
Abstract
The complete chloroplast genome of Secale strictum ssp. strictum (Poaceae: Triticeae) was sequenced and analyzed to support its use in rye and wheat breeding. The genome is 137,063 bp long and includes a pair of inverted repeats (IRs; 21,580 bp each) that separate the small (SSC; 12,817 bp) and large (LSC; 81,086 bp) single-copy regions. It contains 113 genes: 74 protein-coding, 30 tRNA, four rRNA genes, and five conserved open reading frames. A total of 42 repeat sequences were identified, mainly in the LSC region, with direct repeats being most common. All mononucleotide SSRs consisted of A/T motifs. Seven highly variable regions were identified, offering potential as molecular markers for species identification and phylogenetic studies. Phylogenetic analysis based on 73 protein-coding genes confirmed the systematic placement of the species and showed that S. strictum ssp. strictum is closely related to S. cereale and other S. strictum accessions. This study presents the first complete plastome of S. strictum ssp. strictum, now available as a reference genome under GenBank accession number OL979486.
Similar content being viewed by others

Introduction
Rye is a small but highly diverse genus that includes annual and perennial species, as well as self-pollinating, open-pollinating, cultivated, weedy, and wild forms with varied morphologies1. The genus Secale L. comprises four species, among which only S. cereale subsp. cereale is cultivated as a crop2, although S. strictum may have been used as a forage plant3. Various taxonomic classifications have been proposed for this genus1,4,5,6, but a consensus has not yet been reached. The relationships among cultivated, weedy, and wild forms also remain unclear7.
Rye is known for its high tolerance to diverse biotic and abiotic stresses8,9,10,11. Wild and weedy forms are capable of crossbreeding with cultivated rye12 and can serve as valuable genetic resources for breeding programs involving wheat, triticale, and other crops13. Therefore, additional genetic markers are needed to clarify the still-debated phylogenetic relationships within the genus Secale.
Due to their conserved and non-recombinant nature, chloroplast genomes represent a reliable tool for genomic and evolutionary studies14. Certain evolutionary hotspots in the plastid genome, such as single nucleotide polymorphisms (SNPs) and insertions/deletions (indels), can provide critical insights into the phylogenetics of taxonomically unresolved plant groups15,16. Consequently, access to complete chloroplast genomes, which contain novel variable and informative regions, may enable more accurate phylogenetic reconstructions.
To date, only a few chloroplast genome sequences have been reported in GenBank (NCBI)17 for the genus Secale, including the complete cp genomes of S. africanum (OQ700974)18, S. segetale (OL688773)19, and S. sylvestre (MW557517)20; two accessions of S. strictum (KY636137 and KY636138)21 with incomplete plastome sequences; and the cp genome of S. cereale (NC_021761)22, which lacks one IR region. A comprehensive phylogenetic analysis based on complete chloroplast genomes has not yet been published.
We hypothesize that analyses of complete chloroplast genome sequences of Secale species—beginning with S. sylvestre20—may provide a cost-effective and informative approach to evolutionary and phylogenetic studies, as suggested by our previous findings23,24.
Results
Organization of the chloroplast genomes
Sequencing of Secale strictum ssp. strictum chloroplast genome produced 20 795 466 raw reads (150 bp for average read length) which were mapped to the reference genome of Secale cereale. A total 860 406 reads were ultimately mapped with 941.7 × average coverage. The complete chloroplast genome sequence of Secale strictum ssp. strictum was deposited in GenBank (OL979486).
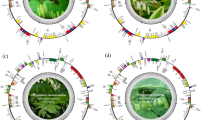
The Secale strictum ssp. strictum chloroplast genome is a typical circular, double-stranded molecule with the length of 137 063 bp. The chloroplast genome displayed a typical quadripartite structure with a pair of IRs (21 580 bp) separated by SSC (12 817 bp) and LSC (81 086 bp) regions (Fig. 1). The overall GC content was 38.24%. It encodes 113 genes which include of 74 protein-coding genes, 30 tRNA genes, four rRNA genes and five conserved chloroplast ORFs (ycf2, ycf3, ycf4, ycf15 and ycf68) (Table 1). Fifty-seven protein-coding genes, 21 tRNA genes and two conserved chloroplast ORFs (ycf3 and ycf4) are located in LSC, whereas the SSC region contained 10 protein-coding genes and one tRNA gene. The IR region contained four rRNA genes, eight tRNA genes, three conserved chloroplast ORFs (ycf2, ycf15 and ycf68) and nine protein-coding genes, including ndhH on the border between IRA/IRB and SSC. The full ndhH sequence is located on the IRA/SSC border, and its incomplete copy on the IRB/SSC border acts as a pseudogene (ψndhH). Eleven genes contained one intron (atpF, ndhA, ndhB, rps16, rpl2, trnI-GAU, trnA-UGC, trnK-UUU, trnG-UCC, trnL-UAA and trnV-UAC), whereas two genes (ycf3 and rps12) consisted of three exons. The first exon of rps12 (5’ end of the sequence) was found in the LSC region, whereas the remaining two exons were located in the IR region. This feature supported the identification of rps12 as a trans-spliced gene. The introns of the following genes, trnK-UUU and trnI-GAU, include coding sequences for matK and ycf68, respectively. Furthermore, the presence of three copies of rpl23 gene were found: two rpl23 gene copies located in the inverted repeats (IRs) and the rpl23 pseudogene located in the large single copy region (LSC).

