
Abstract
Gestational hypothyroxinemia (HTX) is associated with cognitive impairments and autism traits in offspring. However, the underlying mechanisms remain unclear. Prenatal inflammation impairs cortical development and induces diverse neurodevelopmental outcomes. Since thyroid dysfunction elicits inflammation, we sought to investigate whether HTX triggers prenatal pro-inflammatory responses. Using a mouse model of gestational HTX, we found elevated levels of IL-6 and IL-17 A in maternal serum, placental tissues, and embryonic brains at embryonic day (E)14 compared to euthyroid (EUT) dams. We also found increased proportions of dendritic cells, NK cells, M1-like macrophages, and monocytes in the placental tissues of HTX dams. Furthermore, gestational HTX exposure led to reduced Tbr2⁺ progenitors, increased Tbr1⁺ neurons, and an expanded Iba1⁺ microglial population in HTX-exposed embryos compared to EUT-exposed embryos. At postnatal day (P)55, the offspring gestated under HTX exhibited reduced hippocampal dendritic spine density and maturity compared to the progeny gestated under EUT. Notably, restoring T4 levels during HTX induction (HTX + T4 dams) prevented these alterations during pregnancy and in the offspring of HTX + T4 dams. These findings show that gestational HTX causes inflammation during pregnancy and has neurodevelopmental effects on the progeny, opening new pathways related to how maternal HTX impairs neurodevelopment in the offspring.
Similar content being viewed by others

Introduction
Pregnancy is a highly sensitive period of life, given that parental genetic predispositions, along with environmental, nutritional, and hormonal factors, can significantly influence fetal development1,2. Indeed, these factors can lead to in utero adaptations that may induce long-term molecular disruptions in critical tissues and organs of the offspring, potentially increasing their susceptibility to chronic diseases or disorders later in life1,2. In this context, the maintenance of adequate levels of maternal thyroid hormones (TH) [L-3,5,3’,5’-tetraiodothyronine (T4) and L-3,5,3’-triiodothyronine (T3)] is critical for fetal development and growth, as the embryo is unable to synthesize TH until the 16th to 20th week of gestation3. Given this scenario, the transplacental transfer of T4 from the mother to the fetus plays a vital role during early development, as T4 more readily crosses the placenta than T3 through TH transporters, including monocarboxylate transporter 8 (MCT8), organic anion transporting polypeptide 1C1 (OATP1C1), and L-type amino acid transporters LAT1 and LAT24,5. A large portion of maternal T4 is metabolized by placental deiodinase 3 (DIO3) into reverse T3 (rT3) and T2, inactive forms of TH that protect the fetus from excess active T36. Studies in humans suggest that the majority of fetal T3 is produced locally through the conversion of maternal T4 into T3 by fetal deiodinase 2 (DIO2), primarily in the fetal brain7,8,9. The presence of both cytosolic and nuclear T3 in the fetal brain supports a critical role of this hormone in regulating gene expression in the developing brain from the 9th to 10th week of gestation10,11,12. Thus, early neurodevelopmental programming will depend on the adequate maintenance of maternal T4 levels to ensure proper fetal brain maturation and function10,11,12. Nonetheless, the maternal thyroid gland undergoes adaptations in size and function during pregnancy to meet the increased demand for TH, which raises the risk of dysfunction and the subsequent development of thyroid-related conditions that may reduce the circulating T4 levels13.
A frequent and asymptomatic maternal condition during pregnancy is gestational hypothyroxinemia (HTX)14. HTX is clinically defined by reduced blood levels of T4, along with normal T3 and thyroid-stimulating hormone (TSH) levels15. HTX during the first trimester of pregnancy has been associated with numerous effects on the offspring, such as a lower intelligence quotient (IQ)16, impaired motor and auditory skills17, attention-deficit/hyperactivity disorder in children18, and delayed neurobehavioral development19. Rodent models of gestational HTX have shown that even transient reductions in maternal T4 levels can lead to lasting consequences over brain development, including cognitive impairments, particularly in the memory and learning capacities, which are underpinned by hippocampal dysfunction20,21. These deficits have been linked to impaired hippocampal synaptic plasticity, as evidenced by diminished long-term potentiation (LTP) in the CA1 region and abnormal expression of key glutamatergic components20,21. Additionally, HTX disrupts neuronal migration during fetal neocorticogenesis, an effect likely driven by reduced TH responsiveness in the developing neocortex, reinforcing the essential role of maternal TH in early cortical patterning22,23,24. More recently, some retrospective studies in humans have reported that HTX is associated with a 2- to 4-fold increased likelihood of offspring exhibiting social and communicative impairments, as well as repetitive behaviors, which are core features commonly associated with autism spectrum disorder (ASD)25,26. Briefly, ASD is a neurodevelopmental condition characterized by social and communicative impairments and restricted repetitive patterns of interest and behavior27. Using a mouse model of gestational HTX, we have recently reported that HTX-gestated offspring exhibit specific autistic-like behavioral, immune, and synaptic abnormalities28, thereby validating this association and providing a valuable tool for dissecting the cellular and molecular pathways underlying this connection.
The etiology of ASD is highly complex, and it is estimated that 20% to 50% of cases result from intricate interactions during pregnancy between parental genetic predispositions and environmental influences29,30,31,32. In this context, associative studies suggest that pro-inflammatory processes during pregnancy, also referred to as prenatal inflammation, significantly increase the risk of neurodevelopmental disorders in the offspring, including ASD33,34,35,36. This association has been most clearly demonstrated in cases of severe prenatal infections that induce a strong inflammatory response, including Influenza37, Rubella38, Cytomegalovirus39, and SARS-CoV-240, but it has also been related to other maternal conditions, such as exposure to environmental pollutants41, obesity42, maternal chronic stress43, and treatment with neuropsychiatric drugs like valproic acid44. Studies in rodents have successfully reproduced maternal inflammation using an established model called maternal immune activation (MIA), which has been effective in elucidating the mechanisms by which prenatal inflammation affects embryonic neurodevelopment45. MIA and consequent autistic-like abnormalities can be induced in dams via the administration of a synthetic double-stranded RNA (called polyinosinic: polycytidylic acid (poly(I: C)) to mimic a viral infection46, lipopolysaccharide (LPS) to mimic a bacterial infection47, or even a single dose of interleukin 6 (IL-6) during early neurodevelopment, such as embryonic day 12 (E12)35,48. Interestingly, even a mild form of MIA induced by poly(I: C) can be associated with increased self-grooming activity (a stereotyped behavior related to autistic-like traits) in male offspring rats49. It has been found elevated levels of inflammatory cytokines, such as IL-6 and IL-17 A in the maternal serum, placental tissues and fetus of animal models induced with MIA50,51. Whether these cytokines in fetus can cross the placental barrier is still an open question50,51. There is scientific evidence supporting that certain cytokines, such as IL-6, can be rapidly transcribed in the fetal brain following MIA induction50,51. On the other hand, there is evidence showing that IL-6 could cross from the mother to the fetus52. However, the information of crossing cytokines trough the placental barrier depends on the type of cytokine, species, and gestational stage53,54,55. The placenta produces inflammatory mediators that disrupt the immune balance at the maternal-fetal interface by activating natural killer (NK) cells and macrophages, which are primarily located in the decidua basalis, the maternal portion of the placenta56,57. These cytokines can also reach the fetal brain, triggering microglial activation and M1-like polarization58,59,60, which initiates a neuroinflammatory cascade that disrupts cortical development, as previously observed in rats61 and human brain organoid models62. These alterations contribute to abnormal brain development and are associated with autism-like behaviors63,64,65, neuroinflammation66, and reduced spine density and dendritic spine maturation, likely due to impaired synaptic pruning during prenatal development67,68,69.
Thyroid-related conditions, such as hypothyroidism and non-thyroidal illness syndrome, are often accompanied by inflammatory processes, such as increased production of cytokines like IL-6, tumor necrosis factor-alpha (TNF-α), and C-reactive protein (CRP), as well as the activation of NF-κB signaling pathways and immune cell infiltration in target tissues70,71. These findings suggest a potential link between thyroid dysfunction and impaired immune responses. Therefore, we hypothesized that HTX may trigger prenatal pro-inflammatory responses during pregnancy, similar to those observed in MIA models, leading to adverse neurodevelopmental outcomes potentially associated with the autism-like abnormalities recently observed in the HTX-gestated offspring28. In this study, we quantified pro-inflammatory cytokines and evaluated myeloid cell populations in mouse feto-placental units exposed to HTX. Additionally, we quantified Pax6-, Tbr2-, Tbr1-, and Iba1-positive cells in the developing embryonic cortex to assess radial glia, intermediate progenitors, deep-layer neurons, and microglial pool, respectively. Lastly, we investigated the impact of gestational HTX on dendritic spine morphology of the offspring in the offspring by examining dendritic spine density and maturation in the CA1 region of the hippocampus. Our findings revealed that gestational HTX increases the levels of IL-6 and IL-17 A in maternal serum, placental tissues (both maternal and fetal compartments), and embryonic brains at E14, indicating a pro-inflammatory response. This was accompanied by an increased proportion of placental (both maternal and fetal) and uterine myeloid cells, including dendritic cells, NK cells, M1-like macrophages, and monocytes, which is also consistent with the establishment of a pro-inflammatory environment. Additionally, HTX led to a mild reduction in Tbr2⁺ progenitors, an increase in Tbr1⁺ neurons, and an increased Iba1⁺ microglial population in HTX-exposed embryos. These alterations were associated with a decrease in spine density and maturity in the hippocampal CA1 region of male HTX-gestated offspring at P55. In parallel, the restoration of T4 levels during HTX induction prevented these pro-inflammatory responses and long-term detrimental effects, suggesting a critical role for maternal T4 in modulating inflammation during pregnancy.
Results
Methimazole administration in pregnant mice induces gestational hypothyroxinemia
Given the established association between gestational HTX and neurodevelopmental outcomes16,17,18,19,20,21,22,23,24, we aimed to investigate the prenatal impact of HTX on the maternal immune response, embryonic brain development, and postnatal neurostructural alterations using a well-characterized mouse model. Gestational HTX was induced by administering 0.025% w/v methimazole (MMI) in tap drinking water to pregnant mice from E10 to E14, a critical window of embryonic brain development72. We included an untreated control group (EUT) and a reversion group that received daily supplementation of T4 during HTX induction (HTX + T4) to assess baseline physiological parameters and evaluate the potential for hormonal rescue of HTX-induced alterations, respectively (see Materials and methods). This model relies on the mechanism of MMI, which acts as an allosteric inhibitor of the thyroid peroxidase enzyme, thereby blocking the iodination of thyroglobulin tyrosine residues and subsequent coupling reactions essential for T3 and T4 production during TH biosynthesis21,73,74,75,76. To confirm the correct induction of HTX, serum levels of total (t)T4, tT3, and TSH were quantified in pregnant EUT, HTX, and HTX + T4 mice at the end of HTX induction (E14) (Fig. 1). Importantly, the HTX dams exhibited reduced tT4 levels in serum, compared to EUT and HTX + T4 conditions (Fig. 1a). In contrast, the levels of both tT3 (Fig. 1b) and TSH (Fig. 1c) remained similar among the experimental groups. The sole reduction of tT4 in HTX pregnant mice, while maintaining unaltered levels of tT3 and TSH, confirmed the successful induction of HTX.

Total thyroid hormones and TSH levels were measured in serum samples from EUT (euthyroid), HTX (methimazole-treated), and HTX + T4 (methimazole + T4-supplemented) dams at embryonic day 14 (E14) (see Materials and Methods). (a) Total T4 (tT4), (b) Total T3 (tT3), and (c) TSH levels. n = 11 pregnant mice per group. Data are presented as mean ± SEM. Statistical significance was determined by using one-way ANOVA with Tukey’s post hoc test or Kruskal–Wallis test with Dunn’s post hoc test, depending on data normality. ***p ≤ 0.001, ****p ≤ 0.0001. EUT: white box; HTX: red box; HTX + T4: green box.
