
Abstract
Acute myocardial infarction (AMI) triggered cardiomyocyte senescence and impaired cardiac function. Methylation modifications and related enzymes in patients were also significantly altered, but the association between cellular senescence and demethylation remained unclear. This study obtained data from public databases, screened candidate genes by intersecting differentially expressed genes (DEGs) with related genes, identified characteristic genes using three machine learning methods to determine biomarkers, and explored their regulatory mechanisms through gene set enrichment analysis (GSEA) and other approaches. Finally, BCL3, MMP9, NAMPT, and TLR4 were identified as potential biomarkers. GSEA showed they were significantly enriched in 18 common pathways. Thirteen immune cells, such as CD8 T cells, differed significantly between AMI and control samples. A total of 58 miRNAs, 10 lncRNAs, and 242 transcription factors (TFs) were predicted to interact with these biomarkers. Targeted analysis predicted 25 potential therapeutic drugs for AMI, and the biomarkers could target Retinoic acid CTD 00006918. GeneMANIA analysis identified 20 functionally related genes, while Friends analysis showed BCL3 had prominent functional similarity. Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR) results validated the expression of biomarkers, consistent with bioinformatics analyses. These findings may provide valuable clues for AMI treatment.
Similar content being viewed by others
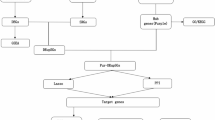


Introduction
Acute myocardial infarction (AMI) is one of the leading cardiovascular diseases with the highest mortality and morbidity rates globally. Its core pathological mechanism is the acute occlusion of coronary arteries leading to ischemic necrosis of the myocardium, which in turn causes serious consequences such as ventricular remodeling and heart failure1. Although timely revascularization (such as thrombolysis and PCI) can save ischemic myocardium, reperfusion injury, microcirculation disorders, and subsequent pathological ventricular remodeling still lead to progressive deterioration of heart function in about 20–30% of patients2. Therefore, exploring the molecular mechanisms of AMI and identifying novel biomarkers is of great significance for early diagnosis, prognostic assessment, and the development of targeted therapies.
In recent years, the role of cellular senescence in cardiovascular diseases has garnered significant attention. At the same time, epigenetic regulation (such as DNA methylation/demethylation) has also been confirmed to participate in the pathological process of myocardial injury. The interaction between these two mechanisms has gradually become a research hotspot in AMI. Cellular senescence refers to a state of irreversible growth arrest that cells enter under stress conditions such as telomere shortening, DNA damage, or oxidative stress, accompanied by the activation of the p53/p16INK4a pathway and the secretion of the senescence-associated secretory phenotype (SASP)3,4. Senescent cells secrete SASP factors (such as IL-6, TNF-α, MMPs), activating pro-inflammatory pathways like NF-κB and recruiting immune cells to infiltrate the infarct area5. Additionally, factors like TGF-β in the SASP can induce senescence in neighboring cells through paracrine signaling, forming a vicious cycle6. Before the occurrence of AMI, the senescence of endothelial cells and cardiomyocytes promotes the development of atherosclerosis. Endothelial cellular senescence increases vascular permeability and inflammation, cell infiltration, and accelerates plaque formation; cardiomyocyte senescence weakens cardiac functional reserve7. Furthermore, the accumulation of senescent cells is closely related to cardiac fibrosis and dysfunction, significantly reducing the heart’s recovery capacity after AMI8.
Demethylation is a key mechanism of epigenetic regulation, where DNA demethylation (such as TET enzyme-mediated oxidation of 5-methylcytosine) regulates transcription by altering the methylation status of gene promoter regions; RNA demethylation (such as the removal of m6A modifications) affects mRNA stability9. In AMI models, the inhibition of demethylation enzymes can improve cardiac function, reduce cardiomyocyte apoptosis, and promote cardiac regeneration10. Moreover, demethylation not only enhances the resistance of cardiomyocytes to ischemia, improving cell survival and function11, but may also influence cardiac remodeling and regenerative capacity by regulating the expression of senescence-related genes8. Although both cellular senescence and demethylation have been confirmed to have independent roles in AMI, how they synergistically regulate cardiac injury and repair remains unclear, lacking a systematic analysis of their dynamic interaction network.
This study, based on transcriptomic data from AMI patients in the GEO database, integrates bioinformatics analysis with experimental validation to systematically screen and identify key biomarkers related to cellular senescence and demethylation regulation. Through multi-omics joint analysis, it reveals the pathways involved with these biomarkers and their association with myocardial repair, providing new evidence for elucidating the interactive mechanisms of cellular senescence and epigenetic regulation in AMI, as well as theoretical support for the development of early clinical diagnostic markers and the design of targeted therapeutic strategies.
Results
Identification of candidate genes related to cellular senescence and demethylation and their gene ontology (GO), kyoto encyclopedia of genes and genomes (KEGG) enrichment analysis
In the current research, a total of 2,218 differentially expressed genes (DEGs) were detected between AMI samples and control samples. In the disease group samples, 849 genes showed up-regulated expression, while 1369 genes showed down-regulated expression. These gene expression differences were visualized by volcano and heat maps (Fig. 1a-b). Additionally, we obtained 866 genes related to cellular senescence and 3741 genes related to demethylation through a literature review. By employing Venn diagram analysis, we successfully identified 79 candidate genes that were associated with both AMI and involved in cellular senescence and demethylation (Fig. 1c). To investigate the functions of these 79 candidate genes and the biological processes they participate in, we performed GO and KEGG enrichment analyses. In the biological process (BP) category, significant enrichment of the genes was observed in processes such as myeloid cell differentiation, mononuclear cell differentiation, muscle cell proliferation, etc. Regarding the cellular-component (CC) category, enrichment predominantly occurred in elements like the RNA polymerase II transcription regulator complex, membrane raft, etc. In terms of the molecular-function (MF) category, they were mainly enriched in DNA-binding transcription factor binding, DNA-binding transcription activator activity etc. (p < 0.05). KEGG enrichment analysis revealed that the most significantly enriched pathways were Hepatitis B, Measles, and Leishmaniasis (p.adj < 0.05) (Fig. 1d-e) (Supplementary Table S1-2). Next, to reveal the protein - level interactions of candidate genes, we constructed PPI networks. After removing isolated nodes that did not interact with other genes, the resulting PPI network graph contained 73 nodes and 631 edges. Among them, IL6, MYC and JUN occupied key positions in the network, with their degree values ranking among the top three. These genes might play key regulatory roles in AMI-related signaling pathways (Fig. 1f).

Screening of differentially expressed genes (DEGs). (a) Volcano plot of DEGs between the acute myocardial infarction (AMI) group and the normal group; (b) Heatmap of DEGs between the AMI group and the normal group; (c) Venn diagram of candidate genes, showing the overlap among DEGs, cellular senescence-related genes, and demethylation-related genes; (d) Circular plot of Gene Ontology (GO) enrichment analysis for candidate genes. The outer ring displayed different GO terms, with colored blocks representing different categories (yellow for biological process (BP) pink for cellular component (CC), blue for molecular function (MF)). The middle ring indicated the number of background genes under specific functional terms. The inner ring’s colored blocks represented the “Rich Factor,” a measure of gene enrichment in specific functional terms; (e) Circular plot of Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment pathways for candidate genes. The outer ring showed different KEGG metabolic pathways, the middle ring indicated the number of background genes under specific pathways, and the inner ring’s colored blocks represented the “Rich Factor” for measuring gene enrichment in specific metabolic pathways; (f) Protein-protein interaction (PPI) network of candidate genes.