Gene map of the Secale strictum ssp. strictum chloroplast genome. Genes drawn inside the circle are transcribed clockwise, and those outside are transcribed counterclockwise (indicated by arrows). Differential functional gene groups are color-coded. GC content variations is shown in the middle circle. Gene map was generated with the OrganellarGenomeDRAW (OGDRAW) 1.3.1. (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html).
Codon usage analysis performed using 74 protein coding gene sequences and five conserved chloroplast ORFs identified a total of 20 774 codons in the Secale strictum ssp. strictum chloroplast genome. The most and least abundant codons (excluding these associated with the initiation and termination of translation) were ATT for isoleucine (4.12%) and TGC for cysteine (0.26%), respectively (Table S1). Furthermore, leucine appeared as the dominant amino acid (10.8%), whereas cysteine was less frequently encountered (1.1%). Since the data for codon usage were not available for the previously published plastomes of S. cereale and S. strictum, these species were also included in the analysis. Secale cereale (NC_021761) shared the same pattern of codon usage: the most abundant codon was ATT for isoleucine (4.18%) whereas the TGC for cysteine was the least abundant (0.27%). For Secale strictum (KY636137 and KY636138) the most abundant codon was ATT for isoleucine (4.17 and 4.16%, respectively), whereas only one codon ATA and one ATG were identified for the coding of methionine in KY636137 and only one codon ATA and one GTG were identified for the coding of methionine in KY636138. Nevertheless, these results should be treated with caution as all these sequences lack of one IR copy.
The boundaries between IR and SSC/LSC regions in Secale strictum ssp. strictum cp genomes were identified (Fig. 2). The IRA/SSC junction was found within the nadhH gene (206 bp from its 5′ end), and the boundary between IRB and LSC region was identified between the sequences for rpl22 and rps19 gene (31 bp from rpl22 5′ end and 52 bp from rps19 3′ end). Consequently, the full ndhH sequence is located only on the IRA/SSC border, and its incomplete copy on IRB/SSC border acts as a pseudogene (ψndhH). The IRA/LSC junction is located within the intergenic spacer betwen the rps19 and psbA gene (52 bp from rps19 3′ end and 80 bp from psbA 3′ end). Identification of boundaries between the IR and SSC/LSC regions in chloroplast genomes of remaining representatives of Secale genus was hampered due to the lack of one IR region in these plastome sequences.

Localization of boundaries between LSC, SSC, and IR regions in Secale strictum ssp. strictum chloroplast genome.
Repetitive sequences and SSRs
A total of 42 repeat structures with length ranging from 30 to 286 bp were observed in the plastome of Secale strictum ssp. strictum (Table S2). Forward (F) repeats dominated among the identified repetitive sequences (73.8%) and were followed by palindromic (P) repeats (26.2%). None reverse and complementary repeats were found. The forward repeats ranged from 30 to 74 bp, the palindromic repeats were 30 bp to 286 bp. Most repeat sequences (83.3%) were detected in the LSC region, followed by IR (14.3%) and SSC regions (2.4%). 57.1% of these sequences were found within coding regions. The highest number of repeats were found within the sequences of the following genes: rpoC2 (9F), rps18 (3F and 1P), rpl23 (2P) and trnfM-AUG (2F).
A total 27 SSRs were detected in the Secale strictum ssp. strictum chloroplast genome, which were composed by a length of at least 12 bp and met thresholds values for chloroplast SSRs identification described in Materials and Methods section. Among them, 13 (48.2%) were mononucleotide SSRs, 5 (18.5%) were trinucleotide SSRs, 6 (22.2%) were tetranucleotide SSRs and 3 (11.1%) were pentanucleotide SSRs (Table S3, Fig. 3). All mononucleotide SSRs were composed of A/T repeat units. Motifs composed of adenine and thymine were also predominant among trinucleotide SSRs, where three AAT/TTA, one AAC/TTG and one AAG/TTC motifs were observed. The majority of SSRs were located in the LSC region (85.2%), followed by SSC and IR regions (in both cases in equal proportions of 7.4%). Furthermore, most of the SSRs were identified within intergenic spacers (16 or 59.3%), while 6 (22.2%) were in the introns and 5 (18.5%) were within the coding sequences.

