hUC-MSCs exhibit therapeutic potential in neurological disorders and serve as an ideal diploid model for senescence studies due to their rapid proliferation-associated senescence17,18,19. Moreover, hUC-MSCs serve as an ideal cell line for investigating eccDNA features during cell senescence, since MSCs are diploidy and presenting senescence status during fast replication.
Extrachromosomal circular DNA (eccDNA) is known as topology circular double stranded DNA molecules that derived from chromosomal DNA. In eukaryotes, the formation of eccDNAs is known as highly correlate to DNA repair during DNA replication12, and transcription20. Since the DNA replication, RNA transcription is reducing as MSCs grow to late passages, the eccDNA number are predicted to reduce. However, the eccDNA number in our dynamic senescence MSCs tends to keep stable, suggesting a balance of eccDNA formation and releasing. The eccDNA abundance in EVs and CMs are significantly higher than that in MSCs cells, and presented most abundant eccDNAs in passage 1, possibly indicate that more eccDNAs are released to extracellular in early passages. Although this is observed in in vitro study, we presume that eccDNA releasing from tumor cells would perform intercellular communication via this genetic information containing eccDNAs. In our study, eccDNAs identified in MSCs (cell pellets) were enriched for genes associated with cell proliferation pathways. While this may suggest a potential role in local cell–cell communication, this hypothesis requires further experimental validation.
We then wondered the form of cell free eccDNAs released by cells. In the comparison of eccDNAs from extracellular vesicles (EVs) and cell culture media (CM), we identified a highly identical size distribution, indicating a shared origin and model of eccDNA release via extracellular vesicles. In details, the eccDNAs in CM and EVs are enriched at size peak of 201 to 204, 362 to 364, 560, and 730 bp, while size of eccDNAs in CP is enriched at 370, 580, 733, and 937 bp. Thus, we concluded that eccDNAs in EVs and CM are possibly from the same origin. In our study, only a subset of eccDNAs were consistently detected across all compartments, and these “universally conserved” species were enriched in shorter fragment sizes. These findings suggest that stringent size or sequence constraints may influence the stability, detectability, or biological roles of such eccDNAs. Further functional studies are needed to determine whether these shorter eccDNAs possess regulatory functions or structural advantages. Furthermore, we observed a significant decrease in eccDNA quantity from young (P1) to old passages (P6, P12) of EVs, consistent with the above concept of educed DNA replication rate and other factors of decreased cell numbers, apoptosis during long-term culture should also be considered12,21,22.
From the perspective of eccDNA-derived gene fragments, we observed a redistribution of 3′UTRs, 5′UTRs, exons, and introns during replicative senescence. During hUC-MSC senescence, the overall abundance of eccDNAs remained stable, while the genomic origins (eccGenes) and structural components, including 3′UTRs, 5′UTRs, exons, and introns, exhibited dynamic redistribution across passages, whereas repeat element composition remained largely unchanged. These findings collectively demonstrate that while global eccDNA quantity and repeat element enrichment are conserved during hUC-MSC senesence, gene-specific eccDNA generation and element-level redistribution exhibit senescence-associated dynamics, implicating eccDNA in fine-tuning genomic regulation during MSC aging.
Limitations
This study has several limitations. First, the relatively small number of donors limits the generalizability of our findings, and potential biological heterogeneity between individuals may influence the observed eccDNA dynamics. Future studies involving larger donor cohorts are warranted to further validate these observations. Second, while human umbilical cord-derived MSCs are known to be multipotent and capable of differentiating into various lineages such as osteoblasts, adipocytes, chondrocytes, and myocytes, we did not perform lineage differentiation assays in this work. Third, although we assessed senescence using multiple morphological and transcriptomic markers (e.g., p21, HMGB1, γH2A.X, SA-β-Gal), we did not validate these markers at the protein level (e.g., by Western blot) or at the RNA level (e.g., by qPCR) due to sample constraints. Future studies will include comprehensive validation of both differentiation capacity and molecular markers to further strengthen the characterization of senescence phenotypes.
In conclusion, our study provides insights into eccDNA features of senescence hUC-MSCs and the first eccDNA profile of extracellular vesicles (EVs). Notably, the eccDNA number remains stable during the senescence of hUC-MSCs. This stability is maintained through the release of eccDNAs into the extracellular environment, likely facilitated by extracellular vesicles as vehicles, suggesting its potential role in intercellular communication. These findings suggest that compartment-specific eccDNA dynamics may hold potential as biomarkers for senescence monitoring, but further functional studies are required to validate this possibility.