Identification and expression validation of biomarkers
To identify characteristic genes associated with AMI by least absolute shrinkage and selection operator (LASSO), Boruta, and support vector machine-recursive feature elimination (SVM-RFE). During the analysis of the LASSO algorithm, the model error rate was minimized when the lambda value was set to a minimum value of 0.001121108. Under this condition, 27 characteristic genes were successfully identified (Fig. 2a). Concurrently, 39 characteristic genes were identified by the Boruta algorithm (Fig. 2b). Based on the SVM-RFE algorithm, 26 characteristic genes were further identified (Fig. 2c). To obtain the most representative characteristic genes, we subjected the genes identified by the three algorithms to Venn diagram analysis, ultimately identifying 13 characteristic genes by taking the intersection set (Fig. 2d). Subsequently, an exploration was carried out on the expression of the 13 characteristic genes within the training and validation sets. These results indicated that the expression levels of BCL3, MMP9, NAMPT, and TLR4 were significantly higher in the AMI group samples compared to the control group samples in both the training set and the validation set (p < 0.05). These genes, which exhibited consistent expression trends and significant differences between the two groups, were identified as potential biomarkers. (Fig. 2e-f).

Screening of key genes. (a) Least absolute shrinkage and selection operator (LASSO) regression analysis. The left panel showed the Lasso coefficient spectrum, with the horizontal axis representing the logarithm of the regularization parameter λ (logλ) and the vertical axis representing variable coefficients; each curve depicted the coefficient trajectory of a variable as λ changed. The right panel showed the LASSO regression regularization path plot, with the horizontal axis representing logλ and the vertical axis representing model prediction error; the curve illustrated how prediction error changed as λ increased; (b) Boruta algorithm analysis, with the horizontal axis indicating gene names and the vertical axis showing feature importance scores; (c) Feature selection results using the support vector machine-recursive feature elimination (SVM-RFE) algorithm. The horizontal axis represented the number of selected variables, and the vertical axis represented cross-validation accuracy; the curve showed fluctuations in model accuracy as the number of variables changed; (d) Venn diagram of characteristic genes, showing the overlap of genes screened by three machine learning methods; (e,f) Expression level validation analysis of characteristic genes in the training and validation sets. Red represented the normal group, and blue represented the AMI group. **** indicated p < 0.0001, ** indicated p < 0.01, and ns indicated no significance; (e) GSE66360; (f) GSE60993.
Gene set enrichment analysis (GSEA)
The involvement of signaling pathways and underlying biological mechanisms linked to the four identified biomarkers was investigated via GSEA. The GSEA findings indicated that 52 pathways were enriched for BCL3, 58 pathways for MMP9, 87 pathways for NAMPT, and 66 pathways for TLR4. Notably, the four biomarkers were co-enriched in 18 pathways, such as leishmania_infection, and fc_gamma_r_mediated_phagocytosis. These results from GSEA provided new perspectives on the mechanisms through which these biomarkers were involved in the pathophysiological process of AMI. (Fig. 3a-d) (Supplementary Table S3-6).

Gene set enrichment analysis (GSEA) of key genes (a) BCL3; (b) MMP9; (c) NAMPT; (d) TLR4.
Association between immune cells and biomarkers
Based on the application of the CIBERSORT algorithm, we carried out immune cell infiltration analysis on the samples within the training set (Fig. 4a). 13 immune cell types exhibited significant differences between the AMI group and the control group, including CD4 memory activated T cells, CD4 memory resting T cells, CD8 T cells, Eosinophils, M0 Macrophages, M2 Macrophages, Monocytes, Neutrophils, activated Dendritic cells, activated Mast cells, follicular helper T cells, resting Mast cells, and resting NK cells (p < 0.05) (Fig. 4b). Once these differential immune cells were pinpointed, a correlation analysis was performed. The findings indicated that monocytes had the most significant positive correlation with activated mast cells (|cor|=0.622, p = 6.506 × 10− 12), while neutrophils and M0 macrophages had the strongest negative correlation (|cor|=0.676, p = 1.571 × 10− 14) (Fig. 4c). Subsequently, we further explored the correlations between the biomarkers (BCL3, MMP9, NAMPT, and TLR4) and the differentially expressed immune cells. The outcomes demonstrated that BCL3 exhibited the highest positive correlation with neutrophils (|cor|=0.538, p = 9.081 × 10− 9) and the most pronounced negative correlation with M0 macrophages (|cor|=0.461, p = 1.534 × 10− 6). MMP9 demonstrated a strong positive correlation with activated mast cells (|cor|=0.544, p = 5.770 × 10− 9) and a significant negative correlation with CD4 memory resting T cells (|cor|=0.409, p = 2.640 × 10− 5). NAMPT displayed the most substantial positive correlation with neutrophils (|cor|=0.696, p = 1.366 × 10− 15) and the greatest negative correlation with M0 macrophages (|cor|=0.490, p = 2.566 × 10− 7). TLR4 was found to have the most notable positive correlation with neutrophils (|cor|=0.621, p = 7.368 × 10− 12) and the strongest negative correlation with M0 macrophages (|cor|=0.448, p = 3.350 × 10− 6) (Fig. 4d). These findings suggested that biomarkers such as BCL3, MMP9, NAMPT, and TLR4 played an essential role in the immune microenvironment of AMI,

Immune microenvironment analysis. (a) Immune infiltration analysis, displaying stacked bar charts of different immune cell abundance proportions colored by cell type. Left: Normal group; Right: AMI group; (b) Box plots of immune cell infiltration differences between the normal and AMI groups. Red represented control samples, and blue represented disease samples. *** indicated p < 0.001, ** indicated p < 0.01, and ns indicated no significance; (c) Correlation analysis between differentially infiltrating immune cells, with red indicating positive correlation and green indicating negative correlation; (d) Correlations between key genes and differentially infiltrating immune cells, with red indicating positive correlation and blue indicating negative correlation. *** indicated p < 0.001, ** indicated p < 0.01, * indicated p < 0.05, and ns indicated no significance.
Molecular regulatory networks, compounds prediction, genemania analysis, friends analysis
To explore the molecular regulatory mechanisms of biomarkers, the study predicted biomarker-targeted miRNAs using several databases, including TargetScan, miRWalk, and miRDB. For BCL3, intersecting predictions from TargetScan (202 miRNAs), miRWalk (181), and miRDB (47) identified five common miRNAs, including hsa-miR-19b-3p. MMP9 predicted 62 miRNAs in TargetScan database, 98 miRNAs in miRWalk database, 5 miRNAs in miRDB database, and 0 intersection miRNAs were obtained. NAMPT predicted 618, 463, 241 miRNAs in TargetScan, miRWalk and miRDB databases, respectively, and there were 31 intersecting miRNAs, such as hsa-miR-206. For TLR4, 1748, 1008, and 95 miRNAs were predicted in these three databases, respectively, and 22 intersecting miRNAs were identified, such as hsa-miR-448. Subsequently, the StarBase database was used to predict lncRNAs targeted by the intersecting miRNAs. A total of 10 lncRNAs were identified, including SNHN1 and RMRP. Finally, an lncRNA-miRNA-mRNA regulatory network diagram was constructed to visually demonstrate the mutual regulatory relationships among the three (Fig. 5a) (Supplementary Table S7). Then, the TFs that played regulatory roles for the biomarkers were predicted. For BCL3, 75 TFs were predicted, for MMP9, 48 TFs were predicted, for NAMPT, 67 TFs were predicted, and for TLR4, 52 TFs were predicted. The predicted TFs included CSRNP1, HLX, MXD1, etc. The TFs-biomarker regulatory network was mapped to show the regulatory relationship between them (Fig. 5b).