The distribution and type of simple sequence repeats (SSRs) in cp genome of Secale strictum ssp. strictum. (A) Number of different SSRs types; (B) Location of different SSRs in IR, SSC and LSC regions; (C) Partition of SSRs among IGS, introns and exons (D); Distribution of SSR motifs in different repeat class types.
Sequence divergence
Comparative analysis of chloroplast genome sequences of Secale strictum ssp. strictum, S cereale (NC_021761) and two sequences for S. strictum (KY636137 and KY636138) available in NCBI database showed their almost identical structure and content. Re-analysis of mentioned above chloroplast genome sequences for S. cereale and S. strictum, allowed us improvement of five protein-coding sequences (petB, rpl16, rps16, rps12 and ycf2) and identification of previously not annotated ycf15 and ycf68 for KY636137 and KY636138 and rpl20 in case of NC_021761 (Table S4). Furthermore, as the NCBI records for NC_021761, KY636137 and KY636138 lack sequence for one IR copy, we did not find there complete sequences for ndhH and ndhG gene in case of S. strictum and any copy of rpl23 gene in S. cereale. On the other hand, analogously to the NC_021761 sequence, we did not annotate ycf1 sequence in reported here chloroplast genome sequence of Secale strictum ssp. strictum.
In order to analyze the overall sequence conservation and to detect the diverged regions in the cp genomes of Secale strictum ssp. strictum, S. strictum and S cereale MAUVE and DnaSP programs were applied. MAUVE results are shown in Fig. 4. Rearrangements (inversions or translocations) were not detected in any of the studied chloroplast genome sequences. According to results obtained in DnaSP, the average nucleotide diversity (π) among the four chloroplast genomes of Secale species was determined at 0.00061, ranging from 0 to 0.00563 (Table S5). In sliding window analysis, the most variable regions were identified, i.e. regions for which π values exceeded 0.003 (Fig. 5). Among the coding regions, the highest differences were found in rps3 locus (0.00313). In non-coding regions divergence was generally higher and the highest π values were observed for rpl32–trnL-UAG (0.00563), ndhF–rpl32 (0.00438), rpl23–psaI (0.00438), trnS-GCU–psbD (0.00417), intron of ndhA gene (0.00333) and psbK–psbI (0.00313). Four of these highly variable regions were identified in LSC whereas remaining three was found in SSC region.

A MAUVE alignment of Secale strictum ssp. strictum (OL979486), Secale cereale (NC_021761), Secale strictum voucher R 853 (KY636138) and Secale strictum R 1108 voucher (KY636137) chloroplast genomes showing the lack of rearrangements between the chloroplast genomes of the four species. The Secale strictum ssp. strictum chloroplast genome is shown at top as the reference. Within each of the alignment, local collinear blocks are represented by blocks of the same color.

Sliding window analysis of the four Secale complete chloroplast genome sequences (window length: 800 bp; step size 50 bp). The Y-axis presents nucleotide diversity of each window, while the X-axis represents position of the midpoint.
Synonymous (Ks) and non-synonymous (Ka) substitution rate analysis
The non-synonymous (Ka) and synonymous (Ks) substitution rate as well as Ka/Ks ratio was calculated for the 76 genes (71 protein-coding and 5 ORFs) shared by all four chloroplast genomes (Table S6). Contrary to nonsynonymous substitutions, synonymous substitution does not change the amino acid within a peptide chain. The substitution rate appeared to be very low across plastome genes in each functional group, and the values of Ka and Ks were determined in the range of 0–0.0084 and 0–0.0233, respectively. The highest average value of Ks (0.0024) was noted for coding sequences associated with the large subunit of ribosome. The average value of Ks was the lowest (0.0) in sequences of RubisCO large subunit (rbcL), translational initiation factor (infA) and seven genes gathered in group called ‘other genes’. The sequences of RubisCO large subunit (rbcL) and translational initiation factor (infA) together with genes associated with small subunit of ribosome, Photosystem I and Photosystem II were also characterized by the lowest average values of Ka (0.0). The highest average values of Ka (0.0010) was noted for genes associated with cytochrome b/f complex. In 50 sequences of plastome genes of the studied Secale species, neither synonymous nor non-synonymous substitutions were observed (Ks = 0 and Ka = 0, respectively). The remaining 26 genes shared 99% similarity, but only synonymous substitutions (Ka = 0) were observed in 15 of those genes. The value of Ka/Ks ratio may indicate whether selective pressure is acting on a particular protein-coding gene. The Ka/Ks > 1 suggest the positive selection, whereas Ka/Ks < 1 points to the influence of purifying selection. In case of all analyzed sequences the Ka/Ks value was less than 1, with the highest value (0.3158) found for ndhF gene in S. strictum ssp. strictum.