Gestational hypothyroxinemia causes a pro-inflammatory environment at the maternal-fetal interface and in the embryonic brain
HTX has been associated with an increased risk of autism in the offspring25,26, and this association was demonstrated to be causal in a mouse model, where reduced maternal T4 levels during pregnancy led to the emergence of autism-like traits28. However, the cellular and molecular mechanisms underlying how gestational HTX leads to these outcomes in offspring remain unclear. Reports available in the literature suggest that maternal T4 crosses the placenta and is locally converted into T3 (the biologically active form of TH)4,5,6, which regulates gene expression in fetal cells during neurodevelopment10,11. Nonetheless, reductions in TH levels have also been linked to enhanced pro-inflammatory responses and stimulation of myeloid cell activation70,71. Therefore, it is pertinent to evaluate whether gestational HTX triggers pro-inflammatory responses that disrupt the immune environment at the maternal-fetal interface, potentially contributing to the neurodevelopmental abnormalities observed in the offspring. For this purpose, pregnant EUT, HTX and HTX + T4 mice were euthanized at E14, and the uterine horn was excised and dissected as detailed in Materials and Methods section. Representative images are shown in Supplementary Figure (SF). 1a. The average number of implantation sites per uterine horn was 8.18 ± 1.66 for EUT, 8.09 ± 1.38 for HTX, and 8.09 ± 1.70 for HTX + T4 pregnant mice (mean ± SD, n = 11 per group). Additionally, both uterine horn length and weight, normalized to the number of implantation sites, were similar across EUT, HTX, and HTX + T4 pregnant mice (SF. 1b, c). To explore whether the potential inflammatory component of gestational HTX, pro-inflammatory cytokine levels were quantified in serum samples, maternal-fetal interface tissues, and embryonic brains (Fig. 2) (see Materials and methods). We found elevated levels of IL-6 and IL-17 A in sera of HTX-induced pregnant mice compared to both EUT and HTX + T4 groups (Fig. 2a). A similar pattern was observed in the fetal compartment of the placenta (hereafter referred to as “placenta”) (Fig. 2b), the maternal compartment hereafter refer to as decidua basalis (Fig. 2c), and the uterine tissues (Fig. 2d), where IL-6 and IL-17 A levels were higher in the HTX group. Embryonic brains from HTX-exposed pregnancies also exhibited increased levels of IL-6 and IL-17 A when compared to the EUT and HTX + T4 counterparts (Fig. 2e). The elevated levels of these pro-inflammatory cytokines in circulation and materno-fetal compartments suggest that HTX may induce the activation of the maternal immune system. The possibility that T4 deficiency is responsible for activation of the maternal immune system is supported by the observation that restoring T4 levels during HTX induction normalizes cytokine levels, making them comparable to those observed in EUT controls.

Pregnant mice from the EUT (offspring gestated under euthyroidism), HTX (offspring gestated under hypothyroxinemia), and HTX + T4 (offspring gestated under MMI+T4 treatment) groups were euthanized at E14. Maternal blood was collected by cardiac puncture, and the serum was isolated. The uterine horns were excised, and the placenta, decidua basalis, uterine tissue, and embryonic brains were dissected. Total protein was extracted from these tissues and used for cytokine quantification by sandwich-type ELISA (see Materials and Methods). The graphs show the levels of IL-6 and IL-17 A in: (a) maternal serum, (b) placenta, (c) decidua basalis, (d) uterus, and (e) embryonic brain. n = 11 pregnant mice per group. In (a), each point represents the cytokine level from an individual pregnant mouse. In (b–e), each point represents the average cytokine level from tissue samples collected from the half of implantation sites per uterine horn, which were processed independently. Data are presented as mean ± SEM. Statistical significance was assessed using two-way ANOVA followed by Tukey’s post hoc test. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. EUT: white box; HTX: red box; HTX + T4: green box.
Another fundamental aspect of prenatal inflammation is the activation of myeloid cells at the maternal-fetal interface77. Among them, macrophages, dendritic cells, and monocytes, are recognized for their key roles in immune regulation and the maintenance of tissue homeostasis during pregnancy78,79. Studies using MIA models have reported that pro-inflammatory stimuli induce a shift toward activated, pro-inflammatory phenotypes in these myeloid populations, contributing to altered immune responses and adverse fetal outcomes77. Since we found that HTX increases the levels of IL-6 and IL-17 A in maternal circulation and the maternal-fetal interface (Fig. 2a–d), we next sought to evaluate the frequency of myeloid cell subsets at the maternal-fetal interface by flow cytometry, aiming to determine whether gestational HTX similarly promotes myeloid cell activation and/or pro-inflammatory-like polarization (Fig. 3) (see Materials and methods). The gating strategies for conventional dendritic cells (cDCs), M1/M2 macrophages, neutrophils, monocytes, and NK cells are illustrated in SF. 2a–c. The analysis revealed a significant increase in cDCs (CD45⁺CD3⁻CD11b⁻CD11c⁺MHC-II⁺) only in the placenta of HTX-induced pregnant mice, as compared to EUT and HTX + T4 groups (Fig. 3a, b). Additionally, NK cells (CD45⁺CD3⁻CD11b⁺CD16/32⁺NK1.1⁺CD49b⁺) were elevated in the uterine tissue of HTX dams compared to EUT and HTX + T4 pregnant mice (Fig. 3c, d). M1-like macrophages (CD45⁺CD3⁻CD11c⁻CD11b⁺CD68⁺CD86⁺CD206⁻) (Fig. 3e, f) and monocytes (CD45⁺CD3⁻CD11c⁻CD11b⁺Ly6C⁺Ly6G⁻) (Fig. 3g, h) were in turn increased in decidua basalis and uterine tissues of HTX dams, as compared to those of the EUT and HTX + T4 groups. Overall, these findings suggest that elevated IL-6 and IL-17 A at the maternal-fetal interface promote a pro-inflammatory environment that favors the polarization of myeloid cells toward a pro-inflammatory phenotype.


Pregnant mice from the EUT (euthyroid), HTX (hypothyroxinemic), and HTX + T4 (T4-supplemented) groups were euthanized at E14. The uterine horns were excised, and the placenta, decidua basalis, and uterine tissues were dissected. Total leukocytes were isolated, and immune cell proportions were assessed by flow cytometry (see Materials and Methods). (a) Representative contour plots of conventional dendritic cells (cDCs); (b) quantification of cDC proportions in the placenta, decidua basalis, and uterus; (c) representative contour plots of uterine NK cells; (d) quantification of NK cell proportions; (e) representative contour plots of M1-like macrophages; (f) quantification of M1 macrophage proportions; (g) representative contour plots of monocytes; (h) quantification of monocyte proportions in the placenta, decidua basalis, and uterus. n = 11 pregnant mice per group. Each point represents the average proportion calculated from tissue samples collected from the half of implantation sites per uterine horn, processed independently. Data are presented as mean ± SEM. Statistical differences were assessed using two-way ANOVA followed by Tukey’s post hoc test. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. EUT: white box; HTX: red box; HTX + T4: green box.
Gestational hypothyroxinemia disrupts embryonic cortical development and expands the microglial pool
It has been demonstrated that the impact of prenatal inflammation extends to embryonic neurodevelopment by altering the biology of neural precursor cells (NPCs) and neuronal migration33,80,81. Similarly, accumulating evidence shows that gestational HTX also alters neuronal migration22,23,24. However, whether HTX affects NPCs or microglial populations remains unexplored. To address this question, we performed immunofluorescence analysis on embryo cortical brain sections to examine NPC populations and microglial pool at E14 (Fig. 4) (see Materials and methods). The study revealed that the number of Pax6+ radial glial cells in the ventricular zone (VZ) of the developing cortex was similar among EUT-, HTX, and HTX + T4-exposed embryos (Fig. 4a, b). However, the number of Tbr2⁺ intermediate progenitors was mildly reduced in the developing cortex of fetuses exposed to HTX compared to those from the EUT and HTX + T4 groups (Fig. 4c, d). As the number of proneurogenic intermediate progenitors was reduced, we asked whether the genesis of neurons could also be affected. To assess this, we immunolabeled Tbr1+ deep-layer neurons and found a significant increase in Tbr1+ neurons in fetuses gestated under HTX, compared to the EUT and HTX + T4 conditions (Fig. 4e, f). These findings suggest that HTX disrupts NPC dynamics and neurogenesis, which can be prevented by T4 supplementation. Since we observed a pro-inflammatory environment in embryonic brains, indicated by increased levels of IL-6 and IL-17 A (Fig. 2e), we next asked whether this inflammatory milieu may lead to an increase in microglial pool, as exacerbated microglial expansion is a known cellular response to elevated pro-inflammatory cytokines and can further influence neurodevelopmental processes67,68,69. Since microglia are relatively sparse in the embryonic cortex compared with neural progenitors, we quantified them within a defined cortical area encompassing a substantial portion of the cortical thickness. This approach minimized variability due to local differences in microglial distribution and ensured that measurements were comparable between experimental groups, as detailed in the Materials and Methods section. We found an increased number of microglial cells (Iba1⁺ cells) in the developing brain cortex of fetuses exposed to HTX, compared to those from EUT and HTX + T4 maternal treatment groups (Fig. 4g, h). This suggests that the pro-inflammatory environment in embryonic brains increases the microglial pool and may lead to pro-inflammatory-like microglial activation in the fetal cortex. Interestingly, this effect can be prevented by restoring T₄ levels during HTX induction, reinforcing the notion of a potential immunomodulatory role for maternal T₄ during pregnancy.

Embryonic brains from EUT (gestated in euthyroidism), HTX (offspring gestated under hypothyroxinemia), and HTX + T4 (offspring gestated undeer MMI+T4 treatment)dams euthanized at E14 were fixed, cryosectioned, and analyzed by immunofluorescence to evaluate neuronal progenitor cells (NPCs) and microglia. (a) Representative images of Pax6⁺ radial glial cells in embryonic cortices from each maternal condition. (b) Quantification of Pax6⁺ cells. (c) Representative images of Tbr2⁺ intermediate progenitor cells in embryonic cortices. (d) Quantification of Tbr2⁺ cells. (e) Representative images of Tbr1⁺ deep-layer neurons. (f) Quantification of Tbr1⁺ neurons. (g) Representative images of Iba1⁺ microglia. (h) Quantification of Iba1⁺ cells. Pax6⁺, Tbr2⁺, and Tbr1⁺ cells were quantified within a rectangular region (200 μm wide x 400 μm long) extending from the ventricular zone to the meninges. Iba1⁺ cells were quantified within a defined area along the cortical wall. For each fetal brain, quantification was performed on three consecutive coronal sections, and the average value was used per animal. Images were acquired at 20x magnification. Scale bar: 200 μm. n = 6–7 per group. CP: cortical plate; VZ: ventricular zone. Data are presented as means ± SEM. Statistical significance was determined using one-way ANOVA followed by Tukey’s post hoc test or Kruskal–Wallis with Dunn’s post hoc test, depending on normality. Mann–Whitney U test was also applied where appropriate. *p ≤ 0.05, **p ≤ 0.01. EUT: white box; HTX: red box; HTX + T4: green box.
Gestational hypothyroxinemia reduces spine density and impairs spine maturation in CA1 pyramidal neurons in male offspring
Aberrant dendritic spine morphology and altered spine density in the hippocampus have been consistently reported in autistic individuals, reflecting disrupted synaptic development and connectivity82. Similarly, MIA models have been associated with aberrations in dendritic spine density and maturation in offspring67,83. On the other hand, we previously reported that offspring gestated under HTX exhibit certain autistic-like behaviors28, and in the present study we show that gestational HTX induces a mild form of prenatal inflammation (Fig. 2), increases placental myeloid cells (Fig. 3), impairs embryonic cortical development (Fig. 4a–f), and increases microglial pool in the developing cortical brain (Fig. 4g, h). Since microglia are known to mediate synaptic pruning during cortical development and thereby influence dendritic spine maturation67,68,69, we sought to determine whether prenatal HTX could lead to long-lasting synaptic alterations in the offspring. Thus, we analyzed the spine density and morphology of dendritic spines in the CA1 region of the adult male hippocampus in offspring gestated under HTX, using Golgi-Cox staining (Fig. 5) (see Materials and methods). We focused this analysis on the CA1 region, as it is a critical area for learning and memory that exhibits well-documented forms of synaptic plasticity, and because previous evidence has shown reduced LTP and abnormal expression of key glutamatergic components in the CA1 region of HTX offspring20,21. Additionally, we focused the analysis on males given the higher prevalence and severity of ASD-related traits reported in males across both human84,85 and animal models86,87.
