
Abstract
Medulloblastoma is the most common malignant pediatric brain tumor. Survival rates vary widely between subgroups, with an average overall survival of 70%. Recurrent medulloblastoma is highly aggressive, treatment-resistant, and usually fatal. In addition, current treatments are highly toxic to the developing brain and surviving patients suffer from lifelong side effects. Therefore, novel therapeutic options are urgently needed. To inform risk-based, personalized therapy, we developed a novel platform called DrugSeq, which allows predictions of drug sensitivities in patients across medulloblastoma subgroups. We used a perturbagen-response dataset to calculate transcriptional response signatures for each drug and compared this to patient medulloblastoma tumor gene expression. We then stratified patients by molecular subgroup and used an ANOVA analysis to identify drugs that selectively targeted each subgroup. We found distinct differences in transcriptional profiles and predicted drug sensitivity for each medulloblastoma subgroup. We identified kinase inhibitors, epigenetic inhibitors, and several drugs that have been investigated in drug repositioning studies for cancer. We posit that DrugSeq may identify novel therapies and facilitate patient stratification in clinical trials, leading to more successful targeted medulloblastoma therapies that improve tumor response while minimizing late toxicities. This computational tool can also be used for other cancers to stratify patients based on any clinical or molecular feature.
Similar content being viewed by others


Introduction
Medulloblastoma is the most common malignant pediatric brain tumor, accounting for approximately 9% of brain cancer diagnoses in children under 14 years of age, with an overall 5-year survival rate of 75.5% that varies widely within patient subgroups1. Typically, current treatment consists of maximal safe surgical resection followed by cranial-spinal radiation and multiagent systemic chemotherapy. However, in younger children (typically 3–5 years of age and under) radiation is omitted or delayed as long as possible given the particularly severe neurocognitive impairment observed after radiation to the developing brain2.
Medulloblastoma has been classified into molecular subgroups based on genomic features and these are the strongest predictors of patient outcomes. These consist of Wingless (WNT), Sonic Hedgehog (SHH), Group 3 (G3), and Group 4 (G4)2,3,4,5. Each subgroup has distinct transcriptional profiles, clinical features, prognoses, and treatment outcomes3,6. Further, these four subgroups have been characterized at the epigenomic and genomic level, allowing for further stratifications within each subgroup3,7,8,9. At relapse, most tumors retain the same classification as their original subgroup and are often resistant to the previously administered first-line therapy5.
WNT subgroup tumors account for 10% of all medulloblastomas and are characterized by activating mutations in the WNT/β-catenin signaling pathway10. WNT-activated tumors have the most favorable prognosis, with approximately a 90% survival rate, and relapse is uncommon7,11. The WNT subgroup has been stratified into two subtypes, α and β. The WNT α subtype is mainly diagnosed in children and frequently harbors monosomy 6, whereas WNT β is mainly comprised of older patients3.
About 30% of medulloblastoma cases are in the SHH subgroup, which is characterized by dysregulation of SHH signaling. SHH tumors are most common in children under 3 years of age. Younger children with this genetic finding have a better prognosis, with a survival rate of 75% in infants and 50% in children. There is heterogeneity in SHH subgroups evident in the different age groups including infants, adolescents, and adults3,12,13,14. The SHH subgroup has been further divided into the subtypes α, β, γ, and δ, each with varying features in histology, gene amplifications, gene deletions, mutations, metastatic dissemination rate, and prognoses3. Among the subtypes, SHH α tumors most often affect children aged 3–16 years and have the worst prognosis. These tumors are enriched for MYCN, GLI2, and YAP1 amplifications. In addition, there is an enrichment of TP53 mutations in SHH α tumors, and these patients have a very poor prognosis15.
While the WNT and SHH subgroups have distinct transcriptional and molecular profiles, G3 and G4 tumors are more difficult to distinguish due to their similar cytogenetic features3,7. G3 medulloblastoma tumors account for approximately 25% of cases. This is the deadliest subgroup of medulloblastoma and the disease is frequently metastatic at the time of diagnosis7,11. These tumors have a poor prognosis and a common feature is MYC amplification9. Group 3 tumors also have higher incidence of isochromosome 17q and OTX2 amplifications, which are linked to poor outcomes16. Treatment is stratified depending on standard and high-risk G3 groups, which are classified based on metastasis rate, level of resection, and MYC amplification status9. G3 tumors have been stratified further into three subtypes including α, β, and γ, which helps differentiate G3 from G43. There is a more favorable prognosis in G3 α and G3 β tumors compared to G3 γ tumors, which have the worst prognosis3.
G4 is the most common subgroup, accounting for 35% of MB diagnoses, with approximately an 80% survival rate. These tumors are stratified into G4 α, β, and γ3. Loss of the X chromosome is common in these tumors, consistent with the high male-to-female ratio of G4 patients17. Like G3 tumors, isochromosome 17q and MYC amplification are more common in G4 tumors. Loss of chromosome 11 or gain of chromosome 17 are correlated with improved survival. Overall, G4 tumors are transcriptionally similar to G3 medulloblastoma; however, the current standard of care regimen shows distinct differences in overall survival between these groups18.
Upon recurrence, medulloblastoma is often treatment-resistant and fatal, with a 5-year survival rate of approximately 25%19. Further, medulloblastoma survivors are frequently burdened with lifelong adverse effects including decreased cognitive abilities, speech difficulties, stunted spinal growth, epilepsy, endocrinopathies, stroke, and even early mortality2,20,21,22, highlighting the need for novel effective medulloblastoma therapies that are more highly targeted and less toxic.
Despite the extensive molecular characterization of medulloblastoma tumors into distinct subgroups, trials based on molecular biomarkers and subgroups are still limited and are critically needed4,23. There is a diverse response to treatments for each subgroup and subtype, indicating the need to stratify future therapy3. Recent efforts have been made to repurpose existing FDA-approved drugs for cancer treatment since this bypasses the time and cost necessary for drug development and toxicity profiling, allowing more rapid approval for clinical trials24. In silico methods have been previously used to identify drugs with desirable predicted target specificity25,26. However, these approaches have not identified potentially patient-specific approaches based on medulloblastoma subgroups. We present a novel means of coupling the transcriptional disease signature of medulloblastoma tumors to compounds that can potentially reverse this signature. To make this pipeline available to the medical and research community, we have created a computational pipeline entitled DrugSeq that will allow patient-specific predictions of the anti-tumor efficacy for both investigational and approved drugs. We posit that DrugSeq will be a useful tool for preclinical and clinical investigations of novel drugs selective for medulloblastoma subgroups.