RNA-editing
Application of PREP suite allowed us to predict a total number of 35 potential editing sites in 15 protein-coding genes in the four analyzed plastome sequences of Secale species; 34 characteristic for all of them and one within the rpl2 sequence unique for S. cereale (NC_021761) and one within the petB sequence identified only for S. strictum (both KY636137 and KY636138) (Table S7). All editing events involved C to U conversion. Four non-synonymous mutations were found at the first position of the codon, 30 mutations were identified at the second position, and none were found at the third position. Serine (S) to leucine (L) changes appeared as the most frequent (15 or 42.9%) mutations, whereas leucine (L) to phenylalanine (F) and proline (P) to serine (S) were least frequently observed (1 or 2.9% for both). Each RNA editing site in the corresponding chloroplast genes of the studied Secale species was generally found at the same nucleotide position. Only in case of petB gene in S. strictum 51 base shift, and 12 base shift in case of rpl2 for S. cereale is observed.
Phylogenetic analysis
The phylogenetic trees generated by BI and ML had a consistent topology. All of the recovered clades of the BI tree were characterized by high nodal support values: in 29 out of 32 nodes (90.6%) Bayesian posterior probability value reached 1.0. The BI tree divided analyzed species into six major clades (Fig. 6). The first, and the biggest cluster contained 20 species which represented Triticinae subtribe. Four other major gathered species representing Hordeinae subtribe. The last major clad consisted of three Littledalea species (Littledaleeae tribe). The most distant position is occupied by Oryza sativa (out group). Reported here chloroplast genome of Secale strictum ssp. strictum appeared to share the highest degree of similarity with plastome of S. strictum and S. cereale. Mentioned above three representatives of genus Secale form separate sub-clad within the Triticinae tribe.

Phylogenetic tree based on sequences of sheared 73 protein-coding genes from three Secale species and 30 other cereal lineages representing Triticodae group within subfamily Pooidae and the cp genome of Oryza sativa as an outgroup, using Bayesian posterior probabilities (PP). Bayesian PP are given at each node.
Discussion
The effective use of genomic information to improve cultivated rye has been hampered by the enormous genome size, high content of repetitive sequences25, and unresolved phylogenetic relationships within the genus Secale. Consequently, breeding progress has been limited, despite the potential for introducing beneficial traits from wild rye species. Compared to cultivated rye, wild Secale strictum (syn. S. montanum), which comprises five subspecies (strictum, kuprijanovii, africanum, anatolicum, ciliatoglume), exhibits perennial growth, large size, profuse tillering, frost and disease resistance, and high protein content26,27. It frequently occurs as a weed in Triticum turgidum fields28. Due to its high forage value, S. strictum has been recommended for the restoration of overgrazed substeppe rangelands29,30. Furthermore, it has been used as a source of disease resistance genes31, cytoplasmic male sterility32,33, and traits related to grain hardness, protein, and arabinoxylan content26. Therefore, understanding the genetic structure and diversity within the genus Secale is of key importance for future crop improvement.
Secale strictum may have been used as a forage crop3. S. strictum and S. sylvestre are, respectively, perennial outcrossers and annual selfers1. Numerous infraspecific taxa have been described for S. cereale and S. strictum, reflecting differences in geographic distribution, growth habit (e.g., weediness), and morphological features such as leaf sheath hairiness or spike brittleness1. Artificial interspecific hybridizations are possible between all rye taxa, although S. sylvestre shows low crossability with other species4,34. Hybrids often exhibit low fertility, likely due to chromosomal translocations35,36. Spontaneous hybridizations between cultivated S. cereale and wild-growing S. strictum populations have been reported in Italy and Anatolia37,38.
Recent research has shown that complete chloroplast genomes contain significantly more informative polymorphic sites than standard cpDNA regions, providing higher resolution for plant phylogenetic analyses38,39,40. Among the nine Secale chloroplast genomes available in GenBank, only three are complete: Secale cereale (137,051 bp)22, S. sylvestre (137,116 bp)20, S. cereale ssp. segetale19, and S. strictum ssp. africanum18.
In the present study, we assembled the complete chloroplast genome of S. strictum ssp. strictum using Illumina whole-genome sequencing data. The plastome exhibits the typical quadripartite structure found in angiosperms, with two inverted repeats (IRs) separating the large and small single-copy regions (LSC and SSC, respectively) (Fig. 1), and gene content and genome size comparable to other Triticinae species22.