Since this experiment used a different cohort of pregnant mice to obtain the respective offspring, we confirmed the induction of HTX by measuring TH and TSH levels in blood samples collected from the facial vein of each pregnant mouse at E14, to avoid interrupting pregnancy. As expected, serum levels of tT4, tT3, and TSH in pregnant EUT, HTX, and HTX + T4 mice confirmed the successful induction of HTX (SF. 3a–c). Additionally, the levels of tT4, tT3, and TSH were also measured in the EUT, HTX, and HTX + T4 offspring at P55 to determine whether gestational HTX can affect or have lasting consequences on offspring thyroid function (SF. 3d–f). TH and TSH levels were similar across groups (SF. 3d–f), suggesting that HTX induction did not impair the offspring’s thyroid function. Additionally, the comparable TH levels across groups suggest that the offspring’s alterations stem not from persistent thyroid dysfunction, but from prenatal HTX exposure during the critical gestational window.
Representative images of Golgi-stained hippocampal sections are shown in SF. 4, in which we provide images of the left hippocampus (SF. 4a), with a focus on the CA1 region (SF. 4b), pyramidal neurons (SF. 4c–e), and dendritic spines (SF. 4f). Representative images of apical dendritic spines are shown for the EUT, HTX, and HTX + T4 offspring (Fig. 5a). We first assessed dendritic spine density, defined as the number of spines per 10 μm of dendritic length, as a proxy for synaptic connectivity. Spine density was lower in the HTX offspring than in EUT and HTX + T4 offspring (Fig. 5b). Furthermore, analysis of spine maturation subtypes revealed that compared to the EUT and HTX + T4 offspring, the HTX offspring exhibited a reduced number of stubby and mushroom-shaped dendritic spines (both considered markers of synaptic maturity) (Fig. 5c). The number of immature thin and filopodia-shaped spines remained similar across EUT, HTX, and HTX + T4 groups (Fig. 5c).

Male EUT (offspring gestated in euthyroidism), HTX (offspring gestated in hypothyroxinemia), and HTX+T4 (offspring gestated in under MMI and T4 treatment) were euthanized at P55. Half of each brain was used for dendritic spine analysis of hippocampal CA1 pyramidal neurons using Golgi-Cox staining. (a) Representative images of dendritic spines in CA1 pyramidal neurons (100x magnification) from EUT, HTX, and HTX + T4 male offspring. (b) Quantification of spine density, defined as the number of spines per 10 μm of dendritic length. (c) Quantification of dendritic spine morphologies. Scale bar = 10 μm. n = 15 animals per group. In (b), each point represents the average of three independent measures (three hippocampal sections per animal). In (c), n = 45 dendritic segments per group (15 animals x 3 dendritic segments per animal); each data point corresponds to an individual dendritic segment measurement. Data are presented as mean ± SEM. Statistical analysis was performed using one-way (b) or two-way (c) ANOVA followed by Tukey’s post hoc test. *p ≤ 0.05, ***p ≤ 0.001, ****p ≤ 0.0001. EUT: blue box; HTX: red box; HTX + T4: green box.
Discussion
This work shows evidence that gestational HTX increases the levels of IL-6 increased levels of IL-6 and IL-17 A in maternal serum, maternal-fetal placental tissues, and embryonic brains (Fig. 2), suggesting that gestational HTX can induce a pro-inflammatory environment during a critical window of pregnancy. These findings are in accordance with the high levels of IL-6 detected in the serum of pregnant hypothyroxinemic women88, reinforcing that gestational HTX can trigger a pro-inflammatory response in the mother. Under physiological conditions, IL-6 plays a crucial role in early pregnancy by supporting implantation and placental development89,90. However, elevated levels of IL-6 can be harmful during pregnancy, inducing an inflammatory state that has been associated with adverse pregnancy outcomes, such as preeclampsia91,92, gestational diabetes mellitus (GDM)93,94, and heightened immune responses to bacterial and viral infections95. Regarding IL-17 A, this cytokine is a key effector of Th17 cells and has been increasingly recognized for its role in mediating inflammatory responses during pregnancy as well as in promoting neurodevelopmental disorders in offspring96. Similar to IL-6, IL-17 A is also often elevated in maternal serum and placental tissues in a MIA model induced by the administration of poly(I: C)97. Our findings of elevated IL-17 A levels in the maternal-fetal interface and embryonic brain (Fig. 2) suggest that gestational HTX may induce a Th17-like immune shift, further contributing to a pro-inflammatory intrauterine environment. In quantitative terms, the levels of IL-6 and IL-17 A detected in the maternal serum of HTX-induced pregnant mice reached nearly 120 pg/mL and 200 pg/mL, respectively (Fig. 2a). At the maternal-fetal interface tissues and fetal brains, these levels ranged from approximately 60–100 pg/mL for IL-6 and 80–200 pg/mL for IL-17 A (Fig. 2b–d). Although the levels of these cytokines are higher than in control, these cytokine concentrations are generally lower than those reported in LPS- and poly(I: C)-induced MIA models, where IL-6 levels typically range from 200 to 400 pg/mL and IL-17 A from 200 to 600 pg/mL36,45,47,56,77,98. The fact that gestational HTX induces a milder pro-inflammatory response compared to LPS-induced MIA does not imply that this response lacks biological impact within the intrauterine environment. Indeed, our results suggest an association between mild MIA induced by gestational HTX and autistic-like outcomes, which could be further tested through complementary approaches, such as IL-6 or IL-17 A neutralization, to determine whether the observed neurodevelopmental effects arise directly from this mild immune activation. The observed concordance of IL-6 and IL-17 A changes across maternal serum, the maternal-fetal interface, and the fetal brain is not unique to gestational HTX, as similar patterns have been reported in classical MIA models induced by poly(I: C) and LPS46,47,48. However, what distinguishes the HTX model is the endocrine origin rather than infectious, providing a different inducer of the inflammatory cascade. This raises a possible mechanism where systemic maternal cytokine is the first changes that increases the cytokine production in the decidual basalis and/or placenta, and finally these cytokines could reach the fetal brain activating fetal microglia. While our data do not establish causal directionality between cytokine production and neurodevelopmental alterations, this framework may be indicative of both the overlap with established MIA models. While our current measurements in unperfused placentas provide an integrated view of maternal and local placental cytokine contributions, future studies could incorporate perfusion or compartment-specific analyses to better distinguish circulating versus locally produced cytokines.
The primary candidates for secreting IL-6 and IL-17 A are myeloid cells99. Macrophages, DCs, monocytes, and NK cells are important mediators of the innate immune response at the maternal-fetal interface, predominantly located at the decidua basalis100,101. Under the context of prenatal HTX, we observed an increased proportion of M1-type macrophages and monocytes in decidua basalis and uterine tissues (Fig. 3e–h). This finding is consistent with the known effects of maternal inflammation, where these cells can shift toward a pro-inflammatory phenotype, promoting the secretion of IL-6 and IL-17 A and contributing to a dysregulated immune environment77. Some reports have shown that TH deficiency modulates the immune response toward a pro-inflammatory phenotype70,71. Evidence from mouse models has demonstrated that T3 administration exerts anti-inflammatory effects, while in vitrostudies have shown that bone marrow-derived monocytes exposed to low TH levels tend to adopt a pro-inflammatory profile70,71. It has been proposed that the pro-inflammatory adaptation triggered by conditions, such as hypothyroidism, is primarily driven by the non-genomic actions of T4, which involve its binding to the plasma membrane integrin αvβ3 and subsequent activation of the PI3K-AKT signaling pathway70,71. This pathway leads to increased ROS production, activation of the NLRP3 inflammasome, and the release of pro-inflammatory cytokines102. Thus, the increased proportion of M1-type macrophages and monocytes aligns with previous studies, which report that TH deficiency promotes macrophage polarization toward a pro-inflammatory M1-like phenotype70,71. Considering that macrophages are major sources of IL-6 during pregnancy77, this finding is also in line with the elevated IL-6 levels detected at the maternal-fetal interface (Fig. 2). This immunological shift may contribute to a sustained pro-inflammatory microenvironment, which has been implicated in altered placental function and impaired fetal brain development, similar to the alterations observed in poly(I: C)-induced MIA models99. Likewise, the observed increase in the monocyte proportion is also consistent with this trend, as maternal monocytes are known to infiltrate gestational tissues during inflammatory states and can differentiate into pro-inflammatory macrophages, further amplifying cytokine release and disrupting immune tolerance79,103,104. In fact, the increase of M1 macrophages in these tissues could be related to the capacity of monocytes to differentiate into M1 macrophages under an inflammatory state. Furthermore, an increase in NK cells was observed only in the uterus (Fig. 3c, d). This finding aligns with previous reports showing alterations in the proportion of uterine and decidual NK cell populations, reduced expression of inflammatory molecules, and impaired trophoblast migration kinetics in rat models of both maternal hyperthyroidism and hypothyroidism105,106. Autoimmune thyroid disorders can also increase the proportion of NK cells and cause adverse outcomes in pregnancy including placental abruption, spontaneous abortion, and premature delivery107,108. Since uterine NK cells are particularly involved in modulating the local immune environment and remodeling uterine vasculature to support pregnancy109,110, the selective increase in uterine NK cells under HTX conditions may represent a compensatory response aimed at maintaining tissue integrity and vascular function in the face of inflammation, similar to what has been observed in LPS-induced MIA model in mice111,112. These findings highlight the potential for gestational HTX to disrupt immune homeostasis in uterine tissues, potentially impairing the tightly regulated processes crucial for successful placental development and fetal tolerance. In addition to NK cells, we observed an increased proportion of cDCs in the fetal portion of the placenta exposed to gestational HTX (Fig. 3a, b). DCs play a dual role at the maternal-fetal interface: under homeostatic conditions, they contribute to maternal immune tolerance and help shape an anti-inflammatory environment necessary for fetal survival113. However, their excessive activation or accumulation has been linked to pro-inflammatory responses and adverse pregnancy outcomes, according to LPS-induced MIA models114. Thus, in our model of maternal HTX, this increase may indicate an immune deviation toward a more activated or pro-inflammatory state, potentially contributing to the inflammatory environment we detected. This increase of DCs in the fetal portion of the placenta of HTX dams is interesting since placental immune cells are typically located in the decidua basalis56,57. This finding is unusual an suggest that the local inflammation can promote the recruitment or differentiation of myeloid cells beyond the decidual compartment. Reviews of the maternal-fetal interface emphasize that uterine and decidual DCs are normally restricted and maintain a tolerogenic phenotype during pregnancy, but inflammatory challenges can alter their phenotype and trafficking, resulting in changes in DC abundance or maturation across placental tissues115. Recent experimental studies using human placenta ex vivo models have similarly demonstrated placental immune remodeling, including increased activation and abundance of otherwise rare myeloid and antigen-presenting cell populations116,117,118. Therefore, it is plausible that our observation may reflect a similar inflammatory remodeling process, supporting the idea that endocrine-driven immune dysregulation could mirror immune-induced alterations at the maternal-fetal interface. Thus, these findings support that there is a milder inflammatory response at maternal-fetal interface tissues during gestational HTX. Moreover, histological or imaging-based approaches will contribute about the spatial and structural context of this pro-inflammatory state.
Overall, gestational HTX causes a mild increased levels of pro-inflammatory cytokines and immune cells at the maternal-fetal interface compared to classical MIA induced by viral infection119, stress120, and obesity121. Since in normal pregnancy there is a tolerogenic environment at the maternal-fetal interface tissues, it is possible that this mild inflammatory state could be one of the cellular mechanisms that promotes the autistic-like traits in the offspring gestated in HTX. Thus, this mild inflammatory state plus other consequences of low T4 in placenta development could result in similar consequences in fetus neurodevelopmental like the classical MIA models36,47. We think that gestational HTX may initially alter the function of circulating immune cells, such as T lymphocytes, monocytes, and dendritic cells, which are directly sensitive to reduced T4 levels. This will lead to changes in their activation profile and promote the early and mild secretion of pro-inflammatory cytokines122,123,124. Then, peripheral maternal tissues that are responsive to TH, such as spleen, adipose tissue, and decidua basalis can react to low T4 levels and will further contribute to the systemic increase of pro-inflammatory cytokines125,126. The resulting alterations on placenta and the inflammatory environment at the maternal-fetal interface may contribute a mild “MIA-like state” that impairs placental function and early fetal brain development. Although this model is currently hypothetical, it offers a unified framework connecting gestational HTX to immune-mediated neurodevelopmental outcomes and identifies mechanisms suitable for future experimental investigation. This interpretation is also supported by evidence showing that rebalancing the immune environment at the maternal-fetal interface can prevent the autism-like outcomes in offspring127.