Prediction network diagrams of key gene-targeted drugs. (a) lncRNA-miRNA-mRNA regulatory network, with left nodes representing mRNAs, middle nodes representing miRNAs, and right nodes representing lncRNAs; (b) Transcription factor (TF) regulatory network of key genes, with red nodes representing key genes and yellow nodes representing TFs targeted by key genes; (c) Network diagram of drugs and key genes, with left nodes representing key genes and right nodes representing predicted drugs for key genes; (d) GeneMANIA network diagram, where different colors represented distinct functional interaction relationships; (e) Friends analysis of key genes, with the vertical axis representing key genes and the horizontal axis representing functional similarity coefficients.
In addition, to explore potential compounds for treating AMI by targeting biomarkers, we searched the CTD database for drugs associated with four biomarkers, namely BCL3, MMP9, NAMPT, and TLR4. These results indicated 1 drug was predicted to be associated with BCL3, 25 with MMP9, 7 with NAMPT, and 24 with TLR4. After removing duplicates, a total of 25 drugs were predicted. Among them, D-glucose BOSS, HEXACHLOROETHANE BOSS, cholesterol BOSS, and resveratrol BOSS were all drugs targeting the three biomarkers NAMPT, MMP9, and TLR4 simultaneously. However, all four biomarkers—BCL3, NAMPT, MMP9, and TLR4 were found to be able to target Retinoic acid CTD 00006918. (Fig. 5c) (Supplementary Table S8). These findings suggested that the above compounds might be worth further investigation as potential therapeutic agents for AMI.
To explore other genes that were functionally associated with the four biomarkers, BCL3, MMP9, NAMPT, and TLR4, we carried out a comprehensive analysis with the help of the GeneMANIA database. The results revealed that these biomarkers were associated with a series of functionally related genes (e.g., TIMP1, LY96, PIR, TICAM2, NFKB2, LCN3, NMNAT2, NMNAT3, NMNAT1, NFKB1, ECSIT, TIRAP, NAPRT, CD180, NOX4, TGFB1, LY86, QPRT, CD14, TLR6, etc.) that had a large number of physical interactions with each other (Fig. 5d). This finding indicated that these genes had a propensity to beco-expressed under comparable circumstances. This, in turn, indicated that they were highly likely to be implicated in the same biological processes and, consequently, played a crucial part in the development of AMI. Subsequently, the GO semantic similarity of the biomarkers was evaluated using the R package GOSemSim. The outcomes revealed that BCL3 shared a significant functional similarity with other genes. (Fig. 5e).
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
RT-qPCR results showed that the expression levels of BCL3, MMP9, NAMPT, and TLR4 were significantly higher in the AMI group. This result was highly consistent with our bioinformatics analysis results, indicating a certain level of reliability of the bioinformatics analysis method. At the same time, it further confirmed that BCL3, MMP9, NAMPT, and TLR4 might have the potential to be used as biomarkers for AMI (Fig. 6).

Expression validation of key genes in clinical tissue samples. The ratio of AMI samples to control samples was 5:5. Statistical analysis was conducted using the t-test, and *indicated p < 0.05.
Discussion
With the intensification of global population aging, the incidence of cardiovascular diseases is rising year by year, and AMI has become one of the deadliest manifestations of cardiovascular diseases12. The occurrence and development of AMI are closely related to cellular senescence and demethylation processes in the heart. The former affects myocardial regeneration capacity through the secretion of pro-inflammatory factors13,14, while the latter influences cell survival and function by regulating gene expression15. This study compares peripheral blood circulating endothelial cell samples from 49 AMI patients and 50 control subjects, identifies candidate genes related to cellular senescence and demethylation, and conducts GO and KEGG enrichment analyses. Additionally, through three machine learning methods (LASSO, Boruta, and SVM) and expression validation, it is determined that BCL3, MMP9, NAMPT, and TLR4 are crucial biomarkers for the occurrence of AMI.
BCL3 (B-cell lymphoma factor 3), as a key regulatory factor in the NF-κB signaling pathway, exhibits multidimensional regulatory effects in the pathological process of AMI. This study confirms that BCL3 is significantly overexpressed in AMI patients, potentially participating in disease progression through the following mechanisms: (1) promoting NF-κB nuclear translocation by enhancing IκBα degradation, upregulating the expression of pro-inflammatory factors such as IL-6 and TNF-α, and driving neutrophil infiltration into the infarct area; (2) inhibiting the polarization of M0 macrophages to repair-type M2, leading to reduced secretion of the anti-inflammatory factor IL-10 and exacerbating myocardial injury16,17. This finding complements previous studies, which have shown that in atherosclerosis models, BCL3 can bi-directionally regulate plaque stability by binding to the p50/p52 dimer18. The innovation of this study lies in suggesting a potential novel mechanism by which BCL3 exacerbates damage during the acute phase of AMI through immune microenvironment reprogramming. Intervention strategies targeting BCL3 in animal models have shown effectiveness in improving myocardial function and reducing cellular senescence19. Combined with the bioinformatics predictions of this study (that BCL3-targeting drug retinoic acid may simultaneously regulate TLR4/MMP9), it suggests that BCL3 may become a potential pivotal node for multi-target combination therapy.
Matrix metalloproteinase-9 (MMP9) promotes myocardial cell migration and tissue remodeling by degrading extracellular matrix components (such as collagen and gelatin)20, with its expression significantly elevated during the acute phase of AMI, positively correlating with the degree of cardiac injury21. Studies have shown that MMP9 is abnormally activated in myocardial cells, endothelial cells, and macrophages, exacerbating myocardial inflammatory responses (such as IL-6 and TNF-α release) and structural damage after ischemia-reperfusion, leading to ventricular dysfunction22,23,24. Recent research suggests that MMP9 interacts with SRGs and may participate in AMI progression by regulating senescence-related pathways, but its specific role in cellular senescence and demethylation remains unclear25. This study finds that MMP9 expression is significantly upregulated in AMI patients, and its level correlates with disease severity, indicating its potential value as a diagnostic marker. Future research should delve into the mechanisms of MMP9 in epigenetic regulation of senescence to expand its clinical translational applications.
Nicotinamide phosphoribosyltransferase (NAMPT) is a key rate-limiting enzyme in NAD + salvage synthesis, participating in energy metabolism, oxidative stress, and inflammatory responses, closely related to myocardial cell apoptosis, inflammation, and remodeling26,27. This study finds that the NAMPT gene is significantly upregulated in AMI patients, consistent with previous research28, suggesting it may serve as a common molecular feature of AMI. However, some studies present contradictory findings, one study found that NAD + levels and NAMPT expression decrease after AMI29, possibly related to cardiac functional impairment; another study indicated that circular RNA CDR1as exacerbates arrhythmias by inhibiting NAMPT, leading to NAD + depletion and mitochondrial dysfunction, while knocking down CDR1as can restore NAMPT expression and improve conduction function30. This contradiction may stem from differences in pathological stages and cell types: NAMPT is stress-upregulated in the early phase of AMI (ischemic stage) but inhibited by CDR1as in the later phase (repair or arrhythmia stage); NAMPT plays a protective role in endothelial cells and macrophages, while it may be suppressed in myocardial cells due to CDR1as overexpression. Future studies should dynamically monitor NAMPT expression patterns and cell specificity at different stages of AMI, combined with functional experiments to elucidate its regulatory network, providing a basis for targeted interventions.
Toll-like receptor 4 (TLR4), as a pattern recognition receptor, activates MyD88-dependent and -independent signaling pathways (such as NF-κB and NLRP3 inflammasome) by recognizing lipopolysaccharides and damage-associated molecular patterns released during myocardial ischemia, driving the release of pro-inflammatory factors (IL-1β, TNF-α) and exacerbating myocardial inflammatory damage31,32,33. In ischemia-reperfusion (I/R), TLR4 induces myocardial cell pyroptosis by activating the NLRP3 inflammasome, expanding tissue damage34. Studies show that TLR4 expression is significantly upregulated in the hearts of elderly AMI patients, correlating with elevated levels of inflammatory factors and the aging process of the heart35, suggesting its involvement in age-related myocardial repair disorders. Current intervention strategies targeting TLR4 focus on antagonist development (such as TAK-242)36 and clinical trials of targeted therapies37, with the aim of blocking its mediated inflammatory cascade and improve AMI prognosis. However, the dual role of TLR4 in myocardial protection and damage still requires in-depth analysis to balance its immune defense and pathological damage effects, providing a basis for precision therapy.