Materials and methods
Isolation and primary culture of umbilical cord mesenchymal stem cells (hUC-MSCs)
Nine sets of stem cell lines were isolated from umbilical cord tissues, and all donors were coded for anonymity (SC-1 to SC-9). The umbilical cord tissues were cut into pieces, and then collected Wharton’s Jelly. After tissue enzymatic digestion by collagenase II, the nine sets of stem cell lines were seeded on nine culture flasks, respectively and cultured in the cell culture incubator with 5% CO2 at 37 °C. Then UltraCULTURE Serum-free medium (Lonza) with serum substitute for animal cell culture (Ultroser™ G, SARTORIUS) and 1% l-Glutamine(Gibco) was changed every 3 ~ 4 days. When the cell confluency reached more than 80%, the cells were collected as P0.
Secondary culture of hUC-MSCs
Cells were continuously passaged every 3–4 days once they reached approximately 80% confluence. For each passage, cells were seeded at a density of ~ 8000–10,000 cells/cm2 and maintained under standard conditions (37 °C, 5% CO2). This serial passaging was carried out over a total period of 43 days to establish a replicative senescence model.
Extraction of extracellular vesicles from human mesenchymal stem cells
The serum-free conditioned media were collected from either 3 or 4-day hUC-MSC culture. The cells and debris were removed by 500 g centrifugation for 10 min. The supernatant was carefully collected and transferred to a new tube, then centrifuged at 3000 g for 20 min to further remove cell debris contaminants. As a result, the treated conditioned media can be stored in -80℃ freezer for longer than 24 h before ultracentrifugation23. A one-step sucrose cushion ultracentrifugation method was applied for the exosomes’ isolation. The samples were centrifugated at 12,000×g for 30 min to remove the microvesicles. 4 ml of 30% sucrose was added to the bottom of the ultracentrifugation tube, approximately 30 ml of the supernatant was added slowly not disturbing the sucrose layer. Then ultracentrifugation at 100,000×g was performed for 90 min at 4 °C using a Sorvall WX + ultracentrifuge in a fixed-angle rotor (Thermo Scientific). Then the sucrose layer containing the exosomes was collected by carefully discarding the supernatant. Phosphate-buffered saline (PBS) was applied to resuspend the sucrose layer, followed by ultracentrifugation of 90 min 100,000×g at 4 °C. Finally, exosome pellets were resuspended in 400 ul of PBS and stored at − 80 °C for long time.
Circle-Seq
For all samples, DNA quantity was normalized based on the number of viable cells. Prior to nucleic acid extraction, live cells were counted using a hemocytometer and trypan blue exclusion. Equal numbers of cells were used for each extraction to ensure consistency across replicates and passages. Total DNA isolation was performed by Magattract HMW DNA kit (Qiagen)for CP-DNA or MGIEasy Circulating DNA Isolation Kit (MGI) for CM-DNA and EV-DNA, following instruction of the manufactures. The DNA concentration was measured by a Qubit dsDNA HS Assay kit (Invitrogen). To remove the linear DNA, the DNA samples were treated with Plasmid-Safe Dnase (Biosearch Technologies) at 37 °C. For DNA of cell pellets, a consecutive five days of Dnase input was performed10,24. For DNA of cell culture media or exosomes, the digestion was performed overnight25. The digestion products were cleaned up from the reaction mixtures using VAHTS DNA clean beads (Vazyme) and resolved in 30 μl DNase-free water. After RCA reaction, the phi29-amplified products were recovered by VAHTS DNA clean beads, resolved in 80 μl DNase-free water and quantified by Qubit dsDNA BR kit (Invitrogen).
Library preparation and sequencing
About 1 ug of phi29-amplified DNA products was sonicated by Covaris LE220 into 300–500 bp DNA fragments. Then 80 ng of fragmented DNA was subjected to library preparation using MGIEasy DNA Library Preparation Kit (MGI-BGI). Bioanalyzer 2100 (Agilent) evaluated the sequencing library’s content and quality. The library was finally sequenced by the MGISeq-T5 sequencing machine, pair-end (PE) 150 bp.
Preprocessing of raw sequencing data
The raw sequencing reads were qualified fastQC (v0.11.3) (https://www.bioinformatics.babraham.ac.uk/projects/fastqc). Then the quality information of all samples was counted by MultiQC (v1.10.1)26. Those reads sequences produced in FASTQ format were pre-processed through fastp (v0.21.0)27. The remaining reads that passed all the filtering steps were counted as clean reads. The index of the reference genome (GRch38.p13) was built using BWA28 (v0.7.12) and clean reads were aligned to the reference genome using BWA mem).
eccDNA detection by circle-map
To detect eccDNAs from the aligned data, we applied Circle-Map (v1.1.4) software (https://github.com/iprada/Circle-Map) to detect the coordinates of each eccDNA29. And then, to improve the accuracy of eccDNA detection, several filtering steps were performed as we previously described25. The specific settings were as follows: (1) the number of split reads is more than two, (2) the score of the circle is more than 200, and (3) the mean base coverage within the circular DNA detection coordinates is greater than the standard deviation of the base coverage vector, (4) Coverage increase in the start/end coordinate is more than 0.3, (5) the fraction of bases not covered by reads within the circular DNA detection coordinates is lower than 0.1.