In Triticeae, complete plastome sizes range from 135,564 bp in S. cereale to 136,886 bp in Triticum monococcum, and from 136,043 bp in Hordeum vulgare ssp. spontaneum to 136,968 bp in H. bogdanii41. In land plants, changes in plastome size are most commonly due to contraction or expansion of the IR regions. Although chloroplast genomes are generally highly conserved, variation at the four junctions (LSC/IRb/SSC/IRa) contributes to differences in plastome structure41,42,43. In the genomes analyzed by Chen et al. (2021)41, the ndhH gene spans the SSC/IRa boundary, with 0–1,007 bp located in the SSC. The rps19 gene is located within the IRa, 1–51 bp from the LSC/IRa boundary, while the rpl2 gene lies in IRa in H. vulgare (590 bp from the junction) but in the LSC in H. vulgare ssp. spontaneum (4 bp from the junction).
Among Triticeae, Secale has one of the largest chloroplast genomes and retains non-functional genes as well as numerous redundant intergenic spacer (IGS) fragments44. Comparative analyses of the S. strictum chloroplast genome with S. cereale (NC_021761)22 and S. strictum sequences (KY636137 and KY636138)21 revealed almost identical gene order and content (Fig. 4). Our analysis improved the annotation quality of these genomes by identifying three previously unannotated genes: ycf15, rpl20, and ycf68 in KY636137, KY636138, and NC_021761, respectively (Table S4). Whole-genome alignments confirmed that no structural rearrangements—such as inversions or gene translocations—occurred among the four genomes (Fig. 4).
The four Secale plastomes were highly conserved, with most variation occurring in non-coding regions (Fig. 5). Among coding regions, rps3 showed the highest nucleotide divergence. Four of the highly variable loci were located in the LSC region, and three in the SSC. The IR/SC boundaries in the S. strictum ssp. strictum plastome were precisely identified (Fig. 2): the IRA/SSC boundary lies within the ndhH gene, while the IRB/SSC junction is located between rpl22 and rps19, consistent with findings in the 31 Triticeae species analyzed by Chen et al. (2021). However, information on the IRA/SSC boundary is missing in these genomes due to the absence of one IR copy.
The LSC/IRb boundary is also located between rpl22 and rps19 in all 31 Triticeae species examined by Chen et al. (2021)41. Comparative analysis of the Agropyron cristatum cp genome suggested that the loss of accD and translocation of rpl23 may represent lineage-specific gene loss or divergence events within Triticeae41.
Non-coding regions were more variable than coding regions, and IRs exhibited lower sequence divergence than SC regions. Hotspot regions of variation were identified in petN–rpoB, rbcL–psaI, and rpl23–ndhB. Evolutionary events such as pseudogenization, indels, intron losses, and IGS variation were mapped onto the Triticeae phylogeny based on complete plastome data. One notable mutation occurred between rbcL and psaI, encompassing the rpl23 and accD genes as well as surrounding intergenic regions. These genes were absent in several species including A. cristatum, A. mongolicum, Eremopyrum triticeum, E. distans, Australopyrum retrofractum, Henradia persica, and Aegilops tauschii. Deletions involving both accD and adjacent IGS regions were also observed in Aegilops speltoides and T. monococcum ssp. aegilopoides41.
Previous studies also confirmed the absence of the accD pseudogene in at least one Triticeae species45, whereas truncated versions (up to 349 bp) have been reported in Secale46.
Phylogenetic reconstruction identified four main clades (I–IV), with Clade I comprising the Aegilops/Triticum complex along with Taeniatherum, Secale, Crithopsis, and Herteranthelium—all of which are distributed across Mediterranean habitats of Triticeae41. The clear clustering of multiple Secale accessions underlines the phylogenetic coherence of the genus, despite its well-documented morphological and ecological diversity1. This observation supports previous reports of hybridization potential and gene flow among Secale species, including between wild and cultivated forms34,35, which may have implications for conservation and breeding.
This study provides the first complete chloroplast genome of Secale strictum ssp. strictum, a wild rye taxon with documented potential for introgression into cultivated cereals.
Materials and methods
Plant material and DNA extraction
Seeds of Secale strictum subsp. strictum (PI 272,338) were obtained from the Botanical Garden of the Polish Academy of Sciences, Warsaw, Poland. Total genomic DNA was isolated from 10-day-old sprouts using the CTAB method47.
Chloroplast genome sequencing, annotation and analysis
Whole-genome sequencing was performed on the DNBSEQ platform (BGI, Shenzhen, China), yielding approximately 40 million high-quality 100 bp paired-end reads. The FastQC tool was used to check the quality of raw reads. Raw reads were trimmed (5 bp of each read end, regions with more than 5% probability of error per base) and mapped to the reference chloroplast genome of Secale cereale (NC_021761) in Geneious v.R7 software with default medium–low sensitivity settings48. Mapped reads were extracted and assembled de novo using SPAdes v3.14.049 with a k-mer size of 85. Contigs were extended by iterative read mapping and manual scaffolding (minimum overlap 50 bp; 97% identity), repeated five times to obtain a circular cpDNA molecule. Finally, the reduced sequences were assembled in the circular chloroplast genome. The chloroplast genome was annotated using MFannot50 and PlasMapper51 with manual adjustments, and based on sequences of complete chloroplast genomes and genes available in GenBank (NCBI) for the genus Secale. The gene map of the annotated cp genome was developed with the OrganellarGenome DRAW tool52.