Functional enrichment analysis and immune cell infiltration: The common pathways significantly enriched by BCL3, MMP9, NAMPT, and TLR4 include 18 pathways, such as leishmania_infection, fc_gammar_mediated_phagocytosis, and chemokine_signaling_pathway. Among them, the chemokine pathway plays a core role in the inflammatory regulation and tissue repair of AMI. First, chemokines recruit immune cells such as neutrophils and monocytes, driving acute inflammatory responses to clear necrotic tissue, but excessive activation can lead to secondary myocardial damage38. Targeting key chemokines (such as CXCL8, CCL2) can inhibit the inflammatory storm and improve myocardial survival39,40. Second, chemokines regulate smooth muscle cell migration and collagen metabolism, affecting plaque fibrous cap integrity, and their imbalance may promote acute AMI onset41. Chemokines (such as the SDF-1/CXCR4 axis) can guide stem cells to home to the infarct area, promoting angiogenesis and myocardial repair, but their timely expression is crucial for efficacy42. This study finds that the aforementioned genes form a synergistic regulatory network through the chemokine pathway, suggesting that targeting this pathway may simultaneously modulate processes of inflammation, plaque stability, and repair processes. Future research should develop multi-target combination intervention strategies based on spatiotemporal-specific expression profiles and assess their potential value in clinical translation.
Given the increasing evidence that immune dysregulation is associated with AMI, understanding the immune landscape can provide deeper insights into the mechanisms of the disease43. Therefore, using the CIBERSORT algorithm, it is analyzed that there are 13 types of differential immune cells between the AMI group and the normal control group, among which monocytes are significantly positively correlated with activated mast cells, while neutrophils are significantly negatively correlated with M0 macrophages. The association analysis of key biomarkers with differential immune cells reveals the following regulatory mechanisms. BCL3 shows the strongest positive correlation with neutrophils and a negative correlation with M0 macrophages. As a key regulatory factor in the NF-κB pathway, BCL3 drives neutrophil infiltration by enhancing the release of chemokines such as IL-8 and MCP-144; at the same time, it inhibits the polarization of M0 macrophages to repair-type M2, hindering myocardial repair45. This bidirectional effect highlights its core position in the inflammation-repair imbalance. MMP9 is positively correlated with activated mast cells and negatively correlated with resting CD4 memory T cells. It promotes mast cell release of histamine, IL-6, and TNF-α by degrading the extracellular matrix, forming a “mast cell-MMP9” positive feedback loop that exacerbates inflammatory damage46. At the same time, high expression of MMP9 may inhibit CD4 memory T cell function, weakening immune regulatory capacity. This bidirectional effect provides a theoretical basis for targeting MMP9 to regulate the mast cell-T cell axis. NAMPT and TLR4 are both positively correlated with neutrophils and negatively correlated with M0 macrophages, but their mechanisms of action differ. NAMPT enhances neutrophil activity through metabolic reprogramming and inhibits the conversion of M0 macrophages to an anti-inflammatory phenotype; TLR4 activates neutrophil inflammatory responses through MyD88-dependent and -independent pathways while downregulating the repair function of M0 macrophages47. Both exacerbate inflammatory damage and inhibit tissue repair.
This study constructed a lncRNA-miRNA-mRNA regulatory network containing 9 miRNAs and 10 lncRNAs, revealing the complexity and synergy of pathological molecular regulation in AMI, with some molecular functions supported by existing research. For example, hsa-miR-19b-3p is known to regulate the PI3K/Akt pathway by targeting PTEN, affecting myocardial apoptosis48. This study found that it targets BCL3, suggesting a possible involvement in inflammation regulation after AMI. hsa-miR-206 is downregulated in AMI, and previous studies have shown that it participates in angiogenesis and apoptosis protection by targeting VEGFA and PTP1B49,50. Its inhibitor protects the heart by activating the AMPK/Nampt signaling pathway51. This study found its association with NAMPT, further expanding its role in energy metabolism regulation. Meanwhile, hsa-miR-448 may inhibit the inflammatory pathway by targeting TLR4 downstream MyD8852. This study verified its direct targeting relationship with TLR4, providing evidence and new directions for its role as a therapeutic target for anti-inflammation in AMI. At the lncRNA level, its multidimensional regulatory role in AMI has also been deepened. MALAT1 is known to be involved in AMI fibrosis, cell aging, and inflammation53,54,55, and can serve as a biomarker for AMI risk and prognosis56. This study further found that it regulates NAMPT and BCL3 through multiple miRNAs, indicating its possible involvement in the regulation of acute inflammatory stages. SNHG1, RMRP, and NEAT1 are known to participate in cardiac repair, promote hypoxic injury, and resist ferroptosis57,58,59. This study found that they all regulate NAMPT, and SNHG1 also targets TLR4, suggesting that non-coding RNAs may form regulatory hubs through core molecules. In addition, this study identified several previously unreported potential regulatory molecules, such as hsa-miR-6807-3p, hsa-miR-6509-3p, and lncRNA AC023509.1. Their targeting relationships with core markers (NAMPT, BCL3) suggest that they may have new regulatory functions in AMI. These findings not only enrich the molecular regulatory network of AMI but also provide potential new targets for early diagnosis and targeted therapy. Future research can further validate the functions of these new molecules and explore their associations with demethylases to promote the translation of research results into clinical applications.
This study predicts through the CTD database that retinoic acid (the main active metabolite of vitamin A) can simultaneously target key biomarkers of AMI, including BCL3, MMP9, NAMPT, and TLR4. It regulates gene expression by activating the RAR/RXR heterodimer60, affecting cell proliferation, differentiation, and immune regulation. Retinoic acid has multiple protective effects in the cardiovascular system, activating cardiomyocyte signaling pathways to resist apoptosis61, inhibiting the TGF-β pathway to reduce cardiac fibrosis62, and regulating cardiac repair through epigenetic modifications63. In the pathological process of AMI, retinoic acid may reduce the release of pro-inflammatory factors (such as TNF-α, IL-1β, IL-6) by inhibiting TLR4 expression and its downstream NF-κB signaling pathway, thereby alleviating the inflammatory response64,65. At the same time, TLR4 inhibition may indirectly reduce MMP9 expression, suppress excessive degradation of the extracellular matrix, and alleviate myocardial remodeling66. Additionally, retinoic acid may influence NAMPT activity by regulating NAD + metabolism, further inhibiting the inflammatory pathway mediated by the TLR4 ligand eNAMPT67. Although the direct action of BCL3 on retinoic acid has not been clarified, as an NF-κB co-activator18, it may participate in the immune regulation process mediated by retinoic acid. Overall, the multi-target characteristics of retinoic acid make it a versatile therapeutic agent suitable for the complex disease of AMI. However, these findings are mostly theoretical speculations and preliminary research results, and future experiments are needed to further verify the direct action mechanisms between retinoic acid and these targets, as well as its synergistic effects in myocardial protection, providing a more solid basis for its clinical application.