The observation of high levels of IL-6 and IL-17 A in the embryonic brain of the offspring gestated in HTX (Fig. 2e) suggest that the inflammatory process extends from the mother to the fetus. In fact, it has been suggested that maternal inflammation may indirectly affect the fetal compartment, potentially through changes in placental permeability or enhanced cytokine transport, rather than direct transplacental passage of pro-inflammatory cytokines128,129. However, beyond the passive diffusion or transport of maternal cytokines, it is increasingly recognized that placental cells themselves can actively participate in propagating inflammation. It has been described that during preeclampsia, different subsets of trophoblasts and decidual immune cells can shift their phenotype and secrete pro-inflammatory cytokines, including IL-6, IL-1β, and TNF-α, into the fetal compartment, thereby contributing to the neuroinflammatory milieu130. A similar phenomenon has been observed during prenatal infections, such as those caused by Listeria monocytogenes131, Zika virus132, Cytomegalovirus (CMV)133, Influenza virus134, and SARS-CoV-2135. Thus, maternal TH deficiency may alter placental immune homeostasis, indirectly contributing to fetal brain inflammation through impaired immune regulation. Additionally, we cannot rule out the possibility that low circulating maternal T4 levels may directly affect fetal brain-resident immune cells. Previous studies have shown that T3 regulates microglial development and activity136,137. Thus, reduced TH availability in the fetal brain may favor microglial priming or activation, further contributing to the elevated IL-6 and IL-17 A levels observed. Together, these findings support the notion that gestational HTX not only triggers maternal inflammation but also may compromise placental immune function and promote fetal neuroinflammation through both systemic and local mechanisms. This issue led us to explore the potential effect of gestational HTX on NPCs and microglial populations (Fig. 4). The increase in Iba1+ microglial pool in the developing cortex of HTX-exposed embryos (Fig. 4g, h) is consistent with the increased IL-6 and IL-17 A levels in the developing brain (Fig. 2e) and with previous reports of MIA models showing microglial activation67,68,69. Interestingly, recent studies have shown that radial glial cells serve not only as neural progenitors and scaffolds for neuronal migration but also as immune-responsive cells during development35. They are capable of secreting IL-6 in both autocrine and paracrine manners in response to inflammatory cues, including MIA challenge35. Although we did not find alterations in the number of Pax6⁺ radial glia populations (Fig. 4a, b), suggesting that gestational HTX may not directly impair the proliferation or maintenance of neural progenitors at this stage, another plausible hypothesis is that their functional state may be affected. Specifically, even if the total number of radial glial cells remains unchanged, their activation state could be altered under HTX conditions. In this context, IL-6 signals originated from the placenta may stimulate radial glial cells in the developing brain to produce and secrete additional IL-6, thereby amplifying local neuroinflammation. This potential mechanism involving placental-derived and brain-intrinsic IL-6 overproduction could represent a critical pathway by which gestational HTX promotes immune-mediated disruptions in cortical development. In parallel, it is well established that a pro-inflammatory environment in the fetal brain can have detrimental effects on neurodevelopment, particularly by disrupting critical processes, such as neurogenesis and neuronal migration33,80,81. Here, we found a mild reduction in Tbr2+ intermediate progenitor cells and a concomitant increase in Tbr1+ deep-layer neurons in the cortical areas of fetuses gestated in HTX at E14 (Fig. 4c–f). These alterations are consistent with impaired corticogenesis, as previously reported in rat models of gestational hypothyroidism138,139. Moreover, Lavado-Autric et al. and Ausó et al. have demonstrated that HTX leads to aberrant neuronal migration in both the somatosensory cortex and the hippocampus22,23,24. Thus, our findings reinforce that gestational HTX during early brain development, a period characterized by intense neurogenic activity140, induces premature differentiation of NPCs. This alteration may lead to exhaustion of the progenitor pool, potentially contributing to long-term structural and functional brain abnormalities. Importantly, T4 supplementation during the period of HTX induction normalized the number of intermediate progenitors, deep-layer neurons, and microglia to levels comparable to those in EUT-exposed fetuses (Fig. 4). These findings highlight the essential role of T4 in maintaining proper neurogenic processes and immune balance during fetal brain development141. These findings further suggest that the neurodevelopmental disruptions observed in HTX may be mediated, at least in part, by a T4-dependent suppression of inflammatory pathways141. The molecular mechanisms by which T4 deficiency leads to impairment of neurogenesis remain to be fully elucidated. However, our results support a model in which reduced T4 levels contribute to altered progenitor cell fate and neuroinflammation, possibly through pro-inflammatory cytokine signaling and microglial activation, ultimately leading to defective corticogenesis.
Conditions that cause prenatal inflammation have been associated with altered dendritic spine density in offspring67,83, which is often linked to microglial expansion and activation during early neocortical development67,68,69. We found that gestational HTX led to long-term implications for synaptic development and connectivity in the adult offspring by reducing spine density (Fig. 5b) and the number of stubby and mushroom-shaped dendritic spines in male offspring, as compared to the EUT and HTX + T4 groups (Fig. 5c). These findings align with impaired synaptic plasticity and cognitive deficits, further emphasizing the long-term impact of maternal inflammation on brain structure and function in offspring67,83 and can also be related to the autistic-like outcomes previously reported in HTX offspring28. Similarly, the reduced total count and mature-shaped dendritic spines in male HTX offspring are consistent with previous observations in the C58/J mouse, a genetic model of ASD characterized by deficits in dendritic spine density and morphology in the hippocampus and prefrontal cortex142,143. Importantly, this works has shown data regarding male offspring dendritic spine morphology. We acknowledge that evaluating female offspring is necessary to assess whether gestational HTX produces sex-specific effects. Hormonal cycles and sex-dependent neurodevelopmental trajectories may differentially affect immune and behavioral outcomes.
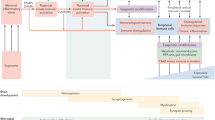
Overall, these observations may represent a turning point in how the molecular and cellular mechanisms by which T4 deficiency impairs CNS development during the first trimester of pregnancy are understood (Fig. 6). While it is well established that maternal TH are essential for fetal neuronal function and differentiation10,11, there remains a significant knowledge gap regarding how low maternal T4 levels early in pregnancy affect fetal CNS development. During early pregnancy, the placenta expresses TH transporters, such as MCT8, OATP1C1, and LAT1/2, which facilitate the transfer of maternal T4 into placental cells4,5. The placenta expresses iodothyronine deiodinases, such as DIO3, which inactivate T4 by converting it into reverse T3 (rT3) and T2, thereby limiting excessive fetal exposure to active T3. To a lesser extent, it also expresses DIO2, which locally converts T4 into the bioactive T3, supporting placental functions, such as growth and vascular development7,8,9. Most of the maternal T4 that bypasses placental metabolism enters the fetal compartment, where DIO2 converts it in the fetal brain into T3 which is essential for neuronal differentiation, migration, and overall brain maturation7,8,9. By as early as 9 weeks of gestation, low levels of T3 have been detected bound to nuclear receptors in the fetal brain10,11,12. Although these low concentrations are thought to play physiological roles, particularly in neuronal migration and corticogenesis, the underlying cellular mechanisms remain incompletely understood10,11,12. In our animal model, gestational HTX was induced by MMI administration from E10 to E1421,73,74,75,76. Although the precise onset of T4 deficiency following MMI treatment is unknown, previous studies and our measurements at E14 confirm that this protocol effectively generates HTX without inducing full hypothyroidism21,73,74,75,76. The selected treatment window ensures that T4 deficiency coincides with critical periods of placental maturation and early fetal neurodevelopment144, allowing both compartments to be exposed to the HTX-induced environment. By initiating HTX at E10, we aimed to approximate the temporal window of classical MIA models, in which the inflammatory challenge occurs during similar neurodevelopmental stages, while considering the relatively slow kinetics of thyroid hormone depletion and the resulting pro-inflammatory response. This strategy enables investigation of mechanisms underlying subsequent changes in fetal brain development. Moreover, it is unknown the metabolism of TH in placenta and fetal tissues under gestational HTX, this knowledge would be highly informative about the fetus exposure to thyroid hormones during critical developmental windows. Given the insights of this work, it is plausible to note that insufficient maternal T4 during early gestation may impair fetal brain development not only through direct hormonal deficiency alone, but also indirectly via the induction of a maternal pro-inflammatory state (Fig. 6). This inflammatory milieu may disrupt immune homeostasis at the maternal-fetal interface and interfere with critical neurodevelopmental processes, leading to persistent neurostructural abnormalities. Notably, these alterations appear preventable by restoring maternal T4 levels during gestation (Figs. 2, 3, 4, 5 and 6), reinforcing the need to investigate the immune-mediated mechanisms by which early gestational HTX impairs neurodevelopment.

Gestational HTX increases circulating levels of IL-6 and IL-17 A, similar to observations in MIA models. These cytokines are also elevated in placental tissues and embryonic brains. It is postulated that this pro-inflammatory environment at the maternal-fetal interface activates myeloid cells, including M1 macrophages, NK cells, and monocytes, thereby contributing to immune imbalance. This immune dysregulation may impair fetal neural progenitor cells (NPCs), particularly by affecting the proportion of intermediate progenitors, which could lead to an altered generation of deep-layer cortical neurons relevant for establishing early cortical circuitry and supporting higher-order cognitive functions. The increased expansion of microglial pool is also consistent with the pro-inflammatory environment at the embryonic developing brain that contribute to long-term neurostructural alterations, including reduced dendritic spine density and a decrease in mature-shaped spines in the hippocampus. These changes are associated with neurocognitive deficits in the offspring. Interestingly, T₄ supplementation during gestation prevents immune imbalance and mitigates these neurodevelopmental alterations, suggesting a critical role of maternal T₄ in modulating maternal-fetal immunity during early embryonic neurodevelopment during early. CP: cortical plate; IZ: intermediate zone; SVZ: subventricular zone; VZ: ventricular zone. Created with BioRender.
Materials and methods
Animals
Wild-type C57BL/6 mice were obtained from The Jackson Laboratory, Bar Harbor, ME, USA. Mice were housed in cages with water, bedding, and chow (Prolab, RMH 3000) under standard conditions (12-hour light/dark cycle, temperature 22 °C) and maintained at the animal facility of the Facultad de Ciencias de la Vida, Universidad Andrés Bello (Bioethics approval certificate number 012/2021). The experimental procedures and care were performed in concordance with the current regulations of the Animal Welfare Committee of the Facultad de Ciencias de la Vida and the National Agency for Research and Development (ANID) (Santiago, Chile). A veterinarian performed daily supervision of the mice. Mice were euthanized by inhalation of 3% isoflurane/O2145. This study is reported in accordance with ARRIVE guidelines.
Gestational hypothyroxinemia and experimental groups
To induce HTX, pregnant mice were treated as described in previous reports28,73,74,75. Six- to eight-week-old C57BL/6 mice were mated overnight. On the next day, the presence of a vaginal plug was evaluated, and this day was designated as embryonic day (E)0. At E10, females were weighed to assess gestational weight gain. A minimum increase of 2 gr was required to consider the mice pregnant, along with the characteristic rounded abdominal shape146. Subsequently, pregnant mice were housed individually and randomly assigned to one of three groups. The first group of dams received 0.025% W/V 2-mercapto-1-methylimidazole (methimazole, MMI) (M8506, Sigma-Aldrich, USA) in tap-drinking water from E10 to E14. The group received the name of “HTX dams”. To reverse the MMI-induced phenotypes, the second group was treated with 0.025% W/V MMI plus a daily subcutaneous dose of T4 (25 µg/kg) from E10 to E14. The group received the name of “HTX + T4 dams”. This treatment also aimed to demonstrate that the effects induced by MMI are exclusively due to maternal T4 reduction and not a side effect of MMI. The third group received regular tap water during the entire pregnancy; thus, gestation was performed under euthyroid-control conditions. The group was named “Euthyroid (EUT) dams”. HTX and HTX + T4 treatments were administered daily from E10 to E14. Fresh water for the EUT dams was also renewed daily from E10 to E14. The bottles containing MMI solution were prepared daily and protected from light. The effect of daily injections per se was not evaluated in a separate vehicle-injected group, as the primary comparison focused on HTX versus EUT dams, neither of which received injections. HTX + T4 pregnant mice were handled prior to injections to minimize stress, ensuring that the observed improvements are attributable to T4 restoration rather than injection-related stress147,148. A second cohort of HTX, HTX + T4, and EUT pregnant mice was employed to evaluate the morphology and density of hippocampal dendritic spines in the resulting adult offspring, which were named according to their maternal treatment: HTX offspring, HTX + T4 offspring, and EUT offspring. To mitigate the impact of the “litter effect” and enhance the reliability of results, experimental groups were formed by randomly selecting 2–3 mice of each sex from different litters149. Mice of the same sex were housed together in cages (4–5 mice per cage) from weaning until postnatal day (P)55.