Methods
Ethics approval and consent to participate
This study utilizes publicly available RNA-sequencing data that were previously published by Cavalli et al. 20173 and Archer et al. 201827 and obtained from GEO GSE85217 and the study’s supplementary materials, respectively. No new experiments involving human participants were conducted for this study. According to the original publications, all experiments were approved by the relevant institutional ethics committee, and informed consent was obtained from all participants and their legal guardians.
Patient gene expression acquisition and processing
Pre-processed microarray or mRNA patient data from Cavalli 2017 and Archer 2018 were obtained3,27. The Cavalli set consists of a total of 763 medulloblastoma samples, comprised of 71 WNT tumors, 233 SHH tumors, 144 G3 tumors, and 326 G4 tumors, and the Archer set consists of 39 medulloblastoma tumors, with 14 SHH, 13 G3, and 12 G4 tumors. A pseudocount of 0.01 was added to each transcript expression value, and transcripts were averaged by external gene name. Control microarray expression was obtained from Allen Brain Atlas (ABA), using samples from the vermis of the cerebellar cortex28. Cavalli and ABA microarrays were normalized using limma, and the log2 fold change value for each patient relative to the median control expression was calculated29. These patient-specific expression profiles, known as the patient disease signatures, were used for downstream analysis (Supplementary File 1).
Transcriptional consensus signature (TCS)
The Library of Integrated Network-Based Cellular Signatures (LINCS) center at the National Institutes of Health developed a perturbagen-response gene expression profiling platform called L1000. The L1000 assay is a high-throughput bead-based fluorescence assay that measures the steady-state levels of 978 gene transcripts30. The L1000 dataset is a compilation of the transcriptional responses induced by over 1,700 drugs across more than 40 cell lines. One previously developed pipeline, SynergySeq, generates a drug-specific transcriptional consensus signature (TCS) containing the L1000 genes that are consistently up or downregulated following treatment with a drug across a multitude of cell lines31. These TCSs were then used to predict drug sensitivity and synergies based on the disease transcriptional profile.
Blood–brain barrier (BBB) penetration predictions
To predict brain penetrance for the L1000 drugs, we utilized LogBB_Pred (http://ssbio.cau.ac.kr/software/logbb_pred/). This is a machine-learning algorithm that is trained on experimentally measured logBB values from the literature. We used Simplified Molecular Input Line Entry System (SMILES) for the L1000 compounds and submitted them to the web server to determine predicted logBB values for each compound32.
Patient-specific drug response predictions using DrugSeq
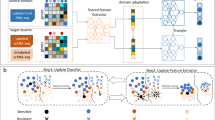
To predict the therapeutic response of compounds across medulloblastoma subtypes, we developed a custom R Shiny application that computes transcriptional discordance scores using compound-specific TCSs from L1000 and patient-specific disease signatures. Log2 fold change normalized patient gene expression was processed using a novel pipeline called DrugSeq to predict drug sensitivity for each patient (Fig. 1). This pipeline uses the previously described compound-specific TCSs and calculates the Spearman correlation between the TCS and the normalized patient gene expression signature, yielding a patient-specific drug discordance score. A more negative score indicates a stronger reversal of the disease signature by the given compound, and therefore the patient is predicted to be more responsive to the drug. The data analysis script is available at https://github.com/AyadLab/DrugSeq and as a user-friendly web portal at https://combin.shinyapps.io/DrugSeq/.

DrugSeq application stratifies individual patient drug response. Patient gene expression is normalized and log2 fold change in gene expression is calculated (Step 1). L1000 transcriptional consensus signatures (TCS) are calculated (Step 2) and discordance between the disease signature and the drug signature is calculated for each patient (Step 3). Patients are stratified by subgroup and differences in drug discordance by subgroup are calculated using an ANOVA with Tukey HSD correction (Step 4).
Subgroup-specific drugseq sensitivity predictions
To identify therapies that are predicted to target tumor cells in a subgroup-specific manner, we performed a one-way ANOVA with Tukey HSD post-hoc test on the patient-specific drug discordance scores for each drug. Patients were stratified into subgroups, including WNT, SHH, G3, and G4 medulloblastoma. A one-way ANOVA test was used to find drugs that vary in patient response by subgroup, then a Tukey HSD test was used to determine which subgroups varied. A separation score was calculated to quantify the degree of separation in predicted response between groups relative to within-group variability. This was defined as the difference between the highest and lowest subtype mean responses, divided by the average within-subtype standard deviation (pooled standard deviation). Results were filtered by minimum size of TCS (≥ 20 genes), predicted blood-brain barrier permeability (logBB > -1), predicted sensitivity (minimum response across all groups < 0). Among these, the most sensitive subtype for the drug was identified and drugs were ranked by descending separation score. The top 20 compounds, as ranked by separation score, were visualized on a heatmap (Fig. 2B-C).

Patient-specific drug discordance reveals distinct drug sensitivities in medulloblastoma subgroups. (A) DrugSeq drug discordance scores were calculated for each patient in the Cavalli dataset and known medulloblastoma therapies were visualized on a boxplot. Dashed lines indicate minimum median subgroup sensitivity. From left to right: SHH-MB inhibitors (vismodegib and taladegib), G3-MB inhibitor (triptolide), and WNT-MB inhibitor (Radiation). One-way ANOVA with Tukey HSD; significance reported as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (B) Heatmap of top hits by subgroup in the Cavalli dataset. DrugSeq drug discordance scores were calculated for each patient. Scores were filtered for drugs that were a hit in any subgroup (minimum mean discordance > 0), and the top anti-cancer drugs were selected using a one-way ANOVA test. (C) Heatmap of top hits for Archer dataset. Drug discordance scores for top inhibitors were plotted with patients separated by medulloblastoma subgroup or recurrence. (D) Identification of protein expression correlated with drug discordance for top compounds in the Archer dataset. Patient-matched protein expression from the Archer dataset was correlated with predicted drug sensitivity. Significant correlations between drug target protein expression and predicted drug sensitivity for selected drugs are shown.