This study systematically identified BCL3, MMP9, NAMPT, and TLR4 as key biomarkers for cellular senescence and demethylation regulation AMI through the integration of multi-omics analysis and experimental validation. It revealed that these biomarkers participate in new mechanisms of myocardial injury through chemokine signaling pathways and immune cell reprogramming, providing important theoretical support for molecular subtyping and targeted therapy of AMI. However, this study has certain limitations: first, the analysis based on public databases may be affected by batch effects and lacks independent validation for different AMI subtypes; second, although a miRNA-TF regulatory network was predicted, the direct regulatory effects of key genes on downstream pathways have not yet been experimentally validated; finally, the drug prediction results require further in vivo and in vitro experimental validation. Based on these limitations, future research could delve into the following aspects: (1) conducting multi-center clinical studies to establish the correlation between BCL3/TLR4 expression profiles and clinical prognosis; (2) utilizing gene editing technology to construct myocardium-specific knockout animal models to elucidate the spatiotemporal regulatory mechanisms of key genes; (3) developing targeted drug delivery systems to evaluate their therapeutic effects on myocardial repair. These studies will help promote the clinical translation of the findings from this research.
Materials and methods
Data collection
The GSE66360 and GSE60993 datasets were retrieved from GEO (https://www.ncbi.nlm.nih.gov/geo/) database. GSE66360 was from the GPL570 platform. It included 49 whole blood circulation endothelial cell samples from AMI and 50 control whole blood circulation endothelial cell samples. The study utilized this dataset as the training set. The GSE60993 dataset, sourced from the GPL6884 platform, encompassed 17 peripheral blood samples from individuals with AMI and 7 control peripheral blood samples, and it was employed as the validation set for this study.
In addition, 866 cellular senescence-related genes (CSRGs) were obtained from the CellAge database (https://genomics.senescence.info/cells/) according to the literature25 (Supplementary Table S9). According to the literature68, 3741 demethylation-related genes (DRGs) were obtained (Supplementary Table S10).
Differential gene expression analysis
The R package“limma” (v 3.58.1)69 was applied to analyze the differential gene expression between AMI and normal samples in the training set. DEGs were filtered using a threshold of p-value < 0.05 & |log2FC| > 0.5. The R package “ggplot2” (v 3.4.1)69 was employed to create volcano plots for visualizing these DEGs, and the top 10 up- and down-regulation DEGs in terms of |log2FC| were marked on the volcano plots. We used the R package “ComplexHeatmap” (v 2.14.0)70 to plot the heatmap of the top 10 DEGs ranked by |log2FC.
Identification and enrichment analysis of candidate genes
DEGs were then further intersected with CSRGs and DRGs using the R package “ggvenn” (v 0.1.9)71 to identify candidate genes. After that, GO analysis with a significance level of p < 0.05 and KEGG analysis with an adj.p < 0.05 were carried out using the R package “clusterProfiler”(v 4.2.2)71,72,73. Moreover, to explore the protein-protein interactions among the candidate genes, a protein-protein interaction (PPI) network was built using the STRING database with a confidence score set ≥ 0.4. Finally, the PPI network was then made visible using Cytoscape software (v 3.10.3)74.
Identification of characteristic genes
For further screening to obtain the characteristic genes, the candidate genes underwent analysis through the LASSO method, utilizing the “glmnet” package (v 4.1.8)75, and 10-fold cross-validation was applied to select the best lambda (λ) value to obtain the characteristic genes. The candidate genes were subjected to Boruta analysis using the R package “Boruta” (v 8.0.0)76, which compares the importance of the real characteristic variables present in the data with the randomly added variables (also known as shadow variables) and ranks them in order of importance to obtain the characteristic genes. The R package “caret” (v 6.0.94)77 was used to perform SVM–RFE analysis on candidate genes with a k-fold cross-validation setting k = 5. It acquired the significance level and corresponding ranking for each gene, along with the error rate and accuracy rate associated with each iteration combination. The combination with the highest accuracy was selected to obtain the corresponding characteristic genes. Finally, the characteristic genes obtained from the above three algorithms were intersected using the R package “ggvenn” (v 0.1.9)71 to obtain the final characteristic genes.
Expression level analysis
In both the training and validation datasets, the disparities in the expression levels of the characteristic genes between the AMI group samples and the control group samples were assessed using the Wilcoxon test. The genes with the same expression trend in the two datasets and with significant differences between the groups were defined as biomarkers for subsequent analyses (p < 0.05).
GSEA
To probe the potential biological mechanisms involved in biomarkers, Spearman correlation analysis was performed between the biomarkers and all genes in all samples of the training set using the R package “psych” (v 2.1.6)78 to obtain the correlation coefficients. The coefficients of the genes were ranked from highest to lowest. From the MSigDB database, the “c2.cp.kegg.v7.4.symbols.gmt” background gene set was obtained. The R package “clusterProfiler” (v 4.2.2)71 was used for GSEA, with thresholds of adj. p < 0.05 and |NES| > 1. The results were sorted in descending order by |NES|. Each biomarker was visualized using the R package “GseaVis” (v 0.1.0)79, with the top 5 significantly enriched results being marked.
Immune infiltration analysis
To explore the relationship between biomarkers and the immune microenvironment, the CIBERSORT algorithm80,81 (v 0.1.0)75 was applied to assess the relative proportions of 22 immune cells in the AMI and control groups within the training set. Then, the R package “ggplot2” (v 3.4.1)82 was utilized to generate visualizations depicting the relative abundances of these 22 immune cells in the two groups. Additionally, the Wilcoxon test was applied to compare the variations in the infiltration proportions of the 22 immune cells between the AMI group and the control group (p < 0.05). The R package “psych” (v 2.1.6)78 was then used to conduct Spearman’s correlation analysis, both among the differential immune cells and between the biomarkers and the differential immune cells, with a correlation threshold of |cor| > 0.3 and p < 0.05.
Regulation network analysis
To explore miRNAs associated with the upstream of biomarkers, the TargetScan (https://www.targetscan.org/vert_80/), miRWalk (http://mirwalk.umm.uni-heidelberg.de/), and miRDB (http://mirdb.org/) databases were used to predict the potential regulatory miRNAs of biomarkers. Then, the miRNAs obtained from the three databases were intersected using the R package “ggvenn” (v 0.1.9)71 to obtain the intersecting miRNAs. Next, the lncRNAs of the intersecting miRNAs were predicted in the StarBase (https://rnasysu.com/encori/), and the filtering threshold was set as clipExpNum greater than 20. Finally, the R package “ggsankey” (v 0.0.9)83 was used to draw the network diagram of the lncRNA-miRNA-mRNA regulatory relationship. To obtain the transcription factors (TFs) with regulatory effects on biomarkers, the biomarker-targeted TFs were predicted by the ChEA3 (https://maayanlab.cloud/chea3/) platform. Then, the TFs-biomarkers regulatory network was graphically represented using the Cytoscape software.
Compounds prediction
To find potential drugs targeting biomarkers for AMI, the Comparative Toxicogenomics Database (CTD) (https://ctdbase.org/) was used to search for potential drugs or molecular compounds related to biomarkers, retaining compounds with Reference Count > = 2. Finally, the biomarkers-drug relationship was visualized using the R package “ggsankey” (v 0.0.9)83.
Genemania analysis and friends analysis
To identify other genes related to the function of the biomarker, the biomarker was imported into the GeneMANIA database, and genes with similar functions were screened. Friends analysis of the biomarkers was performed using the R package “GOSemSim” (v 2.26.1)84 to further understand the functional similarities between the biomarkers.
RT-qPCR
The expression of biomarker was verified by RT-qPCR. 5 AMI and 5 control tissue samples were obtained from the First People’s Hospital of Yunnan Province (Kunming, China). All participants signed and completed an informed consent form, and the ethical approval organization was (KHU2021-229). Results were plotted using GraphPad Prism 10.