Determination of total thyroid hormones and TSH
Blood was collected via cardiac puncture from HTX, HTX + T4, and EUT pregnant mice on E14, and via puncture of the facial vein in the second cohort of dams. The serum was isolated by centrifuging the blood samples at 1,000 xg for 15 min at 4 °C. To verify that HTX was properly induced, total (t)TH and TSH levels were quantified using 100 µL of serum samples. tT3 and TSH levels were determined by sandwich ELISA using mouse CUSABIO® ELISA kits (catalog N°E05086m and N°E05116m, respectively). tT4 serum levels were determined by chemiluminescence at an external certified veterinary laboratory (LQCE, Santiago de Chile). To evaluate the thyroid function of the offspring, the levels of tTH and TSH were also quantified in HTX, HTX + T4, and EUT offspring, using blood samples from the facial vein obtained at P50. The tT4 range for HTX dams was established at 0.1–2.0 (µg/dL), while the range of tT4 in euthyroid and HTX + T4 dams was established at 2.2–4.0 (µg/dL). These ranges were determined based on previous publications of TH levels in pregnant mice150,151.
Isolation of placental tissues and embryonic brains
The maternal-fetal interface tissues were isolated at E14 according to a previous report152. Briefly, the lower abdominal skin and muscular tissues of dams were removed, and the uterine horns with implantation sites were detached from the mesentery. Each implantation site was dissected individually, and the number of implantation sites was counted to determine the average per experimental group. The maternal portion of the placenta (decidua basalis) was carefully separated from the fetal components (the junctional zone and labyrinth) and referred to as “placenta” across the article. The separation of decidua basalis from placenta was performed by using fine forceps under a stereoscopic magnifier. Although separation at this stage is technically challenging, meticulous dissection enabled recovery of decidual tissue suitable for cellular and protein analyses, as previously described153,154,155. Uterine tissues were collected from the mesometrial region associated with each embryo, while embryonic brains were also dissected under magnification. Importantly, E14 corresponds to the end of the MMI treatment window used to induce HTX, making this time point essential for capturing immune and neurodevelopmental alterations as the earliest stage to assess how transient HTX impairs maternal-fetal immune interactions and fetal brain development. Given that each uterine horn contains up to 10 implantation sites, tissues from each horn were divided approximately in half for separate analyses. One portion was allocated for protein extraction and subsequent cytokine measurements using ELISA, while the other was used for cellular analyses by flow cytometry. Fetal brains were also randomly assigned, with half processed for cytokine measurements by ELISA and the remainder fixed in 4% paraformaldehyde (PFA) for immunofluorescence. To avoid litter effect in each assay, implantation sites were randomly selected, and measurements from individual sites were averaged to generate a single representative value per uterine horn for each dam. A total of 11 pregnant dams per group were analyzed across three independent experiments, employing ~ 5 to 6 sites for ELISA and ~ 3 to 4 sites for cellular assays. This random allocation and averaging strategy ensured equal contribution from each dam, minimized confounding from litter size or intra-litter variability, and enabled robust statistical analysis at the maternal level. The approach was designed to assess the global effect of gestational HTX per dam, which is relevant because humans carry a single implantation per pregnancy.
Quantification of cytokines by sandwich-type ELISA
Maternal-fetal interface tissues (placenta, decidua basalis, and uterus) and embryonic brains were homogenized in radioimmunoprecipitation assay (RIPA) buffer composed of 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 0.1% Triton X-100, and supplemented with phosphatase and protease inhibitors (1 mM Na3VO4, 1 mM NaF, 30 mM sodium pyrophosphate, and 1 mM PMSF). The homogenates were incubated on ice for 30 min and then centrifuged at 15,000 xg for 15 min at 4 °C. The supernatants were recovered, and the total protein concentration was quantified using the BCA method (Pierce™, BCA™ 23225). Cytokines were quantified in 100 µL of maternal serum (obtained as previously described) and in 0.5 mg of protein extracted from maternal-fetal interface tissues and embryonic brains, using the following ELISA kits: IL-1β (BioLegend, 432601), IL-6 (BioLegend, 431307), IL-17 A (BioLegend, 432501), TNF-α (BioLegend, 430901), and IL-10 (BioLegend, 555252). The detection was conducted by spectrophotometry using a microplate spectrophotometer (Epoch™). Cytokine levels were normalized to the amount of protein employed for these measurements. The results are presented as pg/mL for cytokines from the serum samples and as pg/mg for cytokines from the maternal-fetal interface tissues and embryonic brains.
Leukocyte isolation
Leukocytes were isolated using a standardized protocol152. Placentas, decidua basalis, and uterine tissues designated for this experiment were transferred to 2 mL tubes containing 500 µL of cold accutase solution (Sigma‒Aldrich A6964). First, the tissues were mechanically digested using sterile surgical scissors. Then, an additional 500 µL of accutase was added to each tube, followed by incubation at 37 °C with gentle orbital agitation (80 rpm) for 30–35 min. The reaction was halted on ice, and the samples were filtered through a 100 μm cell strainer using 20 mL of cold PBS, followed by centrifugation at 1,250 xg for 10 min at 4 °C. After removing the supernatants, cells were resuspended in 1 mL of RPMI-1640 medium. Subsequently, 5 mL of fetal bovine serum (FBS) was slowly added to facilitate phase separation, followed by centrifugation at 1,100 xg for 10 min at room temperature (RT). The cells were then treated with 3 mL of ammonium chloride potassium buffer (pH 7.2) and incubated for 3 min at RT to lyse the red blood cells (RBCs). The reaction was stopped by adding 10 mL of cold PBS and then centrifuging at 460 xg for 5 min at 4 °C. The supernatant was discarded, and the cells were washed with 10 mL of fresh PBS. After centrifugation, the leukocytes were resuspended in 1 mL of PEB buffer (PBS, 0.5% BSA, 2 mM EDTA) and counted using the Countess 3 Automated Cell Counter (Thermo Fisher Scientific) employing the trypan blue exclusion test for cell viability156.
Immune cell assessment by flow cytometry
Leukocytes from each tissue were divided into three groups for the simultaneous detection of myeloid immune cell populations by flow cytometry. Cell staining was performed in a 96-well plate using 50 µl of 1.0 × 107 cells per well. First, the cells were incubated with the fixable viability dye AF700 at a concentration of 0.2 µg/mL (BD Horizon™ 564997) for 15 min at 4 °C in the dark. Subsequently, the cells were centrifuged at 460 xg for 5 min at 4 °C and washed with 100 µL of PBS. After centrifugation, each group of cells was incubated with a specific antibody mixture for extracellular staining (all four were prepared in PEB buffer) for 40 min at 4 °C in darkness. Antibodies are listed in Supplementary Table 1. Antibodies were used at a concentration of 0.2 µg/mL. Following incubation, the cells were centrifuged at 460 xg for 5 min at 4 °C and washed with 200 µL of PEB buffer. Subsequently, the cells were fixed with 200 µL of 2% PFA for 10 min at RT. After washing, the cells were resuspended in 200 µL of PEB buffer for subsequent acquisition in a cytometer. Finally, a total of 250,000 events per sample were recorded using the BD FACSymphony™ A1 Cell Analyzer from the Facultad de Ciencias de la Vida, Universidad Andrés Bello. FlowJo software (version 10.8) was used to analyze the immune cell populations.
Evaluation of radial glia, deep and upper layer neurons, intermediate progenitors, and microglia by Immunofluorescence
Immunostaining of embryonic brain tissue sections was performed as previously described157,158. Briefly, embryonic brains obtained from E14 were submerged in 4% PFA in canonical 50 mL tubes at the time of isolation and maintained for 24 h at 4 °C to ensure proper tissue fixation. Subsequently, the brains were transferred to a 20% sucrose solution dissolved in filtered PBS the following day. After 24 h, the sucrose concentration was increased to 25% and then to 30% 24 h later. The brains were stored at 4 °C for at least seven days until they sank to the bottom of the tube. Subsequently, the dehydrated tissues were embedded in molds using OCT compound for the preparation of coronal Sect. (18 μm in thickness) by cryosectioning. The sections were mounted on glass slides and washed three times with Tris-buffered saline (TBS) for 5 min each. Subsequent permeabilization was conducted at RT using TBS-0.3% Triton X-100 for 30 min, followed by incubation in a 5% BSA-blocking solution for 1 h at RT. Next, the sections were separately probed overnight at 4 °C with the following primary antibodies: anti-Pax6 (2 µg/mL, BioLegend 901301), anti-Tbr1 (1 µg/mL, ab31940), anti-Tbr2 (0.75 µg/mL, ab183991), and anti-Iba1 (0.5 µg/mL, Wako 019–19741) to detect radial glia, deep-layer neurons, intermediate progenitors, and microglia, respectively. The next day, after washing with TBS (six times for 5 min each), the sections were incubated with the secondary antibodies anti-rabbit AF488 (0.5 µg/mL, Jackson 711545152) and anti-rabbit AF555 (0.5 µg/mL, Jackson 711165152) for 1 h at RT. After another round of washing, Hoechst staining was performed in TBS 1 × (1:1,000) for 10 min at RT. Finally, the sections were rinsed twice for 5 min each with TBS and then mounted using Permafluor (ThermoFisher). Image acquisition was performed using a Nikon Eclipse Ti2-E inverted optical microscope equipped for epifluorescence, from the Advanced Microscopy Unit (UMA) at the Pontificia Universidad Católica de Chile. Quantification was conducted using three consecutive coronal sections, and the results were averaged to yield a single value per brain. Iba1+ cells were counted within a predefined region of interest in the embryonic cortex that included a substantial portion of the cortical thickness. The ventral boundary was defined at the tip of the ventricle adjacent to the cortex, and a perpendicular line was drawn to the cortical plate. The region of interest extended dorsally to the area corresponding to the continuation of the neuroepithelium, projected from the ventricular zone to the cortical plate159. This method enabled the inclusion of a sufficient number of microglia, which are relatively sparse in the embryonic cortex compared with neural progenitors, thereby facilitating robust and consistent comparisons between experimental groups. Images were captured at a magnification of 20x, with a scale bar set at 200 μm. Image analysis was conducted using FIJI-ImageJ software (U.S. National Institutes of Health).