Correlation between Archer and cavalli drug sensitivity scores
To compare trends at the subgroup level between the Archer and Cavalli datasets, drug scores were aggregated by molecular subgroup within each dataset. For each drug, the concordance of subgroup trends in sensitivity between datasets was quantified using a Pearson correlation. The distribution of per-drug Pearson correlation coefficients was visualized as a histogram to demonstrate the proportion of drugs exhibiting a strong positive correlation (Supplementary Fig. 1).
Patient protein expression
Protein expression from the Archer 2018 cohort was obtained and normalized to the mean expression across all samples for each protein. Protein targets for top inhibitors were identified using PubChem and correlations between target protein expression and drug discordance were calculated (Fig. 2D).
Mechanism of action enrichment analysis
To determine enriched mechanisms of actions for each subgroup, a list of subgroup- or subtype-specific drugs was generated by filtering for compounds that had the highest mean discordance for each group. A comprehensive list of all MOAs for all compounds was curated and the frequency of each MOA was calculated across the entire set of drugs to establish a background frequency. Next, for each group, drugs were filtered for subgroup-specificity, retaining only those with a predicted mean discordance < − 0.5 to ensure meaningful differential sensitivity. These group-specific drugs were then joined with MOA annotations to establish a group-specific frequency for each MOA. Next, the group-specific frequency was divided by the background frequency for each MOA to calculate the percentage of enrichment for the MOA. Finally, the percentage enrichment was multiplied by the per-group count to weight MOAs recurring within a group. For each subgroup or subtype, the top 10 enriched MOAs were visualized on bar plots (Fig. 3, Supplementary Fig. 2).

Medulloblastoma subgroups have distinct drug targets. DrugSeq drug discordance scores were calculated for each patient in the Cavalli dataset. Top compounds for each subtype were determined by filtering for the most discordant subgroup for each compound. Compounds were compiled based on MOA to determine enrichment of drug targets within the top hits.
In vitro drug response
S47 and Tg(GFAP-Cre; Ptch1fl/fl) are SHH-activated medulloblastoma cell lines derived from mice, and HD-MB03 and D425 cells are derived from human Group 3 medulloblastoma tumors33,34,35. Of note, S47 and Tg(GFAP-Cre; Ptch1fl/fl) cells grow in suspension, D425 cells are semi-adherent, and HD-MB03 cells are adherent. We treated cells with 1:2 serial dilutions of methylstat and measured the ATP content via CellTiter-Glo after a 3-day incubation. Response was normalized to DMSO (negative control) and 10 µM velcade (positive control) (Fig. 4A).

Methylstat, a top-ranked SHH-MB inhibitor, selectively reduces viability of SHH medulloblastoma cells in vitro. A. Methylstat reduces viability in SHH medulloblastoma cells. SHH-MB cell lines (S47 and Tg(GFAP-Cre; Ptch1fl/fl)) and G3-MB cell lines (HD-MB03 and D425) were treated with 1:2 dilutions of methylstat. Dashed line indicates 1 µM dose of methylstat. (A) Dose response curve for methylstat in MB cells. The plot displayed is representative of 3 biological replicates (8 technical replicates). (B, C) SHH-MB cells are more sensitive to methylstat. IC50 values with standard deviation were calculated for each cell line for 3 biological replicates with 8 technical replicates. S47, GFAP-Cre; Ptch1fl/fl, HD-MB03, and D425 cell lines are noted with symbols as in (B). Significance was assessed using nonparametric Mann-Whitney U test to compare the distribution without assuming normality *p = 0.0173 (C).
Drug and radiation synergy in Medulloblastoma
To evaluate possible combination therapies with medulloblastoma drugs, we used the previously developed pipeline SynergySeq31. Briefly, this pipeline takes the normalized patient disease signature and calculates the disease reversal to the first drug to be used in synergy, then ranks the remaining drugs by disease signature reversal. This gives us the LINCS Orthogonality Scores (LOS), where a higher score indicates greater synergy with the reference compound.
Combining monotherapy and combination therapy predictions
To evaluate the performance of our novel platform in predicting drug response compared to the established SynergySeq platform, we analyzed drug sensitivity across recurrent medulloblastoma patients using our DrugSeq discordance scores and compared those results with SynergySeq predictions for radiation synergy in newly diagnosed patients. The DrugSeq algorithm estimates drug sensitivity using an inverse correlation approach, where increasingly negative values denote higher predicted sensitivity. By contrast, the SynergySeq algorithm computes predicted drug synergy based on the ratio of gene reversals, with values approaching 1 indicating greater synergistic potential. Accordingly, discordance scores derived from DrugSeq are expected to exhibit an inverse correlation with those generated by SynergySeq. For this analysis, we used a dataset from the Pediatric Brain Tumor Atlas (PBTA) available from PedcBioPortal, including a total of 81 medulloblastoma samples (7 Group 3 tumors, 44 Group 4 tumors, 24 SHH tumors, 3 WNT tumors, and 8 recurrent tumors). We plotted the average drug discordance score for each drug and calculated the Spearman correlation between DrugSeq-predicted response in recurrent patients to SyngerySeq-predicted radiation synergy in newly diagnosed (untreated) patients (Fig. 5A).

Synergistic combination therapy with radiotherapy in newly diagnosed medulloblastoma tumors correlates with monotherapy discordance in recurrent medulloblastoma. (A) DrugSeq and SynergySeq predictions are significantly correlated. The average drug discordance score for each drug either in monotherapy for recurrent tumors or combination therapy for newly diagnosed tumors (radiation naïve) was plotted and the Spearman correlation between the two cohorts was calculated using the PBTA dataset. (B) SynergySeq reveals drugs that are synergistic with radiation therapy between subgroups. Individual patient SynergySeq LOS scores were calculated for each drug in combination with radiation from the Cavalli dataset. Enrichment within each subtype was calculated for each inhibitor and the top hits were plotted on a heatmap.
Drug synergy predictions with radiation therapy
For each patient, we calculated the LINCS Orthogonality Scores (LOS) for each drug in combination with radiation therapy. LOS scores greater than one were considered to be synergistic, and the proportion of patients across each subtype that were predicted to have a synergistic response to each inhibitor was calculated. To determine combination therapies that would be differentially effective across subgroups and subtypes, we used a one-way ANOVA test to determine significant differences in LOS scores across subtypes. We filtered for ANOVA P < 0.05, subgroup enrichment > 0.75 in at least one group, and known anti-cancer activity. We then selected the top four combination drugs for each reference drug and plotted the subtype-specific enrichment of patients predicted to exhibit synergy for each subgroup (Fig. 5B).