Genomic repeats and SSR analysis
The chloroplast simple sequence repeats (SSRs) in complete plastome of Secale strictum ssp. strictum were detected using Phobos v.3.3.1253. Only perfect SSRs with a motif size of one to six nucleotide units were considered, and the following thresholds for chloroplast SSRs identification were used: ≥ 12 repeat units for mononucleotide SSRs, ≥ 6 repeat units for dinucleotide SSRs, ≥ 4 repeat units for trinucleotide SSRs, and ≥ 3 repeat units for tetra-, penta- and hexanucleotide SSRs54. Furthermore, analysis of long genomic repeats (including forward, reverse, palindromic and complementary sequences) was performed using REPuter software55 with the following settings: (1) hamming distance of 3, (2) sequence identity ≥ 90%, and (3) minimum repeat size ≥ 30 bp. A single IR region was used to eliminate the influence of doubled IR regions. Redundant results in REPuter were deleted manually.
Comparative chloroplast genome analysis
The chloroplast genome sequence of S. strictum ssp. strictum was compared with the three chloroplast genome sequences representing the genus Secale in NCBI, i.e. Secale cereale (NC_021761)22, and two Secale strictum sequences (KY636137 and KY636138)21. In order to assess the complete nucleotide diversity (Pi) among the four analyzed chloroplast genomes, their sequences were aligned in MAFFT v.7.31056, then we performed sliding window analysis using DnaSP v.6.10.0457. The step size was set to 50 base pairs, and window length was set to 800 base pairs. Genome synteny analysis of the four Secale chloroplast genomes was also performed with the use of MAUVE v.1.1.158. Because all NCBI derived Secale sequences do not include one IR region, during mentioned above analyses the appropriate IR region was also removed from our sequence. The 76 protein-coding genes shared by all four analyzed Secale plastomes were extracted and aligned separately using MAFFT v7.310 to estimate the ratio of non-synonymous (Ka) to synonymous (Ks) substitutions. The Secale cereale was the reference species. The values of Ka and Ks in the shared genes were calculated in DnaSP v.6.10.04. Genes with non-applicable (NA) Ka/Ks ratios were changed to zero.
The chloroplast genome borders of LSC, SSC, and IRs for Secale strictum sp. strictum were identified based on their annotations. Unfortunately, due to the lack of one IR region in all Secale plastomes acquired from NCBI database, the comparative analysis of these structures was not possible. The data on the distribution of codon usage for Secale strictum sp. strictum was acquired from the Geneious v.7 statistics panel.
Potential RNA editing sites in the protein-coding genes of four Secale chloroplast genomes were predicted using the Predictive RNA Editor for Plants (PREP) suite59. The cutoff value for the analyzed Seceale plastome sequences was set at 0.8, and 33 out of the 35 reference genes in PREP were used. accD was not included in the analysis because it was not identified within the chloroplast genomes of the studied Seceale species. Furthermore, rps23 gene was also excluded as it was not annotated in S. cereale chloroplast genome.
Phylogenetic analysis
Chloroplast genomes of 36 cereal lineages representing Triticodae group within subfamily Pooidae and the cp genome of Oryza sativa as an outgroup, were downloaded from the NCBI database to investigate the phylogenetic position of Secale strictum sp. strictum. The cp genomes used in phylogenetic analyses are presented in Table 2. The sequences of 73 shared protein coding genes were extracted using custom R script, and they were aligned in MAFFT v.7.310. Finally, concatenated protein-coding gene sequences where used for phylogeny reconstruction by Bayesian Inference (BI) and Maximum-Likelihood (ML) method. The best-fit model of sequence evolution was identified in MEGA v.760, and the GTR + G + I model was selected. The BI analysis was performed in MrBayes v.3.2.661,62, and the ML analysis was conducted in PhyML v.3.063. Parameter settings were previously described by Androsiuk et al.64.
Data availability
The genomic sequence data that support the findings of this study are openly available in the GenBank of NCBI (www.ncbi.nlm.nih.gov/search/all/?term = OL979486) under accession number OL979486 (accessed on 12-SEP-2022).
References
Frederiksen, S. & Petersen, G. A taxonomic revision of the genus Secale (Triticeae, Poaceae). Nordic J. Bot. 18, 399–420 (1998).