Evaluation of the number of CA1 hippocampal dendritic spines by Golgi-cox stain
The morphology and count of hippocampal dendritic spines in the HTX, HTX + T4, and EUT offspring were evaluated using Golgi-cox stain160. Mice were euthanized at P55, brains were removed, and the telencephalon was individually submerged in 10 mL of impregnation solution composed of 5% potassium dichromate (K₂Cr₂O₇), 5% mercuric chloride (HgCl₂), and 5% potassium chromate (K₂CrO₄). After 24 h, the telencephalon was transferred to a fresh impregnation solution and kept in the dark for two weeks. Subsequently, the tissue was handled using plastic forceps, and excess solution was blotted with filter paper. It was then transferred into 50 mL of cryoprotection solution (20% sucrose). The cryoprotection solution was replaced 24 h later with a fresh cryoprotection solution, which was maintained for 4 days until the tissues sank to the bottom. Cryoprotected brains were then embedded in molds using OCT compound for cryosectioning of sagittal sections at −25 °C (150 μm in thickness). Gelatin-coated slides were used as a support medium to enhance the stability and orientation of the tissue sections during cutting. The hippocampus was identified using anatomical landmarks such as the lateral ventricles and hippocampal layers, with sections taken 1.5–3.5 mm posterior to bregma and 1.0–2.5 mm lateral to the midline161. The slides were then washed with double-distilled water and left to dry overnight in the dark at RT. The staining procedure was conducted the following day by submerging the slides in 20% ammonia for 10 min in the dark, followed by 1% sodium thiosulfate for 10 min in the dark. After each step, the slides were washed twice in double-distilled water (4 min each). The slides were dehydrated through a series of ethanol concentrations: 50%, 75%, 95%, and 100% (four times), each for 4 min. Finally, the slides were cleared with xylene four times for 4 min each. Permount Mounting Medium (Fisher Scientific, Waltham, MA, USA, SP15-100) was used to mount the slides, which were kept in darkness at room temperature until microscopic examination. Images were captured using a Leica DM1000 LED microscope (Leica Microsystems GmbH, Wetzlar, Germany) equipped with an MC170 HD camera (Leica Microsystems GmbH). Analysis of dendritic spines focused on pyramidal neurons from the CA1 region, as previously described162. Photographs were taken of three independent hippocampal sections per slide. For dendritic spine analysis, dendritic segments ranging from 10 to 20 μm in length were randomly selected and captured using a 100X oil immersion objective, with a resolution of 1024 × 768 pixels and identical acquisition settings across the compared samples. The total number of dendritic spines and their classification into different morphologies, along with their respective proportions, were analyzed using the Dendritic Spines Counter plugin of Fiji-ImageJ software (NIH, Bethesda, MD, USA, version 1.54f)163. Spine density was defined as the number of spines normalized to 10 μm of dendrite length. Dendritic spine shapes were classified based on established measurements: filopodia were defined as thin spines longer than 2 μm without head protrusions; mushroom was defined by a wide head (> 1 μm width) and short protrusions (< 1 μm); stubby were defined by wide heads without neck (length/width ratio < 1 μm); and thin were identified by thin and short heads (< 2 μm)164.
Statistics and reproducibility
The sample size was determined utilizing G*Power 3.1.9.7 software. For the pregnant mouse groups, one-way ANOVA F-tests were conducted. The effect size was set to 0.80 and the significance level was set to 0.05. This calculation resulted in a total sample size of 33, with 11 pregnant mice allocated to each group. For the offspring, sample size was determined using a two-way model in F-test ANOVA, with an effect size set to 0.5, α set to 0.05, and considering 3 groups. This resulted in a total sample size of 90, with 30 mice allocated to each experimental group, evenly distributed with 15 mice per sex. To account for potential litter effects, offspring were selected from multiple litters, with no more than two animals per litter included in each experimental group. This strategy reduced bias from litter-specific variability and ensured that observed outcomes reflected the treatment effect rather than characteristics unique to a single litter149. Statistical analyses were performed using Prism 9.0.2 software (GraphPad, Inc). Data were assessed for a normal distribution using the Shapiro-Wilk test. For datasets exhibiting a non-normal distribution, the Kruskal-Wallis test was conducted, followed by Dunn’s post hoc analysis. Datasets showing a normal distribution were subjected to parametric one-way ANOVA, followed by Tukey’s post hoc test. Multiple comparisons were evaluated via two-way ANOVA followed by Tukey’s post hoc test. Results are presented as the mean ± SEM. Significance was established at *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001.
Data availability
All data generated during this study are included in the published article and Supplementary materials. The source data can be found in Supplementary Data 1. Additional inquiries can be directed to the corresponding author.
References
Barker, D. J. P. The origins of the developmental origins theory. J. Intern. Med. 261, 412–417 (2007).
Kwon, E. J. & Kim, Y. J. What is fetal programming? A lifetime health is under the control of in utero health. Obstet. Gynecol. Sci. 60, 506–519 (2017).
Moleti, M., Trimarchi, F. & Vermiglio, F. Thyroid physiology in pregnancy. Endocr. Pr. 20, 589–596 (2014).
Leung, A. M. Thyroid function in pregnancy. J. Trace Elem. Med. Biol. 26, 137–140 (2012).
Kilby, M. D., Barber, K., Hobbs, E. & Franklyn, J. A. Thyroid hormone action in the placenta. Placenta 26, 105–113 (2005).
Zuñiga, L. F. F., Muñoz, Y. S. & Pustovrh, M. C. Thyroid hormones: metabolism and transportation in the fetoplacental unit. Mol. Reprod. Dev. 89, 526–539 (2022).
Calvo, R. M. et al. Fetal tissues are exposed to biologically relevant free thyroxine concentrations during early phases of development. J. Clin. Endocrinol. Metab. 87, 1768–1777 (2002).
Costa, A. et al. Thyroid hormones in tissues from human embryos and fetuses. J. Endocrinol. Investig. 14, 559–568 (1991).
Chan, S. et al. Early expression of thyroid hormone deiodinases and receptors in human fetal cerebral cortex. Dev. Brain Res. 138, 109–116 (2002).
Bernal, J. Thyroid hormone receptors in brain development and function. Nat. Clin. Pr Endocrinol. Metab. 3, 249–259 (2007).
Jones, S. A., Thoemke, K. R. & Anderson, G. W. The role of thyroid hormone in fetal and neonatal brain development. Curr. Opin. Endocrinol. Diabetes. 12, 10–16 (2005).
Ferreiro, B., Bernal, J., Goodyer, G. & Branchard, C. L. Estimation of nuclear thyroid hormone receptor saturation in human fetal brain and lung during early Gestation*. J. Clin. Endocrinol. Metab. 67, 853–856 (1988).
Mahadik, K., Choudhary, P. & Roy, P. K. Study of thyroid function in pregnancy, its feto-maternal outcome; a prospective observational study. BMC Pregnancy Childbirth. 20, 769 (2020).
López-Muñoz, E., Mateos-Sánchez, L., Mejía-Terrazas, G. E. & Bedwell-Cordero, S. E. Hypothyroidism and isolated hypothyroxinemia in pregnancy, from physiology to the clinic. Taiwan. J. Obstet. Gynecol. 58, 757–763 (2019).
Alexander, E. K. et al. 2017 guidelines of the American thyroid association for the diagnosis and management of thyroid disease during pregnancy and the postpartum. Thyroid 27, 315–389 (2017).
Levie, D. et al. Thyroid function in early Pregnancy, child IQ, and autistic traits: A Meta-Analysis of individual participant data. J. Clin. Endocrinol. Metab. 103, 2967–2979 (2018).
Grossklaus, R., Liesenkötter, K-P., Doubek, K., Völzke, H. & Gaertner, R. Iodine Deficiency, maternal hypothyroxinemia and endocrine disrupters affecting fetal brain development: A scoping review. Nutrients 15, 2249 (2023).
Modesto, T. et al. Maternal mild thyroid hormone insufficiency in early pregnancy and Attention-Deficit/Hyperactivity disorder symptoms in children. JAMA Pediatr. 169, 838–845 (2015).
Berbel, P. et al. Delayed neurobehavioral development in children born to pregnant women with mild hypothyroxinemia during the first month of gestation: the importance of early iodine supplementation. Thyroid 19, 511–519 (2009).
Wei, W. et al. Development of an animal model of hypothyroxinemia during pregnancy in Wistar rats. Anim. Model. Exp. Med. https://doi.org/10.1002/ame2.12459 (2024).
Opazo, M. C. et al. Maternal hypothyroxinemia impairs Spatial learning and synaptic nature and function in the offspring. Endocrinology 149, 5097–5106 (2008).
Ausó, E. et al. A moderate and transient deficiency of maternal thyroid function at the beginning of fetal neocorticogenesis alters neuronal migration. Endocrinology 145, 4037–4047 (2004).
Cuevas, E. et al. Transient maternal hypothyroxinemia at onset of corticogenesis alters tangential migration of medial ganglionic eminence-derived neurons. Eur. J. Neurosci. 22, 541–551 (2005).
Babu, S. et al. Effect of hypothyroxinemia on thyroid hormone responsiveness and action during rat postnatal neocortical development. Exp. Neurol. 228, 91–98 (2011).
Román, G. C. et al. Association of gestational maternal hypothyroxinemia and increased autism risk. Ann. Neurol. 74, 733–742 (2013).
Andersen, S. L. & Olsen, J. Early pregnancy thyroid function test abnormalities in biobank Sera from women clinically diagnosed with thyroid dysfunction before or after pregnancy. Thyroid 27, 451–459 (2017).
Hodges, H., Fealko, C. & Soares, N. Autism spectrum disorder: definition, epidemiology, causes, and clinical evaluation. Transl Pediatr. 9, S55–S65 (2019).
González-Madrid, E. et al. Gestational hypothyroxinemia induces ASD-like phenotypes in behavior, Proinflammatory markers, and glutamatergic protein expression in mouse offspring of both sexes. Front. Endocrinol. 15, 1381180 (2024).
Cheroni, C., Caporale, N. & Testa, G. Autism spectrum disorder at the crossroad between genes and environment: contributions, convergences, and interactions in ASD developmental pathophysiology. Mol. Autism. 11, 69 (2020).
Persico, A. M. & Merelli, S. Environmental factors in the onset of autism spectrum disorder. Curr. Dev. Disord Rep. 1, 8–19 (2014).
Pugsley, K., Scherer, S. W., Bellgrove, M. A. & Hawi, Z. Environmental exposures associated with elevated risk for autism spectrum disorder May augment the burden of deleterious de Novo mutations among probands. Mol. Psychiatry. 27, 710–730 (2022).
Rylaarsdam, L. & Guemez-Gamboa, A. Genetic causes and modifiers of autism spectrum disorder. Front. Cell. Neurosci. 13, 385 (2019).
Elgueta, D., Murgas, P., Riquelme, E., Yang, G. & Cancino, G. I. Consequences of viral infection and cytokine production during pregnancy on brain development in offspring. Front. Immunol. 13, 816619 (2022).
Yang, G. et al. A Glo1-Methylglyoxal pathway that is perturbed in maternal diabetes regulates embryonic and adult neural stem cell pools in murine offspring. Cell. Rep. 17, 1022–1036 (2016).
Gallagher, D. et al. Transient maternal IL-6 mediates Long-Lasting changes in neural stem cell pools by deregulating an endogenous Self-Renewal pathway. Cell. Stem Cell. 13, 564–576 (2013).
Nudel, R. et al. Maternal pregnancy-related infections and autism spectrum disorder—the genetic perspective. Transl Psychiatry. 12, 334 (2022).
Zerbo, O. et al. Association between influenza infection and vaccination during pregnancy and risk of autism spectrum disorder. JAMA Pediatr. 171, e163609 (2016).
Mawson, A. R. & Croft, A. M. Rubella virus Infection, the congenital Rubella Syndrome, and the link to autism. Int. J. Environ. Res. Public. Heal. 16, 3543 (2019).
Lin, C-H., Chou, I-C., Lee, I-C. & Hong, S-Y. Cytomegalovirus infection in infancy May increase the risk of subsequent epilepsy and autism spectrum disorder in childhood. Children 8, 1040 (2021).
Shook, L.L., Fourman, L.T. & Edlow, A. G. Immune responses to SARS-CoV-2 in pregnancy: implications for the health of the next generation. J. Immunol. (Baltim Md: 1950). 209, 1465–1473 (2022).
Jo, H. et al. Gestational diabetes mellitus, prenatal air pollution exposure, and autism spectrum disorder. Environ. Int. 133, 105110 (2019).
Getz, K. D., Anderka, M. T., Werler, M. M. & Jick, S. S. Maternal Pre-pregnancy body mass index and autism spectrum disorder among offspring: A Population‐Based Case–Control study. Paediatr. Périnat Epidemiol. 30, 479–487 (2016).
Chen, H. J. et al. Prenatal stress causes intrauterine inflammation and serotonergic dysfunction, and long-term behavioral deficits through microbe- and CCL2-dependent mechanisms. Transl Psychiatry. 10, 191 (2020).
Andrade, C. et al. Gestational exposure to valproate and autism spectrum disorder or Attention-Deficit/Hyperactivity disorder in offspring: systematic review and Meta‐Analysis. Acta Psychiatr Scand. 151, 668–679 (2025).