Results
DrugSeq platform identifies drug sensitivity by subgroup of Medulloblastoma
The DrugSeq platform was used to identify drugs that have differential sensitivity across medulloblastoma subgroups (Figs. 1, 2 and 3). To confirm the validity of this approach, several known medulloblastoma therapies were identified and subgroup-specific sensitivity was assessed (Fig. 2A). Additionally, we determined that trends in predicted treatment response were consistent between datasets (Fig. 1, Supplementary Fig. 1). Consistent with the gene expression profiles, G3 and G4 tumors showed the most overlap in predicted drug sensitivity across the subgroups, while SHH and WNT tumors shared similar drug sensitivity profiles (Figs. 2B-C and 3). Next, drugs displaying sensitivity across all subgroups were analyzed based on mechanism of action (MOA) to determine overlapping and specific MOAs (Fig. 3, Supplementary Fig. 2). This showed a high degree of overlap for drug targets across all subgroups, as well as specific vulnerabilities with each subgroup.
WNT-activated drug sensitivities
Several inhibitors were identified as potential therapies for WNT-MB, including decitabine, minoxidil, oxiperomide, and gestrinone (Fig. 2B-C). Top mechanisms of action for WNT α tumors included HDAC inhibitors, dopamine receptor antagonists, and GABA receptor modulators. HDAC inhibitors have been shown to modulate WNT signaling by altering β-catenin acetylation and nuclear localization36,37. Dopamine receptors have been shown to modulate WNT expression and promote cell proliferation, and targeting these receptors has shown promise in clinical trials38,39. Top mechanisms of action for WNT β tumors included oxidative stress inhibitors and cytochrome p450 inhibitors, which has been implicated in WNT-activated cancers40.
SHH-activated drug sensitivities
Vismodegib, a known inhibitor of the SHH pathway, was predicted to be most effective in SHH-MB tumors (Fig. 2A). Furthermore, NFKB1 is upregulated upon SHH activation, and this is revealed as a potential target for vismodegib in SHH-MB (Fig. 2D). Top inhibitors predicted to target SHH-MB tumors include thioguanine, methylstat, and trovafloxacin. Top mechanisms of action for inhibitors included topoisomerase inhibitors for SHH α tumors, histamine receptor ligands for SHH β, microtubule inhibitors for SHH δ, and RNA synthesis inhibitors for SHH γ tumors.
Group 3 and group 4 drug sensitivities
Among the top inhibitors were itraconazole, medetomidine, and lupanine. The top mechanisms of action were glucocorticoid receptor agonists for Group 3 α tumors inhibitors, lactamase inhibitors and estrogen receptor agonists for Group 3 β tumors, and MEK inhibitors for Group 3 γ tumors. Top inhibitors for Group 4 tumors included isradipine, dequalinium, and fenticonazole. Remarkably, inhibitors and MOAs identified for Group 3 and Group 4 medulloblastoma tumors at the subgroup level displayed a very high degree of overlap, highlighting the similarities between these subgroups (Fig. 2B, Supplementary Fig. 2).
Drug sensitivity correlates with protein expression to provide insight into drivers of drug response
To investigate the potential drivers of drug response in medulloblastoma, we explored the correlation of predicted drug sensitivity and protein expression across a panel of patients. Using proteomic profiles and drug sensitivity predictions, we performed correlation analyses to identify proteins whose expression levels were significantly associated with response to methylstat. Two proteins, FAM60A and SCML2, were found to be strongly and significantly correlated with drug sensitivity (Fig. 2D). High expression of both proteins was associated with increased sensitivity to methylstat across the patient cohort. These proteins are both part of complexes that regulate histone methylation41,42, suggesting a potential mechanistic link to the drug’s mode of action.
In vitro cell viability reflects subgroup-specific drug sensitivity predictions
As a proof of concept, we selected methylstat for further testing. Methylstat is ranked very highly in our analysis for top inhibitors that vary significantly between subgroups. According to the separation score, it is ranked 6th out of 94 inhibitors that meet the set criteria for the SHH subgroup (TCS > 20, logBB > -1, minimum sensitivity < 0). Additionally, we prioritized testing this drug as the mechanism of action was relevant to cancer treatment. Methylstat was tested in a panel of medulloblastoma cell lines, including two SHH-activated cell lines (S47 and Tg(GFAP-Cre; Ptch1fl/fl)) and two Group 3 cell lines (HD-MB03 and D425). We found that SHH-activated cell lines had reduced IC50 and reduced viability in vitro, compared to Group 3 cell lines (Fig. 4A). The IC50 values for S47 and Tg(GFAP-Cre; Ptch1fl/fl)) (1.06, 0.85 µM) were significantly lower than those for HD-MB03 and Tg(GFAP-Cre; Ptch1fl/fl) (1.36, 4.83 µM), indicating increased sensitivity to methylstat (Fig. 4B-C). Cell viability assays showed a dose-dependent decrease in proliferation in all SHH cell lines, with over 80% reduction at 1 µM. By contrast, Group 3 cell lines exhibited modest changes in viability, with only 25% reduction in viability at 1 µM (Fig. 4A, dashed line at 1 µM).
Drug sensitivity for recurrent patients correlates with predicted synergy with radiation in newly diagnosed patients
To assess the ability of the DrugSeq platform to predict drug sensitivity with radiation therapy, we looked at the correlation between predicted drug sensitivity in recurrent patients and drug synergy with radiation in newly diagnosed patients. To do this, we used the disease discordance scores from the SynergySeq predictions with radiation in newly diagnosed patients and the DrugSeq drug discordance score in recurrent patients. Discordance scores were averaged across all patients, and a Spearman correlation was performed. We found a strong correlation between these drug sensitivity predictions, with a Spearman ρ of -0.74 and a P-value < 0.0001, suggesting that DrugSeq may be a valuable tool for determining synergistic combinations with radiotherapy (Fig. 5A).