Genomic and sequence features of eccDNAs
After mapping the overall population of plasma-derived eccDNA in the human genome, the numbers of eccDNA molecules whose starting positions map to genomic obtained. The theoretical distribution of plasma eccDNA was predicted as the percentage of the genome covered. The normalized genomic coverage of plasma eccDNA was then calculated using the following formula:
We used the “observed/expected ratio of genomic elements” for statistics of seven major classes of genomic elements (i.e., 3′UTR, 5′UTR, CpG island, exon, Gene2KbD, Gene2KbU, and intron), and the “observed/expected ratio of genomic elements” was calculated according to the following formula:
Additionally, using bedtools (v2.25.0), we counted the number of reads that map to a specific repeat element. The normalized mapping ratio was calculated as the percentage of reads that mapped to a specific repeat element divided by the percentage of the specific repeat element in the nuclear genome.
Genome annotation of eccDNA fragment
To calculate the bias of each gene to produce eccDNA, we defined the abundance of eccDNA on a gene using the number of eccDNA on each gene and the gene length and the eccDNA abundance were calculated as follows30:
Genes with differential eccDNA abundance were then identified based on a Wilcoxon rank-sum test, and differential genes with significant eccDNA abundance were filtered out using the threshold of p < 0.05 and |log2 FC|≥ 1. GO and KEGG31,32 enrichment analysis was implemented by the WEB-based gene set analysis toolkit (http://www.webgestalt.org/).
Statistics
All statistical analyses were performed by R (v4.2.2) or perl(v5.32.1). Pairwise comparisons between two groups were assessed using the Wilcoxon rank-sum test. For analyses involving more than two groups (e.g., comparisons across passages P1, P6, and P12, or across different compartments such as CP, CM, and EV), p-values were adjusted for multiple comparisons using the Benjamini–Hochberg false discovery rate (FDR) correction, unless otherwise specified. Differential eccDNA abundance across multiple genes was also subjected to FDR adjustment. For pathway enrichment analyses, significance was defined as FDR-adjusted p < 0.05. In all cases, corrected p-values < 0.05 were considered statistically significant.
Data availability
The data supporting this have been deposited into the CNGB Sequence Archive (CNSA) of China National GeneBank DataBase (CNGBdb) with accession number CNP0004523 and are available from the corresponding author on reasonable request.
Abbreviations
- UTR:
-
Untranslated region
- Gene2kbD:
-
2Kb downstream of genes
- Gene2kbU:
-
2Kb upstream of genes
- SINE:
-
Short interspersed nuclear element
- srpDNA:
-
Signal recognition particle DNA repeats
- rDNA:
-
Ribosomal DNA repeats
- tDNA:
-
Transport DNA repeats
- scDNA:
-
Small conditional DNA repeats
- snDNA:
-
Small nuclear DNA repeats
- LTR:
-
Long terminal repeat
- LINE:
-
Long interspersed nuclear element
- RC:
-
Rolling circle repeats
References
Andrzejewska, A., Lukomska, B. & Janowski, M. Concise review: Mesenchymal stem cells: From roots to boost. Stem Cells Dayt. Ohio 37, 855–864 (2019).
Tkach, M. & Théry, C. Communication by extracellular vesicles: Where we are and where we need to go. Cell 164, 1226–1232 (2016).
Pittenger, M. F. et al. Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–147 (1999).
Mackay, A. M. et al. Chondrogenic differentiation of cultured human mesenchymal stem cells from marrow. Tissue Eng. 4, 415–428 (1998).
Neri, S. Genetic stability of mesenchymal stromal cells for regenerative medicine applications: A fundamental biosafety aspect. Int. J. Mol. Sci. 20, 2406 (2019).
Turinetto, V., Vitale, E. & Giachino, C. Senescence in human mesenchymal stem cells: Functional changes and implications in stem cell-based therapy. Int. J. Mol. Sci. 17, 1164 (2016).
Li, Z. et al. Epigenetic dysregulation in mesenchymal stem cell aging and spontaneous differentiation. PLoS ONE 6, e20526 (2011).
de Witte, S. F. H. et al. Aging of bone marrow– and umbilical cord–derived mesenchymal stromal cells during expansion. Cytotherapy 19, 798–807 (2017).
Noer, J. B., Hørsdal, O. K., Xiang, X., Luo, Y. & Regenberg, B. Extrachromosomal circular DNA in cancer: History, current knowledge, and methods. Trends Genet. 38, 766–781 (2022).
Møller, H. D. et al. Circular DNA elements of chromosomal origin are common in healthy human somatic tissue. Nat. Commun. 9, 1–12 (2018).
Turner, K. M. et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543, 122–125 (2017).