GRIN. Germplasm Resources Information Network. USDA-ARS. https://www.ars-grin.gov (2019).
Hammer, K., Skolimowska, M. & Knüpffer, H. Collection and evaluation of wild and weedy rye. Plant Genet. Resour. Newsl. 71, 5–10 (1987).
Khush, G. S. Cytotaxonomy of the genus Secale. Bot. Gaz. 123, 188–195 (1962).
Sencer, R. & Hawkes, J. G. On the origin and domestication of rye. J. Biogeogr. 7, 457–461 (1980).
Hammer, K. Vorarbeiten zur Monographie von Secale L. Kulturpflanze 38, 33–41 (1990).
Marques, I. et al. Cytogenetic and genome size variation in Iberian Secale taxa. Plant Syst. Evol. 299, 135–145 (2013).
Alptekin, B., Langridge, P. & Budak, H. Abiotic stress signaling and transcriptome regulation in wheat. Funct. Integr. Genomics 17, 145–170 (2017).
Madej, L. Disease resistance in wild rye. Biul. IHAR 197, 117–122 (1996).
Mago, R. et al. Characterization and mapping of resistance genes in rye. Theor. Appl. Genet. 104, 1317–1324 (2002).
Hurni, S. et al. The powdery mildew resistance gene Pm8 derived from rye is suppressed by its wheat orthologue Pm3. Plant J. 74, 798–810 (2013).
Broda, Z. et al. Interspecific hybridization between rye and wheat: Molecular evidence for translocations. J. Appl. Genet. 57, 1–9 (2016).
Schlegel, R. Encyclopedia of Plant Breeding and Genetics (CRC Press, 2006).
Cho, M. S. et al. Utility of chloroplast genome sequences in plant systematics: Comparative analysis in Asteraceae. Genes Genomics 37, 529–540 (2015).
Eguiluz, M., Garcia, J. & Aguinagalde, I. Plastid genome variation in Quercus: Insights from nucleotide substitution patterns. Plant Syst. Evol. 303, 1359–1373 (2017).
Ruhfel, B. R. et al. Phylogeny of the clusioid clade (Malpighiales): Evidence from plastome and nuclear data. Am. J. Bot. 101, 1491–1506 (2014).
GenBank. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 2004. https://www.ncbi.nlm.nih.gov (accessed on 6.06.2025).
Skuza, L. et al. The first complete chloroplast genome of Secale strictum ssp. africanum.. Curr. Issues Mol. Biol. 47, 64 (2025).
Skuza, L. et al. Molecular structure, comparative and phylogenetic analysis of the complete chloroplast genome sequences of weedy rye Secale cereale ssp. segetale. Sci. Rep. 13, 5412 (2023).
Skuza, L. et al. The complete chloroplast genome of Secale sylvestre. J. Appl. Genet. 63, 115–117 (2022).
Bernhardt, N., Brassac, J., Kilian, B. & Blattner, F. R. Dated tribe-wide whole chloroplast genome phylogeny indicates recurrent hybridizations within Triticeae. BMC Evol. Biol. 17, 141 (2017).
Middleton, C. P. et al. Sequencing of chloroplast genomes from wheat, barley, rye and their relatives provides a detailed insight into the evolution of the Triticeae tribe. PLoS ONE 9, e85761 (2014).
Skuza, L. et al. DNA barcoding in selected species and subspecies of rye using three chloroplast loci. Not. Bot. Horti Agrobot. Cluj-Napoca 47, 54–62 (2018).
Skuza, L. et al. Genetic diversity among rye species revealed by cpDNA and mtDNA non-coding regions. PLoS ONE 14, e0213023 (2019).
Haseneyer, G. et al. A sequence ready physical map of barley chromosome 1H. Plant J. 65, 902–913 (2011).
Schneider, A. et al. Genetic diversity and agronomic performance of perennial rye populations. Crop Sci. 56, 1944–1953 (2016).
Kubiczek, J. Systematics and geographical distribution of the genus Secale. Prace Inst. Gen. Hod. Zwierząt PAN 21, 135–142 (1981).
Davis, P. H. Flora of Turkey and the East Aegean Islands Vol. 9 (Edinburgh University Press, 1985).
Yanchenko, V. Y. Use of wild perennial rye for pasture rehabilitation. Forage Res. Bull. 2, 15–20 (1984).
Andersen, B., Landgridge, P. & Mujeeb-Kazi, A. Wild rye as forage grass in substeppe environments. Rangeland J. 14, 51–58 (1992).
Andriyash, Y. V. Use of Secale strictum in breeding disease-resistant rye. Plant Breed. Abstr. 59, 624 (1989).