Synergistic drug responses with radiation across Medulloblastoma subtypes
Drug synergy with radiation therapy was evaluated across the twelve molecular subtypes of MB, as determined by Cavalli et al.3. Hierarchical clustering of drugs reveals distinct patterns of synergy. Several cyclooxygenase inhibitors (epirizole, diclofenac, indobufen, and indoprefen) displayed strong synergy in Group 3 and Group 4 subtypes. This suggests a potential vulnerability in these subtypes to combined inhibition of inflammatory pathways and radiation, which is consistent with literature demonstrating the radiosensitizing and radioprotective effects of COX-2 inhibitors43,44. SHH subtypes exhibited enrichment for DNA alkylating agents (mitomycin C) and kinase inhibitors (BI-2536). Mitomycin C has been evaluated in clinical trials as an adjunct treatment to radiation therapy with positive outcomes45. BI-2536, a PLK1 inhibitor, has been evaluated for combination with radiation in SHH-MB with positive preclinical results46. Collectively, these results underscore the potential for subtype-specific therapies in combination with conventional therapy to enhance treatment efficacy (Fig. 5B).
Discussion
We report distinct drug sensitivity profiles across medulloblastoma subgroups. We developed a platform called DrugSeq, which is freely available for use at https://combin.shinyapps.io/DrugSeq. DrugSeq users can upload a panel of patient gene expression profiles with associated metadata subgroups to assess predicted drug sensitivity (Fig. 6A). DrugSeq compares the patient transcriptional profile to the L1000 perturbagen-response transcriptional consensus scores and predicts drug sensitivity for each individual patient (Fig. 6B). DrugSeq then calculates drugs that vary significantly across groups of patients (Fig. 6C). Finally, users can generate boxplots of drug sensitivity scores for drugs of interest separated by subgroups (Fig. 6D).

DrugSeq web portal. (A) User uploads disease signature and metadata files, then selects the L1000 drug dataset to analyze. (B, C) DrugSeq calculates the drug discordance for each patient (B) and statistics across subgroups (C) for each drug. (D) User selects drugs to visualize on boxplots.
For our analysis of medulloblastoma patient tumors, we identified compounds that were predicted to target patients in distinct molecular subgroups. As expected, SHH-MB tumors were found to be most sensitive to the hedgehog pathway inhibitors vismodegib and taladegib47,48. Triptolide, which was recently described in Group 3-MB, was found to be most sensitive for Group 3 and Group 4 tumors33. Finally, radiation therapy was found to be most effective in WNT-MB, which is consistent with the literature49,50.
Top inhibitors for WNT-MB included decitabine, minoxidil, oxiperomide, and gestrinone. Decitabine, a DNA methyltransferase inhibitor, has been evaluated in combination with chemotherapy, radiotherapy, and immunotherapy51,52. Minoxidil, a KATP channel opener, has been shown to significantly reduce cell invasion in a model of breast cancer53. Additionally, a recent study used minoxidil-loaded nanoparticles to induce tumor vasodilation and increase drug delivery to brain tumor tissue54. Other repositioned agents highlight how drugs may have anticancer potential55,56.
Several inhibitors were predicted to target SHH-MB, including thioguanine, methylstat, trovafloxacin. Thioguanine, a purine analog, is FDA-approved for AML treatment. Further, it has been shown to inhibit GLI2, which is part of the SHH-pathway57. Methylstat, a selective inhibitor of Jumonji C domain-containing histone demethylase, has been shown to inhibit angiogenesis in several cancer types and has been linked to SHH signaling via JMJD358,59. Trovofloaxin, an antibiotic, has recently been shown to target p90 ribosomal S6 kinase 4 (RSK4), a potential target for SHH-activated MB60,61.
Group 3 and Group 4 MB tumors shared similar drug sensitivity profiles, consistent with their similar transcriptional profiles. Predictions for G3-MB included itraconazole, which induces autophagy in glioblastoma and medulloblastoma62,63, and dexmedetomidine, which has been shown to induce cancer cell death and sensitize tumors to immunotherapy64. Subgroup-specific predictions for G4-MB included isradipine, a calcium channel blocker, has been implicated as a FAK-1 inhibitor in a computational model, with potential anticancer effects65. Additionally, dequalinium, an antimicrobial, and fenticonazole, an antifungal, have shown promise in preclinical cancer models66,67.
Our data provide valuable information that is critical for stratifying patients into treatment groups for clinical trials. Using this platform, we can predict subgroup-specific drug vulnerabilities. Indeed, our data suggest that methylstat, which is predicted to target SHH-MB preferentially induces heightened cell killing of SHH-subtype cells in vitro and ex vivo. Collectively, these studies suggest that there is a significant correlation between what we predict using DrugSeq and what is seen in vitro or in animal models of MB.
Drug repositioning is a powerful tool that can be used to fast-track drug development for brain cancers. This process involves taking an already FDA-approved drug, or a drug in advanced clinical trials, and expanding the treatment criteria to include cancers such as medulloblastoma. This is advantageous because safety profiles and dosing regimens are already established for both animal studies and eventual clinical trials. Furthermore, brain penetrance and toxicities are often already determined. Prior drug repositioning studies have relied heavily on observational data, such as associations between patient survival and previously prescribed medication for cardiovascular diseases68,69, as well as large in vitro drug screens leading to discoveries such as SHH pathway components being targeted by antifungal drugs62,70. By contrast, in silico approaches to drug repositioning often rely on structure-based modeling to identify drugs that target a known oncogene, for instance targeting smoothened (SMO), the positive regulator of the hedgehog pathway, for SHH subgroup tumors in medulloblastoma. However, these inhibitors have often failed in the clinic due to downstream mutations in the SMO pathway and acquired resistance71,72. Other in silico models, such as ours, focus on finding new mechanisms of action for known drugs using transcriptional profiles73,74,75.
Using the DrugSeq pipeline, drugs can then be filtered by known anti-cancer relevance, blood-brain barrier permeability, or mechanism of action. For instance, psychiatric drugs have frequently been found to be effective at inhibiting brain tumor cell growth76,77,78. Dopamine antagonists have been shown to reduce proliferation and viability in several cancer types, including CNS tumors79,80,81. We were also able to predict synergistic combinations, suggesting potential combination therapy with decitabine, a hypomethylating agent, and radiation in WNT, Group 3, and Group 4 tumors. Furthermore, we were able to compare compounds predicted to target recurrent tumors and compare these to compound synergy predictions with radiation in newly diagnosed (non-irradiated) tumors. We found a strong correlation between the drug predictions, suggesting that DrugSeq may provide a way to predict drug sensitivity in recurrent medulloblastoma, as well as in combination with radiation therapy. While these results will need to be validated, we believe this is a powerful tool to identify drugs that may synergize with the current standard of care therapy.