Total RNA from 10 samples was isolated with TRIzol (Novozymes, Nanjing, China) following the manufacturer’s instructions. The concentration and purity of total RNA were determined by ultraviolet absorbance (A260/A280 ratio) to ensure that RNA quality met the experimental requirements. Subsequently, the Hifair® Ⅲ 1st Strand cDNA Synthesis SuperMix for qPCR kit (Next Sage, Shanghai, China) was employed to reverse- transcribe the total RNA into cDNA as per the manufacturer’s guidelines. 2xUniversal Blue SYBR Green qPCR Master Mix (Servicebio, Wuhan, China) was used for RT-qPCR. The reaction system contained 3 µl of cDNA template, 5 µl of 2× Universal Blue SYBR Green qPCR Premix, 1 µl of 10 µM forward primer, and 1 µl of 10 µM reverse primer (primers were synthesized by a biotechnology company, with sequences provided in Table 1). The reaction was carried out on a CFX96 Real-Time Quantitative PCR Detection System (BIO-RAD, USA; manufactured in Shanghai, China) under the following conditions: pre-denaturation at 95 °C for 1 min, followed by 40 cycles of denaturation at 95 °C for 20 s, annealing at 55 °C for 20 s, and extension at 72 °C for 30 s. In the experimental design, 5 biological replicates were set for each group, and 3 technical replicates were performed for each sample to ensure the reliability of results. The expression of biomarker genes was normalized using GAPDH as the reference gene, and the relative expression levels of biomarkers were calculated by the 2−ΔΔCt method. Finally, Graphpad Prism 10 software was used to generate graphs, and p-values were statistically calculated using t-tests.
Statistical analysis
R software was used to carry out statistical analysis. The Wilcoxon test was applied to examine the differences between the two groups, with a significance threshold of P < 0.05.
Data availability
The datasets generated analysed during the current study are available in the [GEO (GSE66360 and GSE60993)] repository, [https://www.ncbi.nlm.nih.gov/gds], and [CellAge database] repository, [https://genomics.senescence.info/cells/]
References
Reed, G. W., Rossi, J. E. & Cannon, C. P. Acute myocardial infarction. Lancet 389, 197–210. https://doi.org/10.1016/s0140-6736(16)30677-8 (2017).
Frantz, S., Hundertmark, M. J., Schulz-Menger, J., Bengel, F. M. & Bauersachs, J. Left ventricular remodelling post-myocardial infarction: pathophysiology, imaging, and novel therapies. Eur. Heart J. 43, 2549–2561. https://doi.org/10.1093/eurheartj/ehac223 (2022).
Gorgoulis, V. et al. Cellular senescence: defining a path forward. Cell 179, 813–827. https://doi.org/10.1016/j.cell.2019.10.005 (2019).
Giuliani, A. et al. Senescent endothelial cells sustain their senescence-associated secretory phenotype (SASP) through enhanced fatty acid oxidation. Antioxidants. 12 https://doi.org/10.3390/antiox12111956 (2023).
Li, T. et al. Pathological implication of camkii in NF-κB pathway and SASP during cardiomyocytes senescence. Mech. Ageing Dev. 209, 111758. https://doi.org/10.1016/j.mad.2022.111758 (2022).
Rana, T. et al. PAI-1 regulation of TGF-β1-induced alveolar type II cell senescence, SASP Secretion, and SASP-mediated activation of alveolar macrophages. Am. J. Respir Cell. Mol. Biol. 62, 319–330. https://doi.org/10.1165/rcmb.2019-0071OC (2020).
Bochenek, M. L., Schütz, E. & Schäfer, K. Endothelial cell senescence and thrombosis: ageing clots. Thromb. Res. 147, 36–45. https://doi.org/10.1016/j.thromres.2016.09.019 (2016).
Lodrini, A. M. & Goumans, M. J. Cardiomyocytes cellular phenotypes after myocardial infarction. Front. Cardiovasc. Med. 8, 750510. https://doi.org/10.3389/fcvm.2021.750510 (2021).
Traube, F. R. & Carell, T. The chemistries and consequences of DNA and RNA methylation and demethylation. RNA Biol. 14, 1099–1107. https://doi.org/10.1080/15476286.2017.1318241 (2017).
Zhang, Z. et al. Prmt1 upregulated by Hdc deficiency aggravates acute myocardial infarction via NETosis. Acta Pharm. Sin B. 12, 1840–1855. https://doi.org/10.1016/j.apsb.2021.10.016 (2022).
Booth, L. K. et al. Heart disease and ageing: the roles of senescence, mitochondria, and telomerase in cardiovascular disease. Subcell. Biochem. 103, 45–78. https://doi.org/10.1007/978-3-031-26576-1_4 (2023).
Mozaffarian, D. et al. Heart disease and stroke statistics-2016 update: A report from the American Heart Association. Circulation 133, e38–360. https://doi.org/10.1161/cir.0000000000000350 (2016).
Wang, L. et al. Dexmedetomidine prevents cardiomyocytes from hypoxia/reoxygenation injury via modulating tetmethylcytosine dioxygenase 1-mediated DNA demethylation of Sirtuin1. Bioengineered 13, 9369–9386. https://doi.org/10.1080/21655979.2022.2054762 (2022).
Merdji, H., Schini-Kerth, V., Meziani, F. & Toti, F. Long-term cardiovascular complications following sepsis: is senescence the missing link? Ann. Intensive Care. 11, 166. https://doi.org/10.1186/s13613-021-00937-y (2021).
Gao, X. et al. Downregulation of ALKBH5 rejuvenates aged human mesenchymal stem cells and enhances their therapeutic efficacy in myocardial infarction. Faseb j. 37, e23294. https://doi.org/10.1096/fj.202301292R (2023).
Xu, Z. et al. Storax attenuates cardiac fibrosis following acute myocardial infarction in rats via suppression of AT1R-Ankrd1-P53 signaling pathway. Int. J. Mol. Sci. 23 https://doi.org/10.3390/ijms232113161 (2022).
Shao, M., Ren, Z. & Zhang, R. MYBL2 protects against H9c2 injury induced by hypoxia via AKT and NF–κB pathways. Mol. Med. Rep. 17, 4832–4838. https://doi.org/10.3892/mmr.2018.8387 (2018).
Seaton, G., Smith, H., Brancale, A., Westwell, A. D. & Clarkson, R. Multifaceted roles for BCL3 in cancer: a proto-oncogene comes of age. Mol. Cancer. 23, 7. https://doi.org/10.1186/s12943-023-01922-8 (2024).
Han, B. J. et al. Cardioprotective effects of tetrahydropalmatine on acute myocardial infarction in rats. Am. J. Chin. Med. 50, 1887–1904. https://doi.org/10.1142/s0192415x2250080x (2022).
Scannevin, R. H. et al. Discovery of a highly selective chemical inhibitor of matrix metalloproteinase-9 (MMP-9) that allosterically inhibits zymogen activation. J. Biol. Chem. 292, 17963–17974. https://doi.org/10.1074/jbc.M117.806075 (2017).
Nordeng, J. et al. TIMP-1 expression in coronary thrombi associate with myocardial injury in ST-elevation myocardial infarction patients. Coron. Artery Dis. 33, 446–455. https://doi.org/10.1097/mca.0000000000001128 (2022).
Chen, R. et al. SGLT2 inhibitor dapagliflozin alleviates intramyocardial hemorrhage and adverse ventricular remodeling via suppressing hepcidin in myocardial ischemia-reperfusion injury. Eur. J. Pharmacol. 950, 175729. https://doi.org/10.1016/j.ejphar.2023.175729 (2023).
Domínguez-Rodríguez, A. et al. Early treatment of acute myocardial infarction with melatonin: effects on MMP-9 and adverse cardiac events. J. Clin. Med. 11 https://doi.org/10.3390/jcm11071909 (2022).