Lapinski, B. Materiały do cytogenetyki żyta dzikiego (Secale montanum Guss.). Biul. Inst. Hod. Aklim. Roślin 3, 29–38 (1972).
Lapinski, B. Genetic resources of Secale species and their use in breeding. Proc. Conf. Rye Genet., 1–15 (1991).
Khush, G. S. & Stebbins, G. L. Cytogenetic and phylogenetic relationships among the species of Secale. Am. J. Bot. 48, 723–730 (1961).
Singh, R. J. & Röbbelen, G. Cytogenetics of interspecific hybrids in Secale. Caryologia 30, 275–284 (1977).
Stutz, H. Chromosomal translocations in rye hybrids. Genetics 42, 253–265 (1957).
Perrino, P., Hammer, K. & Hanelt, P. Ecogeographical differentiation and distribution of wild rye species. Plant Genet. Resour. Newsl. 59, 19–26 (1984).
Zohary, D. Studies on the origin of cultivated rye. Evolution 14, 444–450 (1960).
Hatmaker, E. A. et al. Chloroplast genome variation and evolution across the genus Pinus. BMC Genomics 21, 1–14 (2020).
Skuza, L. et al. Molecular structure and comparative analysis of the cp genome of weedy rye. Sci. Rep. 13, 5412 (2023).
Chen, Z. H. et al. Comparative chloroplast genomics reveals phylogeny and adaptive evolution of Triticeae. Front. Plant Sci. 12, 635133 (2021).
Saski, C. et al. The complete chloroplast genome sequence of Gossypium hirsutum: Organization and phylogenetic relationships to other angiosperms. BMC Genomics 8, 1–14 (2007).
Choi, K. S. & Park, S. The chloroplast genome of Aster spathulifolius (Asteraceae): Comparison to other Asteraceae and characterization of the triplication of rps12. PLoS ONE 10, e0119141 (2015).
Skuza, L., Filip, E. & Szućko, I. Intergenic Spacer Length Variability in Cultivated, Weedy and Wild Rye Species. Open Life Sci. https://doi.org/10.1515/biol-2015-0020 (2015).
Ogihara, Y. et al. Structural features of a wheat plastome as revealed by complete sequencing of chloroplast DNA. Mol. Genet. Genomics 266, 740–746 (2002).
Aagesen, L., Greilhuber, J., Košnar, J., Temsch, E. M. & Vít, P. AccD loss and variability in plastid genome structure across the Triticeae. Plant Syst. Evol. 254, 183–194 (2005).
Doyle, J. J. & Doyle, J. L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15 (1987).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Bankevich, A. et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
MFannot Tool. https://megasun.bch.umontreal.ca/cgi-bin/dev_mfa/mfannotInterface.pl (accessed on 6 May 2020).
Dong, X., Stothard, P., Forsythe, I. J. & Wishart, D. S. PlasMapper: A web server for drawing and auto-annotating plasmid maps. Nucleic Acids Res. 32, W660–W664 (2004).
Greiner, S., Lehwark, P. & Bock, R. OGDRAW version 1.3.1: Improved visualization of organelle genomes. Nucleic Acids Res. 47, W59–W64 (2019).
Mayer, C. Phobos Version 3.3.11. A tandem repeat search program. 2006–2010. http://www.rub.de/spezzoo/cm/cm_ phobos.htm (2019).
Sablok, G. et al. ChloroMitoSSRDB 200: More genomes, more repeats, unifying SSRs search patterns and on-the-fly repeat detection. Database 2015, av084 (2015).
Kurtz, S. et al. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642 (2001).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol. Biol. Evol. 34, 3299–3302 (2017).
Darling, A. E., Mau, B. & Perna, N. T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403 (2004).
Mower, J. P. The PREP suite: Predictive RNA editors for plant mitochondrial and chloroplast genes. Nucleic Acids Res. 37, W253–W259 (2009).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Huelsenbeck, J. P. & Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Androsiuk, P. et al. Comparative analysis of plastid genomes within the genus Chenopodium (Amaranthaceae). Int. J. Mol. Sci. 21, 4956 (2020).
Funding
This study was co-financed by the Minister of Science under the “Regional Excellence Initiative” Program for 2024–2027.
Author information
Authors and Affiliations
Contributions
Conceptualization, L.S.; methodology, L.S., R.G. and P.A.; writing—original draft preparation, L.S. and P.A.; writing—review and editing, L.S., A.K. and P.A.; bioinformatic analyses, P.A., Ł.P. and J.P.J.; supervision, L.S.; and funding acquisition, L.S. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Skuza, L., Androsiuk, P., Gastineau, R. et al. The complete chloroplast genome of Secale strictum ssp. strictum provides insights into Triticeae evolution and breeding. Sci Rep 15, 42882 (2025). https://doi.org/10.1038/s41598-025-27062-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-27062-1