Large-scale drug screening assays to assess drug sensitivity in vivo are less powerful for medulloblastoma since the majority of cell and animal models of medulloblastoma are representative of the Group 3 or SHH subgroups82. Most established human MB cell lines are derived from MYC-amplified Group 3 tumors, the majority of widely used animal models are SHH-driven, which does not accurately represent the diversity in molecular features among MB tumors83. We believe that DrugSeq can help bridge this gap in screening by prioritizing drugs for distinct transcriptional subgroups of medulloblastoma, allowing clinicians to stratify patients in clinical trials. Further preclinical studies of drug candidates will need to be performed in vitro and in vivo to assess the effect on tumor growth.
In conclusion, we have developed a powerful computational platform that allows drug sensitivity predictions for individual patients, paired with statistical analysis to identify drugs that target subsets of patients. The drugs predicted in this study need to be further validated in preclinical models of medulloblastoma to assess the accuracy of the platform for stratifying tumors. This pipeline can be used for additional cancer types and various molecular features, allowing predictions of drug sensitivities for various features in cancer patients.
Conclusions
Medulloblastoma is the most common malignant pediatric brain tumor. Current standard-of-care includes surgical resection, multi-agent chemotherapy, and radiation, but outcomes vary widely by molecular subgroup. Additionally, survivors often face severe long-term neurocognitive deficits. Therefore, novel therapeutic options are urgently needed to address intertumor heterogeneity and limit toxicity. To address this, we developed DrugSeq, a novel computational tool, to predict drug sensitivity based on patient-specific gene expression profiles. DrugSeq was able to accurately identify established treatments for MB subgroups, including triptolide for G3-MB, radiation therapy for WNT-MB, and vismodegib for SHH-MB. Additionally, as a proof of concept, we validated the sensitivity of SHH-MB cell lines to the drug methylstat, demonstrating the predictive power of DrugSeq. Overall, DrugSeq provides a strategy to personalize treatment, guide clinical trial stratification, and offers a practical route toward precision oncology.
Data availability
The data analysis script is available at https://github.com/AyadLab/DrugSeq and as a user-friendly web portal at https://combin.shinyapps.io/DrugSeq/.
References
Li, Q., Dai, Z., Cao, Y. & Wang, L. Comparing children and adults with medulloblastoma: a SEER based analysis. Oncotarget 9 (53), 30189–30198 (2018).
Millard, N. E., De Braganca, K. C. & Medulloblastoma J. Child. Neurol. ;31(12):1341–1353. (2016).
Cavalli, F. M. G. et al. Intertumoral heterogeneity within Medulloblastoma subgroups. Cancer Cell. 31 (6), 737–54e6 (2017).
Amayiri, N. et al. Molecular subgroup is the strongest predictor of Medulloblastoma outcome in a Resource-Limited country. JCO Global Oncol. 7, 1442–1453 (2021).
Ramaswamy, V. et al. Recurrence patterns across Medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol. 14 (12), 1200–1207 (2013).
Endersby, R. et al. Small-molecule screen reveals synergy of cell cycle checkpoint kinase inhibitors with DNA-damaging chemotherapies in Medulloblastoma. Sci. Transl Med. 13, 577 (2021).
Taylor, M. D. et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 123 (4), 465–472 (2012).
Cho, Y. J. et al. Integrative genomic analysis of Medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J. Clin. Oncol. 29 (11), 1424–1430 (2011).
Hovestadt, V. et al. Medulloblastomics revisited: biological and clinical insights from thousands of patients. Nat. Rev. Cancer. 20 (1), 42–56 (2020).
Morrissy, A. S. et al. Spatial heterogeneity in Medulloblastoma. Nat. Genet. 49 (5), 780–788 (2017).
Northcott, P. A., Dubuc, A. M., Pfister, S. & Taylor, M. D. Molecular subgroups of Medulloblastoma. Expert Rev. Neurother. 12 (7), 871–884 (2012).
Northcott, P. A. et al. Medulloblastoma comprises four distinct molecular variants. J. Clin. Oncol. 29 (11), 1408–1414 (2011).
Kool, M. et al. Genome sequencing of SHH Medulloblastoma predicts genotype-related response to smoothened Inhibition. Cancer Cell. 25 (3), 393–405 (2014).
Lafay-Cousin, L. et al. Clinical, Pathological, and molecular characterization of infant Medulloblastomas treated with sequential High-Dose chemotherapy. Pediatr. Blood Cancer. 63 (9), 1527–1534 (2016).
Ramaswamy, V., Nör, C. & Taylor, M. D. p53 and meduloblastoma. Cold Spring Harb Perspect. Med. 6 (2), a026278 (2015).
Clifford, S. C. et al. Biomarker-driven stratification of disease-risk in non-metastatic medulloblastoma: results from the multi-center HIT-SIOP-PNET4 clinical trial. Oncotarget 6 (36), 38827–38839 (2015).
Menyhárt, O., Giangaspero, F. & Győrffy, B. Molecular markers and potential therapeutic targets in non-WNT/non-SHH (group 3 and group 4) Medulloblastomas. J. Hematol. Oncol. 12 (1), 29 (2019).
Pastori, C. et al. BET bromodomain proteins are required for glioblastoma cell proliferation. Epigenetics 9 (4), 611–620 (2014).
Dressler, E. V., Dolecek, T. A., Liu, M. & Villano, J. L. Demographics, patterns of care, and survival in pediatric Medulloblastoma. J. Neurooncol. 132 (3), 497–506 (2017).
Xu, W. et al. Endocrine outcome in children with Medulloblastoma treated with 18 Gy of craniospinal radiation therapy. Neuro Oncol. 6 (2), 113–118 (2004).
Chevignard, M., Câmara-Costa, H., Doz, F. & Dellatolas, G. Core deficits and quality of survival after childhood medulloblastoma: a review. Neurooncol Pract. 4 (2), 82–97 (2017).
Mulhern, R. K. et al. Neurocognitive consequences of risk-adapted therapy for childhood Medulloblastoma. J. Clin. Oncol. 23 (24), 5511–5519 (2005).
Henssen, A. et al. BET bromodomain protein Inhibition is a therapeutic option for Medulloblastoma. Oncotarget ;4. (2013).
He, B., Hou, F., Ren, C., Bing, P. & Xiao, X. A review of current in Silico methods for repositioning drugs and chemical compounds. Front. Oncol. ;11. (2021).