Brunton-O’Sullivan, M. M. et al. Examining variation and temporal dynamics of extracellular matrix biomarkers following acute myocardial infarction. Biomark. Med. 16, 147–161. https://doi.org/10.2217/bmm-2021-0531 (2022).
Xiang, J., Shen, J., Zhang, L. & Tang, B. Identification and validation of senescence-related genes in circulating endothelial cells of patients with acute myocardial infarction. Front. Cardiovasc. Med. 9, 1057985. https://doi.org/10.3389/fcvm.2022.1057985 (2022).
Garten, A. et al. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat. Rev. Endocrinol. 11, 535–546. https://doi.org/10.1038/nrendo.2015.117 (2015).
Tsai, K. L. et al. Anti-IL-20 antibody protects against ischemia/reperfusion-impaired myocardial function through modulation of oxidative Injuries, inflammation and cardiac remodeling. Antioxidants. 10. https://doi.org/10.3390/antiox10020275 (2021).
Li, X. et al. Uncovering Hippo pathway-related biomarkers in acute myocardial infarction via scRNA-seq binding transcriptomics. Sci. Rep. 15, 10368. https://doi.org/10.1038/s41598-025-94820-6 (2025).
Habeichi, N. et al. (ed, J.) Nicotinamide riboside: A promising treatment for type 1 cardiorenal syndrome in myocardial Infarction-Induced acute kidney injury by upregulating nicotinamide phosphoribosyltransferase-mediated nicotinamide dinucleotide levels. J. Am. Heart Assoc. https://doi.org/10.1161/jaha.124.038603 (2025).
Liu, Y. et al. CDR1as promotes arrhythmias in myocardial infarction via targeting the NAMPT-NAD + pathway. Biomed. Pharmacother. 165, 115267. https://doi.org/10.1016/j.biopha.2023.115267 (2023).
Kim, H. J., Kim, H., Lee, J. H. & Hwangbo, C. Toll-like receptor 4 (TLR4): new insight immune and aging. Immun. Ageing. 20, 67. https://doi.org/10.1186/s12979-023-00383-3 (2023).
Kawai, T. & Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650. https://doi.org/10.1016/j.immuni.2011.05.006 (2011).
Younis, N. S. & Mohamed, M. E. Anethole’s effects against myocardial infarction: the role of TLR4/NFκB and Nrf2/HO1 pathways. Chem. Biol. Interact. 360, 109947. https://doi.org/10.1016/j.cbi.2022.109947 (2022).
Mehta, A. A., Patel, P., Thakur, V. R. & Beladiya, J. V. Effect of soya phosphatidylcholine and possible underlying mechanism on ischemia/reperfusion injury in isolated perfused rat heart: an experimental and computational study. Can. J. Physiol. Pharmacol. 100, 252–258. https://doi.org/10.1139/cjpp-2021-0056 (2022).
Wang, Y. et al. The serum soluble Klotho alleviates cardiac aging and regulates M2a/M2c macrophage polarization via inhibiting TLR4/Myd88/NF-κB pathway. Tissue Cell. 76, 101812. https://doi.org/10.1016/j.tice.2022.101812 (2022).
Ramirez-Carracedo, R. et al. Targeting TLR4 with ApTOLL improves heart function in response to coronary ischemia reperfusion in pigs undergoing acute myocardial infarction. Biomolecules. 10 https://doi.org/10.3390/biom10081167 (2020).
Xue, X. et al. Hydroxysafflor yellow A, a natural compound from Carthamus tinctorius L with good effect of alleviating atherosclerosis. Phytomedicine 91, 153694. https://doi.org/10.1016/j.phymed.2021.153694 (2021).
Heinrichs, M. et al. The healing myocardium mobilizes a distinct B-cell subset through a CXCL13-CXCR5-dependent mechanism. Cardiovasc. Res. 117, 2664–2676. https://doi.org/10.1093/cvr/cvab181 (2021).
Feng, G. et al. CCL17 aggravates myocardial injury by suppressing recruitment of regulatory T cells. Circulation 145, 765–782. https://doi.org/10.1161/circulationaha.121.055888 (2022).
Wen, J., Guan, Y., Niu, H., Dang, Y. & Guan, J. Targeting cardiac resident CCR2 + macrophage-secreted MCP-1 to attenuate inflammation after myocardial infarction. Acta Biomater. https://doi.org/10.1016/j.actbio.2024.08.025 (2024).
Gao, R. et al. ALKBH5 regulates macrophage senescence and accelerates atherosclerosis by promoting CCL5 m6A modification. Arterioscler. Thromb. Vasc Biol. https://doi.org/10.1161/atvbaha.125.322508 (2025).
Wagner, M. J., Khan, M. & Mohsin, S. Healing the broken heart; the Immunomodulatory effects of stem cell therapy. Front. Immunol. 11, 639. https://doi.org/10.3389/fimmu.2020.00639 (2020).
Kyaw, T. et al. Alarmin-activated B cells accelerate murine atherosclerosis after myocardial infarction via plasma cell-immunoglobulin-dependent mechanisms. Eur. Heart J. 42, 938–947. https://doi.org/10.1093/eurheartj/ehaa995 (2021).
Collins, P. E. et al. Mapping the interaction of B cell leukemia 3 (BCL-3) and nuclear factor κB (NF-κB) p50 identifies a BCL-3-mimetic anti-inflammatory peptide. J. Biol. Chem. 290, 15687–15696. https://doi.org/10.1074/jbc.M115.643700 (2015).
Ahsan, T., Shoily, S. S., Ahmed, T. & Sajib, A. A. Role of the redox state of the Pirin-bound cofactor on interaction with the master regulators of inflammation and other pathways. PLoS One. 18, e0289158. https://doi.org/10.1371/journal.pone.0289158 (2023).
Huang, K. K., Zheng, H. L., Li, S. & Zeng, Z. Y. Identification of hub genes and their correlation with immune infiltration in coronary artery disease through bioinformatics and machine learning methods. J. Thorac. Dis. 14, 2621–2634. https://doi.org/10.21037/jtd-22-632 (2022).
Wu, D. et al. Mechanism of Xue-Jie-San treating Crohn’s disease complicated by atherosclerosis: network pharmacology, molecular docking and experimental validation. Phytomedicine 135, 156169. https://doi.org/10.1016/j.phymed.2024.156169 (2024).
Li, K., Ya, X., Duan, X., Li, Y. & Lin, X. miRNA-19b-3p stimulates cardiomyocyte apoptosis induced by myocardial ischemia reperfusion via downregulating PTEN. Dis. Markers. 2021, 9956666. https://doi.org/10.1155/2021/9956666 (2021).
Sun, T. et al. The role of MicroRNAs in myocardial infarction: from molecular mechanism to clinical application. Int. J. Mol. Sci. 18 https://doi.org/10.3390/ijms18040745 (2017).
Yan, Y., Dang, H., Zhang, X., Wang, X. & Liu, X. The protective role of MiR-206 in regulating cardiomyocytes apoptosis induced by ischemic injury by targeting PTP1B. Biosci. Rep. 40 https://doi.org/10.1042/bsr20191000 (2020).
MicroRNA-206 inhibition. And activation of the AMPK/Nampt signalling pathway enhance sevoflurane post-conditioning-induced amelioration of myocardial ischaemia/reperfusion injury. J. Drug Target. 30, 1136. https://doi.org/10.1080/1061186x.2022.2078565 (2022).
Shi, H. et al. Astragaloside IV prevents acute myocardial infarction by inhibiting the TLR4/MyD88/NF-κB signaling pathway. J. Food Biochem. 45, e13757. https://doi.org/10.1111/jfbc.13757 (2021).
Huang, S. et al. Long noncoding RNA MALAT1 mediates cardiac fibrosis in experimental postinfarct myocardium mice model. J. Cell. Physiol. 234, 2997–3006. https://doi.org/10.1002/jcp.27117 (2019).