Arrey, G., Keating, S. T. & Regenberg, B. A unifying model for extrachromosomal circular DNA load in eukaryotic cells. Semin. Cell Dev. Biol. https://doi.org/10.1016/j.semcdb.2022.03.002 (2022).
Noer, J. B., Hørsdal, O. K., Xiang, X., Luo, Y. & Regenberg, B. Extrachromosomal circular DNA in cancer: history, current knowledge, and methods. Trends Genet. 20, 1–16 (2022).
Zou, S. et al. Extrachromosomal circular MiR-17-92 amplicon promotes hepatocellular carcinoma. Hepatology https://doi.org/10.1097/HEP.0000000000000435 (2023).
Prada-Luengo, I. et al. Replicative aging is associated with loss of genetic heterogeneity from extrachromosomal circular DNA in Saccharomyces cerevisiae. Nucleic Acids Res. https://doi.org/10.1093/nar/gkaa545 (2020).
Henriksen, R. A. et al. Circular DNA in the human germline and its association with recombination. Mol. Cell https://doi.org/10.1016/j.molcel.2021.11.027 (2022).
Sun, J. M. et al. Infusion of human umbilical cord tissue mesenchymal stromal cells in children with autism spectrum disorder. Stem Cells Transl. Med. 9, 1137–1146 (2020).
Lee, J. et al. Efficacy of intravenous mesenchymal stem cells for motor recovery after ischemic stroke: A neuroimaging study. Stroke 53, 20–28 (2022).
Gu, J. et al. Therapeutic evidence of umbilical cord-derived mesenchymal stem cell transplantation for cerebral palsy: a randomized, controlled trial. Stem Cell Res. Ther. 11, 43 (2020).
Dillon, L. W. et al. Production of extrachromosomal MicroDNAs is linked to mismatch repair pathways and transcriptional activity. Cell Rep. 11, 1749–1759 (2015).
Wang, Y. et al. eccDNAs are apoptotic products with high innate immunostimulatory activity. Nature https://doi.org/10.1038/s41586-021-04009-w (2021).
Paulsen, T. et al. MicroDNA levels are dependent on MMEJ, repressed by c-NHEJ pathway, and stimulated by DNA damage. Nucleic Acids Res. https://doi.org/10.1093/nar/gkab984 (2021).
Bojmar, L. et al. Extracellular vesicle and particle isolation from human and murine cell lines, tissues, and bodily fluids. STAR Protoc. 2, 100225 (2021).
Gupta, S. et al. An improvised one-step sucrose cushion ultracentrifugation method for exosome isolation from culture supernatants of mesenchymal stem cells. Stem Cell Res. Ther. 9, 180 (2018).
Lv, W. et al. Circle-Seq reveals genomic and disease-specific hallmarks in urinary cell-free extrachromosomal circular DNAs. Clin. Transl. Med. 12, e817 (2022).
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics https://doi.org/10.1093/bioinformatics/btw354 (2016).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics https://doi.org/10.1093/bioinformatics/bty560 (2018).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM (2013) https://doi.org/10.48550/arxiv.1303.3997.
Prada-Luengo, I., Krogh, A., Maretty, L. & Regenberg, B. Sensitive detection of circular DNAs at single-nucleotide resolution using guided realignment of partially aligned reads. BMC Bioinform. https://doi.org/10.1186/s12859-019-3160-3 (2019).
Quinlan, A. R. & Hall, I. M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics https://doi.org/10.1093/bioinformatics/btq033 (2010).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016).
Ogata, H. et al. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27, 29–34 (1999).
Acknowledgements
Foremost, we express our gratitude to all volunteers who consented to participate in this study. And we would also like to thank for China National GeneBank (CNGB) for the support to this research.
Author information
Authors and Affiliations
Contributions
Q.Y. and D.M.F. proposed the scientific hypothesis and designed the study. Q.Y. and Q.X. provided clinical support and relevant information. Y.J.Y., D.W., W.J.Z., and Y.W. conducted the experiments. C.L.L., Z.X., and D.M.F. supervised the study. Z.X. analyzed the genomic data. Q.Y., Y.J.Y., and Z.X. drafted the manuscript, while Q.X. and D.M.F. reviewed and revised it. All authors contributed to the study design, discussions, and manuscript revisions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The collection of umbilical cords was approved by the Human Research Ethics Committee of BGI (BGI-IRB 21080). Informed consent was obtained from all donors. All procedures were conducted in accordance with the relevant guidelines and regulations of Chinese Academy of Sciences, Qingdao University and BGI-Qingdao.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yan, Q., Xu, Q., Yu, Y. et al. Compartment-specific eccDNA patterns reveal senescence associated biomarkers in hUC-MSCs. Sci Rep 16, 1980 (2026). https://doi.org/10.1038/s41598-025-25377-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-25377-7