Dubus, E., Ijjaali, I., Barberan, O. & Petitet, F. Drug repositioning using in Silico compound profiling. Future Med. Chem. 1 (9), 1723–1736 (2009).
Cheng, F. et al. Prediction of drug-target interactions and drug repositioning via network-based inference. PLoS Comput. Biol. 8 (5), e1002503 (2012).
Archer, T. C. et al. Proteomics, Post-translational Modifications, and integrative analyses reveal molecular heterogeneity within Medulloblastoma subgroups. Cancer Cell. 34 (3), 396–410e8 (2018).
Sunkin, S. M. et al. Allen brain atlas: an integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res. 41 (Database issue), D996–d1008 (2013).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43 (7), e47–e (2015).
Subramanian, A. et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 171 (6), 1437–52e17 (2017).
Stathias, V. et al. Drug and disease signature integration identifies synergistic combinations in glioblastoma. Nat. Commun. 9 (1), 5315 (2018).
Shaker, B. et al. A machine learning-based quantitative model (LogBB_Pred) to predict the blood–brain barrier permeability (logBB value) of drug compounds. Bioinformatics ;39(10). (2023).
Rodriguez-Blanco, J. et al. Triptolide and its prodrug Minnelide target high-risk MYC-amplified Medulloblastoma in preclinical models. J. Clin. Invest. ;134(15). (2024).
Milde, T. et al. HD-MB03 is a novel group 3 Medulloblastoma model demonstrating sensitivity to histone deacetylase inhibitor treatment. J. Neurooncol. 110 (3), 335–348 (2012).
Bigner, S. H., Friedman, H. S., Vogelstein, B., Oakes, W. J. & Bigner, D. D. Amplification of the c-myc gene in human Medulloblastoma cell lines and Xenografts1. Cancer Res. 50 (8), 2347–2350 (1990).
Li, Y., Zhang, X., Polakiewicz, R. D., Yao, T. P. & Comb, M. J. HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J. Biol. Chem. 283 (19), 12686–12690 (2008).
You, H. et al. The interaction of canonical Wnt/β-catenin signaling with protein lysine acetylation. Cell. Mol. Biol. Lett. 27 (1), 7 (2022).
Prabhu, V. V. et al. Cancer stem cell-related gene expression as a potential biomarker of response for first-in-class Imipridone ONC201 in solid tumors. PLoS One. 12 (8), e0180541 (2017).
Pierce, S. R. et al. Targeting dopamine receptor D2 as a novel therapeutic strategy in endometrial cancer. J. Experimental Clin. Cancer Res. 40 (1), 61 (2021).
Park, Y. S., Kwon, Y. J. & Chun, Y. J. CYP1B1 activates Wnt/β-Catenin signaling through suppression of Herc5-Mediated isgylation for protein degradation on β-Catenin in HeLa cells. Toxicol. Res. 33 (3), 211–218 (2017).
Nabeshima, R. et al. Loss of Fam60a, a Sin3a subunit, results in embryonic lethality and is associated with aberrant methylation at a subset of gene promoters. eLife 7, e36435 (2018).
Maezawa, S. et al. Polycomb protein SCML2 facilitates H3K27me3 to Establish bivalent domains in the male germline. Proc. Natl. Acad. Sci. 115 (19), 4957–4962 (2018).
Komaki, R., Liao, Z. & Milas, L. Improvement strategies for molecular targeting: Cyclooxygenase-2 inhibitors as radiosensitizers for non-small cell lung cancer. Semin Oncol. 31 (1 Suppl 1), 47–53 (2004).
Laube, M., Kniess, T. & Pietzsch, J. Development of antioxidant COX-2 inhibitors as radioprotective agents for radiation Therapy—A. Hypothesis-Driven Rev. Antioxid. 5 (2), 14 (2016).
Haffty, B. G. et al. Mitomycin C as an adjunct to postoperative radiation therapy in squamous cell carcinoma of the head and neck: results from two randomized clinical trials. Int. J. Radiat. Oncol. Biol. Phys. 27 (2), 241–250 (1993).
Markant, S. L. et al. Targeting Sonic hedgehog-associated Medulloblastoma through Inhibition of Aurora and Polo-like kinases. Cancer Res. 73 (20), 6310–6322 (2013).
Kian, W. et al. Vismodegib as First-Line treatment of mutated Sonic Hedgehog pathway in adult Medulloblastoma. JCO Precision Oncol. (4):437–441. (2020).
Robinson, G. W. et al. Vismodegib exerts targeted efficacy against recurrent Sonic Hedgehog-Subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC-025B and PBTC-032. J. Clin. Oncol. 33 (24), 2646–2654 (2015).
Cohen, K. J. et al. A pilot study omitting radiation in the treatment of children with newly diagnosed Wnt-Activated Medulloblastoma. Clin. Cancer Res. 29 (24), 5031–5037 (2023).
Gottardo, N. G. & Gajjar, A. Verschlimmbesserung: craniospinal radiotherapy is essential in WNT Medulloblastoma patients. Clin. Cancer Res. 29 (24), 4996–4998 (2023).
Gringmuth, M. et al. Enhanced survival of High-Risk Medulloblastoma-Bearing mice after multimodal treatment with Radiotherapy, Decitabine, and abacavir. Int. J. Mol. Sci. ;23(7). (2022).
Konkankit, V. V. et al. Decitabine immunosensitizes human gliomas to NY-ESO-1 specific T lymphocyte targeting through the Fas/Fas ligand pathway. J. Transl Med. 9, 192 (2011).
Qiu, S., Fraser, S. P., Pires, W. & Djamgoz, M. B. A. Anti-invasive effects of Minoxidil on human breast cancer cells: combination with Ranolazine. Clin. Exp. Metastasis. 39 (4), 679–689 (2022).
Mohammadzadeh, V. et al. Enhancing targeted delivery and efficacy of pegylated liposomal doxorubicin with liposomal minoxidil: comprehensive in silico, in vitro, and in vivo tumor model studies. Drug Deliv. 32 (1), 2536802 (2025).
Jeon, H. M. et al. Dopamine receptor D2 regulates glioblastoma survival and death through MET and death receptor 4/5. Neoplasia 39, 100894 (2023).
Ciou, H. H. et al. Repurposing gestrinone for tumor suppressor through P21 reduction regulated by JNK in gynecological cancer. Transl Res. 243, 21–32 (2022).