McEwan, F., Glazier, J. D. & Hager, R. The impact of maternal immune activation on embryonic brain development. Front. Neurosci. 17, 1146710 (2023).
Hameete, B. C. et al. The poly(I:C)-induced maternal immune activation model; a systematic review and meta-analysis of cytokine levels in the offspring. Brain Behav. Immun. - Heal. 11, 100192 (2021).
Simões, L. R. et al. Maternal immune activation induced by lipopolysaccharide triggers immune response in pregnant mother and fetus, and induces behavioral impairment in adult rats. J. Psychiatr Res. 100, 71–83 (2018).
Smith, S. E. P., Li, J., Garbett, K., Mirnics, K. & Patterson, P. H. Maternal immune activation alters fetal brain development through Interleukin-6. J. Neurosci. 27, 10695–10702 (2007).
Lan, X-Y. et al. Poly(I:C)-induced maternal immune activation causes elevated self-grooming in male rat offspring: involvement of abnormal postpartum static nursing in dam. Front. Cell. Dev. Biol. 11, 1054381 (2023).
Woods, R. M. et al. Maternal immune activation and role of placenta in the prenatal programming of neurodevelopmental disorders. Neuronal Signal. 7, NS20220064 (2023).
Liverman, C. S. et al. Altered expression of pro-inflammatory and developmental genes in the fetal brain in a mouse model of maternal infection. Neurosci. Lett. 399, 220–225 (2006).
Dahlgren, J., Samuelsson, A-M., Jansson, T. & Holmäng, A. Interleukin-6 in the maternal circulation reaches the rat fetus in Mid-gestation. Pediatr. Res. 60, 147–151 (2006).
Zaretsky, M. V., Alexander, J. M., Byrd, W. & Bawdon, R. E. Transfer of inflammatory cytokines across the placenta. Obstet. Gynecol. 103, 546–550 (2004).
Aaltonen, R., Heikkinen, T., Hakala, K., Laine, K. & Alanen, A. Transfer of Proinflammatory cytokines across term placenta. Obstet. Gynecol. 106, 802–807 (2005).
Keelan, J. A. & Pugazhenthi, K. Trans-placental passage and anti-inflammatory effects of solithromycin in the human placenta. Placenta 35, 1043–1048 (2014).
Zawadzka, A., Cieślik, M. & Adamczyk, A. The role of maternal immune activation in the pathogenesis of autism: A review of the Evidence, proposed mechanisms and implications for treatment. Int. J. Mol. Sci. 22, 11516 (2021).
Yang, F., Zheng, Q. & Jin, L. Dynamic function and composition changes of immune cells during normal and pathological pregnancy at the Maternal-Fetal interface. Front. Immunol. 10, 2317 (2019).
Zhao, Q. et al. Maternal immune activation-induced PPARγ-dependent dysfunction of microglia associated with neurogenic impairment and aberrant postnatal behaviors in offspring. Neurobiol. Dis. 125, 1–13 (2019).
Ozaki, K. et al. Maternal immune activation induces sustained changes in fetal microglia motility. Sci. Rep. 10, 21378 (2020).
Ostrem, B. E. L. et al. Fetal brain response to maternal inflammation requires microglia. Development 151. https://doi.org/10.1242/dev.202252 (2024).
Baines, K. J. et al. Maternal immune activation alters fetal brain development and enhances proliferation of neural precursor cells in rats. Front. Immunol. 11, 1145 (2020).
Sarieva, K. et al. Human brain organoid model of maternal immune activation identifies radial glia cells as selectively vulnerable. Mol. Psychiatry. 28, 5077–5089 (2023).
Yasumatsu, K. et al. Bacterial-induced maternal interleukin-17A pathway promotes autistic-like behaviors in mouse offspring. Exp. Anim. 69, 250–260 (2020).
Tartaglione, A. M. et al. Maternal immune activation induces autism-like changes in behavior, neuroinflammatory profile and gut microbiota in mouse offspring of both sexes. Transl Psychiatry. 12, 384 (2022).
Horváth, G. et al. P2X7 Receptors Drive Poly(I:C) Induced Autism-like Behavior in Mice. J. Neurosci. 39, 2542–2561 (2019).
Cieślik, M. et al. Maternal immune activation induces neuroinflammation and cortical synaptic deficits in the adolescent rat offspring. Int. J. Mol. Sci. 21, 4097 (2020).
Coiro, P. et al. Impaired synaptic development in a maternal immune activation mouse model of neurodevelopmental disorders. Brain Behav. Immun. 50, 249–258 (2015).
Ostrem, B. E. L. et al. Fetal brain response to maternal inflammation requires microglia. Development 151, dev202252 (2024).
Ball, J. B., Green-Fulgham, S. M. & Watkins, L. R. Mechanisms of microglia-mediated synapse turnover and synaptogenesis. Prog Neurobiol. 218, 102336 (2022).
Lasa, M. & Contreras-Jurado, C. Thyroid hormones act as modulators of inflammation through their nuclear receptors. Front. Endocrinol. 13, 937099 (2022).
Luca, R. D. et al. Thyroid hormones interaction with immune Response, inflammation and Non-thyroidal illness syndrome. Front. Cell. Dev. Biol. 8, 614030 (2021).
Chen, V. S. et al. Histology atlas of the developing prenatal and postnatal mouse central nervous System, with emphasis on prenatal days E7.5 to E18.5. Toxicol. Pathol. 45, 705–744 (2017).
Haensgen, H. et al. Gestational hypothyroxinemia affects its offspring with a reduced suppressive capacity impairing the outcome of the experimental autoimmune encephalomyelitis. Front. Immunol. 9, 1257 (2018).
Opazo, M. C. et al. Gestational hypothyroxinemia imprints a switch in the capacity of astrocytes and microglial cells of the offspring to React in inflammation. Mol. Neurobiol. 55, 4373–4387 (2018).
Rivera, J. C. et al. Transient gestational hypothyroxinemia accelerates and enhances ulcerative colitis-like disorder in the male offspring. Front. Endocrinol. 14, 1269121 (2024).
Roy, G. & Mugesh, G. Bioinorganic Chemistry in Thyroid Gland: Effect of Antithyroid Drugs on Peroxidase-Catalyzed Oxidation and Iodination Reactions. Bioinorg Chem Appl ; 2006: 23214. (2006).
Hsiao, E. Y. & Patterson, P. H. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav. Immun. 25, 604–615 (2011).
Sun, F., Wang, S. & Du, M. Functional regulation of decidual macrophages during pregnancy. J. Reprod. Immunol. 143, 103264 (2021).
True, H., Blanton, M., Sureshchandra, S. & Messaoudi, I. Monocytes and macrophages in pregnancy: the good, the bad, and the ugly*. Immunol. Rev. 308, 77–92 (2022).
Jonakait, G. M. The effects of maternal inflammation on neuronal development: possible mechanisms. Int. J. Dev. Neurosci. 25, 415–425 (2007).
Boulanger-Bertolus, J., Pancaro, C. & Mashour, G. A. Increasing role of maternal immune activation in neurodevelopmental disorders. Front. Behav. Neurosci. 12, 230 (2018).
Phillips, M. & Pozzo-Miller, L. Dendritic spine dysgenesis in autism related disorders. Neurosci. Lett. 601, 30–40 (2015).
Pendyala, G. et al. Maternal immune activation causes behavioral impairments and altered cerebellar cytokine and synaptic protein expression. Neuropsychopharmacology 42, 1435–1446 (2017).
Schuck, R. K., Flores, R. E. & Fung, L. K. Brief report: Sex/Gender differences in symptomology and camouflaging in adults with autism spectrum disorder. J. Autism Dev. Disord. 49, 2597–2604 (2019).
Cruz, S. et al. Is there a bias towards males in the diagnosis of autism? A systematic review and Meta-Analysis. Neuropsychol. Rev. 35, 153–176 (2025).
Jaber, M. Genetic and environmental mouse models of autism reproduce the spectrum of the disease. J. Neural Transm. 130, 425–432 (2023).
Wang, Z. et al. Effects of Fmr1 gene mutations on sex differences in Autism-Like behavior and dendritic spine development in mice and transcriptomic studies. Neuroscience 534, 16–28 (2023).
Pop, V. J. M., Krabbe, J. G., Broeren, M., Wiersinga, W. & Rayman, M. P. Hypothyroxinaemia during gestation is associated with low ferritin and increased levels of inflammatory markers. Eur. Thyroid J. 13, e230163 (2024).
Gómez-Chávez, F. et al. IκBNS and IL-6 expression is differentially established in the uterus of pregnant healthy and infected mice. Heliyon 6, e04122 (2020).
Dutta, S., Sengupta, P. & Liew, F. F. Cytokine landscapes of pregnancy: mapping gestational immune phases. Gynecol. Obstet. Clin. Med. 4, e000011 (2024).
Xiao, J. P. et al. The increased maternal serum levels of IL-6 are associated with the severity and onset of preeclampsia. Cytokine 60, 856–860 (2012).
Michalczyk, M., Celewicz, A., Celewicz, M., Woźniakowska-Gondek, P. & Rzepka, R. The role of inflammation in the pathogenesis of preeclampsia. Mediat Inflamm. 2020, 3864941 (2020).
Amirian, A., Mahani, M. B. & Abdi, F. Role of interleukin-6 (IL-6) in predicting gestational diabetes mellitus. Obstet. Gynecol. Sci. 63, 407–416 (2020).
Zgutka, K. et al. Gestational diabetes Mellitus-Induced inflammation in the placenta via IL-1β and Toll-like receptor pathways. Int. J. Mol. Sci. 25, 11409 (2024).
Kumar, M., Saadaoui, M. & Khodor, S. A. Infections and pregnancy: effects on maternal and child health. Front. Cell. Infect. Microbiol. 12, 873253 (2022).
Andruszewski, D. et al. Embryo-restricted responses to maternal IL-17A promote neurodevelopmental disorders in mouse offspring. Mol. Psychiatry. 30, 1585–1593 (2025).
Choi, G. B. et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 351, 933–939 (2016).
Zerbo, O. et al. Maternal infection during pregnancy and autism spectrum disorders. J. Autism Dev. Disord. 45, 4015–4025 (2015).
Onore, C. E., Schwartzer, J. J., Careaga, M., Berman, R. F. & Ashwood, P. Maternal immune activation leads to activated inflammatory macrophages in offspring. Brain Behav. Immun. 38, 220–226 (2014).
Krop, J. et al. Imaging mass cytometry reveals the prominent role of myeloid cells at the maternal-fetal interface. iScience 25, 104648 (2022).
van der Zwan, A., van Beyrend, U. V., Laban, G., Keur, S. & van der Kapsenberg, C. Visualizing dynamic changes at the Maternal-Fetal interface throughout human pregnancy by mass cytometry. Front. Immunol. 11, 571300 (2020).
Davis, P. J., Mousa, S. A. & Lin, H-Y. Nongenomic actions of thyroid hormone: the integrin component. Physiol. Rev. 101, 319–352 (2021).
Shi, C. & Pamer, E. G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 11, 762–774 (2011).
Tang, M-X., Hu, X-H., Liu, Z-Z., Kwak-Kim, J. & Liao, A-H. What are the roles of macrophages and monocytes in human pregnancy? J. Reprod. Immunol. 112, 73–80 (2015).
Santos, L. C., de Souza, C. A., Silva, J. F., Ocarino, N. M. & Serakides, R. Maternal hyperthyroidism alters the immunological mediators profile and population of natural killers cells in decidua of rats. Acta Histochem. 125, 152026 (2023).
Silva, J. F., Ocarino, N. M. & Serakides, R. Maternal thyroid dysfunction affects placental profile of inflammatory mediators and the intrauterine trophoblast migration kinetics. Reproduction 147, 803–816 (2014).
Crnčić, T. B. et al. Innate immunity in autoimmune thyroid disease during pregnancy. Int. J. Mol. Sci. 24, 15442 (2023).
Perricone, R., Perricone, C., Carolis, C. D. & Shoenfeld, Y. NK cells in autoimmunity: A two-edg’d weapon of the immune system. Autoimmun. Rev. 7, 384–390 (2008).
Santoni, A., Carlino, C. & Gismondi, A. Uterine NK cell development, migration and function. Reprod. Biomed. Online. 16, 202–210 (2008).