Eshraghi, R. et al. Unveiling the role of long non-coding RNA MALAT1: a comprehensive review on myocardial infarction. Front. Cardiovasc. Med. 11, 1429858. https://doi.org/10.3389/fcvm.2024.1429858 (2024).
Chang, F. et al. Malat1 promotes macrophage-associated inflammation by increasing PPAR-γ methylation through binding to EZH2 in acute myocardial infarction. Int. Immunopharmacol. 123, 110695. https://doi.org/10.1016/j.intimp.2023.110695 (2023).
Li, R., Jin, J., Liu, E. & Zhang, J. A novel circulating biomarker lnc-MALAT1 for acute myocardial infarction: its relationship with disease risk, features, cytokines, and major adverse cardiovascular events. J. Clin. Lab. Anal. 36, e24771. https://doi.org/10.1002/jcla.24771 (2022).
Zhu, W., Luo, L., Ye, G. & Ou, J. Potential diagnostic value of N1LR and SNHG1 in acute myocardial infarction. BMC Med. Genomics. 16, 71. https://doi.org/10.1186/s12920-023-01501-2 (2023).
Teng, Y. et al. LncRNA RMRP accelerates hypoxia-induced injury by targeting miR-214-5p in H9c2 cells. J. Pharmacol. Sci. 142, 69–78. https://doi.org/10.1016/j.jphs.2019.07.014 (2020).
Yu, Q. et al. Silencing of LncRNA NEAT1 alleviates acute myocardial infarction by suppressing miR-450-5p/ACSL4-mediated ferroptosis. Exp. Cell. Res. 442, 114217. https://doi.org/10.1016/j.yexcr.2024.114217 (2024).
Wang, S. & Moise, A. R. Recent insights on the role and regulation of retinoic acid signaling during epicardial development. Genesis 57, e23303. https://doi.org/10.1002/dvg.23303 (2019).
Da Silva, F. et al. Retinoic acid signaling is directly activated in cardiomyocytes and protects mouse hearts from apoptosis after myocardial infarction. Elife. 10 https://doi.org/10.7554/eLife.68280 (2021).
Teodorescu, P. et al. Transforming growth factor β-mediated micromechanics modulates disease progression in primary myelofibrosis. J. Cell. Mol. Med. 24, 11100–11110. https://doi.org/10.1111/jcmm.15526 (2020).
Han, W. et al. A review on regulation of DNA methylation during post-myocardial infarction. Front. Pharmacol. 15, 1267585. https://doi.org/10.3389/fphar.2024.1267585 (2024).
Gu, B., Miao, J., Fa, Y., Lu, J. & Zou, S. Retinoic acid attenuates lipopolysaccharide-induced inflammatory responses by suppressing TLR4/NF-kappaB expression in rat mammary tissue. Int. Immunopharmacol. 10, 799–805. https://doi.org/10.1016/j.intimp.2010.04.022 (2010).
Li, T. et al. Cardiac repair after myocardial infarction: A two-sided role of inflammation-mediated. Front. Cardiovasc. Med. 9, 1077290. https://doi.org/10.3389/fcvm.2022.1077290 (2022).
Jing, N., Fang, B., Li, Z. & Tian, A. Exogenous activation of cannabinoid-2 receptor modulates TLR4/MMP9 expression in a spinal cord ischemia reperfusion rat model. J. Neuroinflammation. 17, 101. https://doi.org/10.1186/s12974-020-01784-7 (2020).
Dai, Y. et al. NAMPT/NAD(+)/PARP1 pathway regulates CFA-induced inflammatory pain via NF-κB signaling in rodents. Adv. Biol. (Weinh). 8, e2400028. https://doi.org/10.1002/adbi.202400028 (2024).
Kang, K., Huang, H. & Chen, Z. Identification and validation of the prognostic signature of a novel demethylation-related gene associated with the clinical features of colon cancer. Int. Immunopharmacol. 139, 112798. https://doi.org/10.1016/j.intimp.2024.112798 (2024).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. https://doi.org/10.1093/bioinformatics/btw313 (2016).
Zhou, W. et al. Identification and mechanism analysis of biomarkers related to butyrate metabolism in COVID-19 patients. Ann. Med. 57, 2477301. https://doi.org/10.1080/07853890.2025.2477301 (2025).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–462. https://doi.org/10.1093/nar/gkv1070 (2016).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. https://doi.org/10.1101/gr.1239303 (2003).
Zhang, H., Zheng, C., Xu, Y. & Hu, X. Comprehensive molecular and cellular characterization of endoplasmic reticulum stress-related key genes in renal ischemia/reperfusion injury. Front. Immunol. 15, 1340997. https://doi.org/10.3389/fimmu.2024.1340997 (2024).
Yang, J., Li, W., Lin, X. & Liang, W. A lactate metabolism-related gene signature to diagnose osteoarthritis based on machine learning combined with experimental validation. Aging. 16 https://doi.org/10.18632/aging.205873 (2024).
Mahmoudian, M., Venäläinen, M. S., Klén, R. & Elo, L. L. Stable Iterative Variable Selection Bioinformatics 37, 4810–4817 https://doi.org/10.1093/bioinformatics/btab501 (2021).
Harrison, A. M. & Goozee, R. Psych-related iPhone apps. J. Ment Health. 23, 48–50. https://doi.org/10.3109/09638237.2013.869575 (2014).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22 (2010).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 12, 453–457. https://doi.org/10.1038/nmeth.3337 (2015).
Chen, B., Khodadoust, M. S., Liu, C. L., Newman, A. M. & Alizadeh, A. A. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol. Biol. 1711, 243–259. https://doi.org/10.1007/978-1-4939-7493-1_12 (2018).
Gustavsson, E. K., Zhang, D., Reynolds, R. H., Garcia-Ruiz, S. & Ryten, M. ggtranscript: an R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinformatics. 38, 3844–3846. https://doi.org/10.1093/bioinformatics/btac409 (2022).
Li, M. et al. Integrating network pharmacology to investigate the mechanism of quercetin’s action through AKT inhibition in co-expressed genes associated with polycystic ovary syndrome and endometrial cancer. Int. J. Biol. Macromol. 139468. https://doi.org/10.1016/j.ijbiomac.2025.139468 (2025).
Yang, L., Yu, X., Liu, M. & Cao, Y. A comprehensive analysis of biomarkers associated with synovitis and chondrocyte apoptosis in osteoarthritis. Front. Immunol. 14, 1149686. https://doi.org/10.3389/fimmu.2023.1149686 (2023).
Funding
This work was supported by the National Natural Science Foundation of China (81960065), the Applied Basic Research Foundation of Yunnan Province (202101AY070001-268, 202301AT070095), the Clinical Medical Center research open project of Yunnan Provincial Health committee (2022LCZXKF-XY07) and the Candidate Talents Training Fund of Yunnan Province (H-2024069), Zhang Cuntai Expert Workstation of Yunnan Province (202405AF140057).
Author information
Authors and Affiliations
Contributions
WHS and QH designed the whole study. HH, QH, HZ and QRR prepared the figures and tables. WHS, SQL, JHD and YZ wrote the manuscript. All the authors reviewed the manuscript and approved the manuscript for submission.
Corresponding author
Ethics declarations
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Ethical statement
The Ethics Committee of The Affiliated Hospital of Kunming University of Science and Technology (Kunming, China) approved the study protocol (KHU2021-229), which complied with the principles of the Declaration of Helsinki.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Su, W., Huang, H., Ruan, Q. et al. Identification and validation of biomarkers associated with cellular senescence and demethylation in acute myocardial infarction. Sci Rep 15, 41385 (2025). https://doi.org/10.1038/s41598-025-25391-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-25391-9