Johnson, R. W. et al. 6-Thioguanine Inhibition of parathyroid hormone-related protein expression is mediated by GLI2. Anticancer Res. 31 (9), 2705–2712 (2011).
Kacı, F. N., Kiraz, Y., Çekdemir, D. & Baran, Y. Synergistic apoptotic effects of bortezomib and Methylstat on multiple myeloma cells. Arch. Med. Res. 51 (3), 187–193 (2020).
Schwarz, F. M. et al. Methylstat sensitizes ovarian cancer cells to PARP-inhibition by targeting the histone demethylases JMJD1B/C. Cancer Gene Ther. 32 (3), 286–296 (2025).
Chrysostomou, S. et al. Repurposed Floxacins targeting RSK4 prevent chemoresistance and metastasis in lung and bladder cancer. Sci. Transl Med. 13, 602 (2021).
Pambid, M. R. et al. Overcoming resistance to Sonic Hedgehog Inhibition by targeting p90 ribosomal S6 kinase in pediatric Medulloblastoma. Pediatr. Blood Cancer. 61 (1), 107–115 (2014).
Kim, J. et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell. 17 (4), 388–399 (2010).
Liu, R. et al. Itraconazole suppresses the growth of glioblastoma through induction of autophagy: involvement of abnormal cholesterol trafficking. Autophagy 10 (7), 1241–1255 (2014).
Zhao, L. et al. Dexmedetomidine induces Immunogenic cancer cell death and sensitizes tumors to PD-1 Blockade. J. Immunother. Cancer. 13 (6), e010714 (2025).
Mohapatra, P. K., Srivastava, R., Varshney, K. K. & Babu, S. H. Formulation and evaluation of Isradipine nanosuspension and exploring its role as a potential anticancer drug by computational approach. Anticancer Agents Med. Chem. 22 (10), 1984–2001 (2022).
Gamboa-Vujicic, G., Emma, D. A., Liao, S. Y., Fuchtner, C. & Manetta, A. Toxicity of the mitochondrial poison dequalinium chloride in a murine model system. J. Pharm. Sci. 82 (3), 231–235 (1993).
Cipolletti, M. et al. A new Anti-Estrogen discovery platform identifies FDA-Approved imidazole Anti-Fungal drugs as bioactive compounds against ERα expressing breast cancer cells. Int. J. Mol. Sci. ;22(6). (2021).
Nielsen, S. F., Nordestgaard, B. G. & Bojesen, S. E. Statin use and reduced Cancer-Related mortality. N. Engl. J. Med. 367 (19), 1792–1802 (2012).
Hammoud, H. et al. Drug repurposing in medulloblastoma: challenges and recommendations. Curr. Treat. Options Oncol. 22 (1), 6 (2020).
Giambelli, C., Fei, D. L., Wang, H. & Robbins, D. J. Repurposing an old anti-fungal drug as a Hedgehog inhibitor. Protein Cell. 1 (5), 417–418 (2010).
Atwood, S. X. et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 27 (3), 342–353 (2015).
Danial, C., Sarin, K. Y., Oro, A. E. & Chang, A. L. An Investigator-Initiated Open-Label trial of sonidegib in advanced basal cell carcinoma patients resistant to vismodegib. Clin. Cancer Res. 22 (6), 1325–1329 (2016).
Lamb, J. et al. The connectivity map: using Gene-Expression signatures to connect small Molecules, Genes, and disease. Science 313 (5795), 1929–1935 (2006).
Chang, M., Smith, S., Thorpe, A., Barratt, M. J. & Karim, F. Evaluation of phenoxybenzamine in the CFA model of pain following gene expression studies and connectivity mapping. Mol. Pain. 6, 1744–8069 (2010).
March-Vila, E. et al. On the integration of in Silico drug design methods for drug repurposing. Front. Pharmacol. ;8. (2017).
Tan, S. K. et al. Drug repositioning in glioblastoma: A pathway perspective. Front. Pharmacol. 9, 218 (2018).
Lee, J. K., Nam, D. H. & Lee, J. Repurposing antipsychotics as glioblastoma therapeutics: potentials and challenges. Oncol. Lett. 11 (2), 1281–1286 (2016).
Diamandis, P. et al. Chemical genetics reveals a complex functional ground state of neural stem cells. Nat. Chem. Biol. 3 (5), 268–273 (2007).
Allen, J. E. et al. Discovery and clinical introduction of first-in-class Imipridone ONC201. Oncotarget 7 (45), 74380–74392 (2016).
Weissenrieder, J. S., Neighbors, J. D., Mailman, R. B. & Hohl, R. J. Cancer and the dopamine D(2) receptor: A Pharmacological perspective. J. Pharmacol. Exp. Ther. 370 (1), 111–126 (2019).
Arrillaga-Romany, I. et al. A phase 2 study of the first Imipridone ONC201, a selective DRD2 antagonist for oncology, administered every three weeks in recurrent glioblastoma. Oncotarget 8 (45), 79298–79304 (2017).
Li, Z. & Langhans, S. A. In Vivo and ex vivo pediatric brain tumor models: an overview. Front. Oncol. ;11. (2021).
Ivanov, D. P., Coyle, B., Walker, D. A. & Grabowska, A. M. In vitro models of medulloblastoma: choosing the right tool for the job. J. Biotechnol. 236, 10–25 (2016).
Acknowledgements
We would like to thank all members of the Ayad Lab for the helpful discussion and editing of this manuscript, including. Simon Kaeppeli, Jordan Walter, Matthew D’Antuono, and Emma Rowland.
Funding
This work was supported by the National Institutes of Health grants T32 CA009686 and NS118023.
Author information
Authors and Affiliations
Contributions
Project design, manuscript preparation: NGA. Clinical significance: NKL, DH. Computational design: RKS. Data analysis and figure preparation: LR. Project design, computational processing and data analysis, figure preparation: AMJ.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jermakowicz, A.M., Ruiz, L., Chu, J. et al. In silico drug sensitivity predicts subgroup-specific therapeutics in medulloblastoma patients. Sci Rep 15, 42373 (2025). https://doi.org/10.1038/s41598-025-26411-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-26411-4
Keywords
This article is cited by
-
Drug and single-cell gene expression integration identifies sensitive and resistant glioblastoma cell populations
Nature Communications (2026)