Zhou, J., Yan, P., Ma, W. & Li, J. Cytokine modulation and immunoregulation of uterine NK cells in pregnancy disorders. Cytokine Growth Factor. Rev. 81, 40–53 (2025).
Fraser, R. et al. Impaired decidual natural killer cell regulation of vascular remodelling in early human pregnancies with high uterine artery resistance. J. Pathol. 228, 322–332 (2012).
Murphy, S. P., Fast, L. D., Hanna, N. N., Sharma, S. & Uterine, N. K. Cells mediate Inflammation-Induced fetal demise in IL-10-Null mice. J. Immunol. 175, 4084–4090 (2005).
Mahajan, D. et al. Dendritic cells and the establishment of fetomaternal tolerance for successful human pregnancy. Arch. Immunol. Ther. Exp. 72, 20240010 (2024).
Akira, S., Uematsu, S. & Takeuchi, O. Pathogen recognition and innate immunity. Cell 124, 783–801 (2006).
Tagliani, E. & Erlebacher, A. Dendritic cell function at the maternal–fetal interface. Expert Rev. Clin. Immunol. 7, 593–602 (2011).
Talayev, V. Y. et al. The effect of human placenta cytotrophoblast cells on the maturation and T cell stimulating ability of dendritic cells in vitro. Clin. Exp. Immunol. 162, 91–99 (2010).
Toothaker, J. M. et al. Immune landscape of human placental villi using single-cell analysis. Development; 149. https://doi.org/10.1242/dev.200013
Kim, C. J., Romero, R., Chaemsaithong, P. & Kim, J-S. Chronic inflammation of the placenta: definition, classification, pathogenesis, and clinical significance. Am. J. Obstet. Gynecol. 213, S53–S69 (2015).
Guma, E., Plitman, E. & Chakravarty, M. M. The role of maternal immune activation in altering the neurodevelopmental trajectories of offspring: A translational review of neuroimaging studies with implications for autism spectrum disorder and schizophrenia. Neurosci. Biobehav Rev. 104, 141–157 (2019).
Li, Y. et al. Maternal immune activation mediated prenatal chronic stress induces Th17/Treg cell imbalance May relate to the PI3K/Akt/NF-κB signaling pathway in offspring rats. Int. Immunopharmacol. 126, 111308 (2024).
Davis, J. & Mire, E. Maternal obesity and developmental programming of neuropsychiatric disorders: an inflammatory hypothesis. Brain Neurosci. Adv. 5, 23982128211003484 (2021).
Wenzek, C. et al. The interplay of thyroid hormones and the immune system – where we stand and why we need to know about it. Eur. J. Endocrinol. 186, R65–R77 (2022).
Luca, R. D. et al. Thyroid hormones interaction with immune Response, inflammation and Non-thyroidal illness syndrome. Front. Cell. Dev. Biology. 8, 614030 (2021).
Jara, E. L. et al. Modulating the function of the immune system by thyroid hormones and Thyrotropin. Immunol. Lett. 184, 76–83 (2017).
Kunz, H. E. et al. Adipose tissue macrophage populations and inflammation are associated with systemic inflammation and insulin resistance in obesity. Am. J. Physiol-Endocrinol Metab. 321, E105–E121 (2021).
Segovia, S. A., Vickers, M. H., Gray, C. & Reynolds, C. M. Maternal Obesity, Inflammation, and Developmental Programming. BioMed Res Int ; 2014: 418975. (2014).
Shen, C. et al. The rebalancing of the immune system at the maternal-fetal interface ameliorates autism-like behavior in adult offspring. Cell. Rep. 43, 114787 (2024).
Zhou, J. et al. Placental inflammatory cytokines mRNA expression and preschool children’s cognitive performance: a birth cohort study in China. BMC Med. 21, 449 (2023).
Yockey, L. J. & Iwasaki, A. Interferons and Proinflammatory cytokines in pregnancy and fetal development. Immunity 49, 397–412 (2018).
Lockwood, C. J. et al. Preeclampsia-Related inflammatory cytokines regulate Interleukin-6 expression in human decidual cells. Am. J. Pathol. 172, 1571–1579 (2008).
Megli, C., Morosky, S., Rajasundaram, D. & Coyne, C. B. Inflammasome signaling in human placental trophoblasts regulates immune defense against Listeria monocytogenes infection. J. Exp. Med. 218, e20200649 (2020).
Torres, L. R. et al. ZIKV replication is differential in explants and cells of human placental which is suppressed by HSV-2 coinfection. Virology 570, 45–56 (2022).
Kovács, I. J., Hegedűs, K., Pál, A. & Pusztai, R. Production of Proinflammatory cytokines by syncytiotrophoblasts infected with human cytomegalovirus isolates. Placenta 28, 620–623 (2007).
Liong, S. et al. Influenza A virus causes maternal and fetal pathology via innate and adaptive vascular inflammation in mice. Proc. Natl. Acad. Sci. 117, 24964–24973 (2020).
Hecht, J. L. et al. SARS-CoV-2 can infect the placenta and is not associated with specific placental histopathology: a series of 19 placentas from COVID-19-positive mothers. Mod. Pathol. 33, 2092–2103 (2020).
Lima, F. R. et al. Regulation of microglial development: a novel role for thyroid hormone. J. Neurosci: Off J. Soc. Neurosci. 21, 2028–2038 (2001).
Mori, Y. et al. Effects of 3,3’,5-triiodothyronine on microglial functions. Glia 63, 906–920 (2015).
Mohan, V. et al. Maternal thyroid hormone deficiency affects the fetal neocorticogenesis by reducing the proliferating pool, rate of neurogenesis and indirect neurogenesis. Exp. Neurol. 237, 477–488 (2012).
O’Shaughnessy, K. L. et al. A transient window of hypothyroidism alters neural progenitor cells and results in abnormal brain development. Sci. Rep. 9, 4662 (2019).
Zeiss, C. J. Comparative milestones in rodent and human postnatal central nervous system development. Toxicol. Pathol. 49, 1368–1373 (2021).
Salloum-Asfar, S. et al. The potential role of thyroid hormone therapy in neural progenitor cell differentiation and its impact on neurodevelopmental disorders. Mol. Neurobiol. 61, 3330–3342 (2024).
Barón-Mendoza, I. et al. Altered hippocampal neurogenesis in a mouse model of autism revealed by genetic polymorphisms and by atypical development of newborn neurons. Sci. Rep. 14, 4608 (2024).
Barón-Mendoza, I. et al. Changes in the number and morphology of dendritic spines in the hippocampus and prefrontal cortex of the C58/J mouse model of autism. Front. Cell. Neurosci. 15, 726501 (2021).
Elmore, S. A. et al. Histology atlas of the developing mouse placenta. Toxicol. Pathol. 50, 60–117 (2021).
Guidelines, A. AVMA guidelines for the euthanasia of animals: 2020 Edition*. J. Am. Vet. Med. Assoc. 1972 (160), 761–772 (2020).
Heyne, G. W. et al. A simple and reliable method for early pregnancy detection in inbred mice. J. Am. Assoc. Lab. Anim. Sci: JAALAS. 54, 368–371 (2015).
Deutsch-Feldman, M., Picetti, R., Seip-Cammack, K., Zhou, Y. & Kreek, M. J. Effects of handling and vehicle injections on adrenocorticotropic and corticosterone concentrations in Sprague-Dawley compared with Lewis rats. J. Am. Assoc. Lab. Anim. Sci: JAALAS. 54, 35–39 (2015).
Drude, S. et al. Side effects of control treatment can conceal experimental data when studying stress responses to injection and psychological stress in mice. Lab. Anim. 40, 119–128 (2011).
Jiménez, J. A. & Zylka, M. J. Controlling litter effects to enhance rigor and reproducibility with rodent models of neurodevelopmental disorders. J. Neurodev Disord. 13, 2 (2021).
Feng, X. et al. Exposure of pregnant mice to perfluorobutanesulfonate causes hypothyroxinemia and developmental abnormalities in female offspring. Toxicol. Sci. 155, 409–419 (2017).
Hua, X., Cao, X-Y., Wang, X-L., Sun, P. & Chen, L. Exposure of pregnant mice to triclosan causes insulin resistance via thyroxine reduction. Toxicol. Sci. 160, 150–160 (2017).
Arenas-Hernandez, M., Sanchez-Rodriguez, E. N., Mial, T. N., Robertson, S. A. & Gomez-Lopez, N. Isolation of Leukocytes from the Murine Tissues at the Maternal-Fetal Interface. J Vis. Exp e52866 (2015)
Ottersbach, K. & Dzierzak, E. Analysis of the mouse placenta as a hematopoietic stem cell niche. Methods Mol. Biol. (Clifton NJ). 538, 335–346 (2009).
Gekas, C., Rhodes, K. E. & Mikkola, H. K. A. Isolation and Visualization of Mouse Placental Hematopoietic Stem Cells. Curr Protoc Stem Cell Biol. 6: 2A.8.1-2A.8.14. (2008).
Liang, G., Huang, B., Wang, F. & Liu, F. Protocols for isolation and characterization of mouse placental hemogenic endothelial cells. STAR. Protoc. 2, 100884 (2021).
Strober, W. Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol. 111, A3B1–A3B3 (2015).
Cancino, G. I., Fatt, M. P., Miller, F. D. & Kaplan, D. R. Conditional ablation of p63 indicates that it is essential for embryonic development of the central nervous system. Cell. Cycle. 14, 3270–3281 (2015).
Tomita, H. et al. The protein tyrosine phosphatase receptor delta regulates developmental neurogenesis. Cell. Rep. 30, 215–228e5 (2020).
Kumaraguru, S., Morgan, J. & Wong, F. K. Activity-dependent regulation of microglia numbers by pyramidal cells during development shape cortical functions. Sci. Adv. 11, eadq5842 (2025).
Zhong, F., Liu, L., Wei, J-L. & Dai, R-P. Step by step Golgi-Cox staining for Cryosection. Front. Neuroanat. 13, 62 (2019).
Ortiz, C. et al. Molecular atlas of the adult mouse brain. Sci. Adv. 6, eabb3446 (2020).
Flores-Muñoz, C. et al. The Long-Term pannexin 1 ablation produces structural and functional modifications in hippocampal neurons. Cells 11, 3646 (2022).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 9, 676–682 (2012).
Risher, W. C., Ustunkaya, T., Alvarado, J. S. & Eroglu, C. Rapid golgi analysis method for efficient and unbiased classification of dendritic spines. Plos One. 9, e107591 (2014).
Acknowledgements
Millennium Institute on Immunology and Immunotherapy (MIII), Programa ICM-ANID “ICN2021_045”, ANID-FONDECYT #1210507, #1250762, #11241567, #1241072, #1211959, #1231866, #1251980, #1240971, #1231905, #1231851, ANID-Postdoctoral project 3200846, and ANID Doctoral Scholarships 21202356 and 21202085.
Author information
Authors and Affiliations
Contributions
Conceptualization: E.G-M., M.A.R-R., M.C.O., and C.A.R. Methodology: E.G-M., M.A.R-R., D.E., and S.A.E. Investigation: E.G-M., and M.A.R-R. Data analysis: E.G-M., M.A.R-R., S.A.E., D.E., M.C.O., and C.A.R. Statistical analysis: E.G-M., M.A.R-R., S.A.E., D.E., M.C.O., and C.A.R. Writing—original draft: E.G-M. Writing—review & editing: E.G-M., M.A.R-R., M.C.O., S.A.E., D.E., G.I.C., E.B., A.O.A., L.F, D., J.A.S., F.S., S.M.B., P.A.G., A.M.K., C.A.R.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics declarations
The protocols employed in this study were approved by the Bioethics committee of the Facultad de Ciencias de la Vida, Universidad Andrés Bello (Bioethics approval certificate number 012/2021). The experimental procedures and care were performed in concordance with the current regulations of the Animal Welfare Committee of the Facultad de Ciencias de la Vida and the National Agency for Research and Development (ANID) (Santiago, Chile). This study is reported in accordance with ARRIVE guidelines.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
González-Madrid, E., Rangel-Ramírez, M.A., Opazo, M.C. et al. Gestational hypothyroxinemia causes an inflammatory environment at maternal-fetal tissues and fetal brain with impaired hippocampal dendritic spine maturation in the offspring. Sci Rep 15, 42160 (2025). https://doi.org/10.1038/s41598-025-26206-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-26206-7
